

Abstract
Although next generation sequencing has revolutionised the ability to associate variants with human diseases, diagnostic rates and development of new therapies are still limited by our lack of knowledge of function and pathobiological mechanism for most genes. To address this challenge, the International Mouse Phenotyping Consortium (IMPC) is creating a genome- and phenome-wide catalogue of gene function by characterizing new knockout mouse strains across diverse biological systems through a broad set of standardised phenotyping tests, with all mice made readily available to the biomedical community. Analysing the first 3328 genes reveals models for 360 diseases including the first for type C Bernard-Soulier, Bardet-Biedl-5 and Gordon Holmes syndromes. 90% of our phenotype annotations are novel, providing the first functional evidence for 1092 genes and candidates in unsolved diseases such as Arrhythmogenic Right Ventricular Dysplasia 3. Finally, we describe our role in variant functional validation with the 100,000 Genomes and other projects.
INTRODUCTION
With its extensive toolkit for genome modification and capacity for recapitulating human disease, the laboratory mouse is arguably the preferred model organism for studying and validating the effect of genetic variants in Mendelian disease, as well as identifying previously unsuspected disease genes. Null mouse mutations have hitherto been generated and described in the literature for approximately one-half of the genes in the genome1. However, hypothesis-driven phenotyping of these mutants reveals discoveries in areas that largely reflect the expertise and specific research questions of individual investigators. As a result, the extent of functional annotation, potential to fully uncover pleiotropy, and opportunity to exploit mutant mouse models for disease-agnostic interrogation is limited. Furthermore, the lack of replicability in knockout experiments is a well-documented challenge in drug target development, behavioural and other translational studies 2,3 and is commonly due to using poorly defined statistical methods, performing studies in only one sex, and practicing bias in animal selection4. The development of a comprehensive reference phenotype database employing fully validated, standardised and automated phenotyping procedures across all body systems that are applied to mutants of both sexes provides a robust dataset to corroborate disease-causing factors in humans.
The IMPC is creating just such a catalogue of mammalian gene function that systematically associates mouse genotype-to-phenotype data and enables researchers to formulate hypotheses for biomedical and translational research, and purpose-driven preclinical studies5,6. The IMPC adult phenotyping pipeline analyses cohorts of male and female knockouts on an isogenic C57BL/6N background from Embryonic Stem (ES) cell resources produced by the International Knockout Mouse Consortium comprising targeted null mutations with reporter gene elements7–9. Homozygotes are characterised, except in those strains (approximately 30%) where gene inactivation necessitates the use of heterozygotes to study mice that are embryonic/perinatal lethality or subviable10,11. The pipeline measures a total of 509 phenotyping parameters that encompass diverse biological and disease areas including neurological, behavioural, metabolic, cardiovascular, pulmonary, reproductive, respiratory, sensory, musculoskeletal, and immunological. Standardised and harmonised protocols developed by the IMPC are used to reduce phenotype variance across the centers and builds off experience from the pilot EUMODIC project where a limited 7% discordant phenotype rate was observed for a large set of 22 common reference mutant lines6. Rigorous data quality control is applied to the captured data from the 10 phenotyping centers and an automated statistical analysis pipeline (PhenStat12, see Methods) identifies mutants with statistically significant phenotype abnormalities.
In the current IMPC data release 5.0 (2nd August 2016), 3,328 genes have been fully or partially phenotyped, generating over 20 million data points and 28,406 phenotype annotations. Complementing the physiological, behavioural and structural phenotype datasets, the IMPC also provides annotated expression of LacZ data across multiple organ and tissue systems for 1413 genes13 and extensive histopathology analysis of adult tissues for 333 genes14. The IMPC portal is the single point of access to phenotype data, ES cell and Cas9-RNA-guided nuclease resources and mutant mouse strains. Sophisticated query interfaces of both gene and phenotype data are provided, as well as tools to visualise phenotypes encompassing quantitative, categorical, and image data15. Periodic data releases provide the latest genotype-phenotype associations.
In our current analysis, we identify 1) new mouse models for human Mendelian disorders with a known genetic basis, 2) uncover candidate disease genes for human Mendelian disorders where to date only a genomic location is associated, and 3) identify new mouse disease models involving genes with previously little or no functional annotation. A summary of the results is presented in Figure 1 and described in more detail in the following sections.
Figure 1. IMPC Mutant Models of Human Disease and Gene Function.
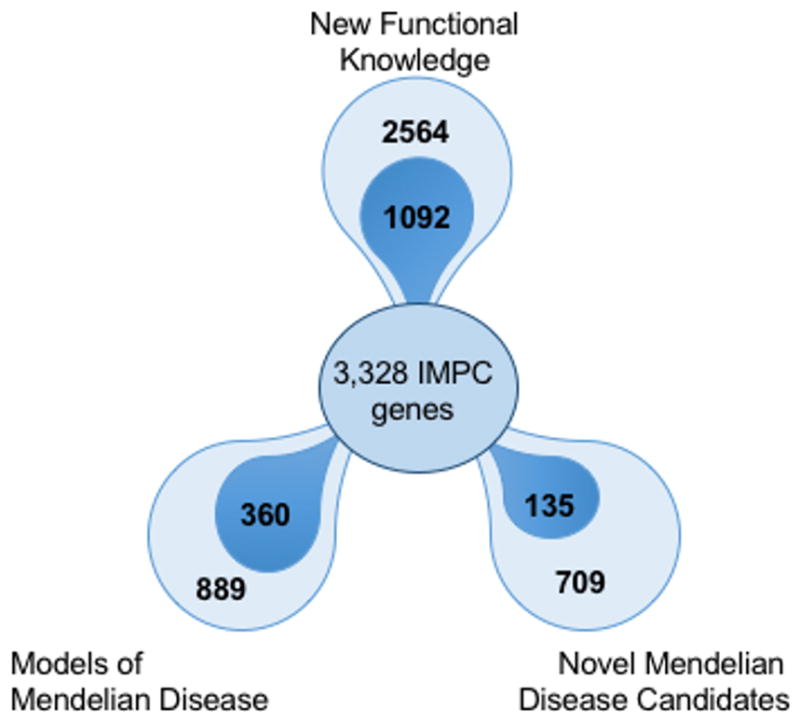
Human disease models were identified by measuring the degree of phenotype similarity between IMPC null mutant mouse strains and their orthologous human disease genetic loci. Models of Mendelian Disease- of 889 potential disease models, 360 mutant strains had both phenotype overlap and an orthologous null allele to diseases with known mutations as described in OMIM and Orphanet; Novel Mendelian Disease Candidates- 135 strains had phenotype overlap and null alleles syntenic to linkage or cytogenetic regions associated with human diseases with unknown molecular mechanisms; New Functional Knowledge- of 2564 genes with a non-lethal IMPC phenotype, IMPC data provide the first functional experimental evidence for 1092 of these genes based on Gene Ontology Annotation.
RESULTS
Comparison of IMPC findings with previous knowledge
We first investigated how concordant our phenotype annotations are with previously reported data for mouse lines involving the same genes. 621 genes assessed by us have previous mouse models annotations from literature review of knockout lines by the Mouse Genome Informatics (MGI) group1. An assessment of the corresponding 2547 MGI gene-phenotype associations that have already been assessed by an IMPC procedure revealed 958 (38%) were detected, with 62% (385 out of 621) of the genes having at least one phenotype reproduced (Supplement Table 1). This is in line with previous reports describing reproducibility of biomedical models16. Non-reproduced phenotypes could be due to several factors including different genetic backgrounds and variations in experimental technique and statistical methods e.g. evidence for previously reported increased circulating glucose for Gad2 mice is seen in our data (see URLs) but not considered statistically valid by our robust methods. There are also an additional 10,068 MGI phenotypes for these genes that we have not assessed despite our best efforts to be as broad-based as feasible in the context of a high-throughput project, or that require a different type of allele to be introduced to observe the effect. However, we show below that our pipeline covers all the major disease areas, MGI’s literature curation of over 1300 publications published to date using our resources is generating numerous additional annotations, and upcoming changes to our pipeline such as phenotyping a subset of genes at later ages (12–18 months) and new behavioural tests will increase our coverage. Furthermore, we generate extensive new knowledge, as discussed further below, with 90% (8984 of 9942) gene-phenotype annotations described by IMPC having not been described in the literature before.
Models of Human Mendelian Disease
The high volume and complexity of data produced by the IMPC presents challenges for finding relevant human disease models. To facilitate discovery, we developed a translational pipeline to automatically detect phenotype similarities between the IMPC strains and over 7000 rare diseases described in the Online Mendelian Inheritance in Man (OMIM)17 and Orphanet databases18. The pipeline utilises the human phenotype ontology (HPO)19 annotations for rare diseases maintained by the Monarch Initiative20 and our Mammalian Phenotype Ontology (MP)21 annotations of phenotype abnormalities and the PhenoDigm algorithm, also developed by the Monarch Initiative22. The results provide a quantitative measure of how well an adult mouse model recapitulates clinical features of a disease and is based on previous work that demonstrated superior identification of disease models compared to defining mouse strains solely by orthology or by other methods of calculating phenotype similarity22.
From the ~15% of mouse protein-coding genes phenotyped thus far by IMPC, 889 known rare disease-gene associations represented within OMIM and Orphanet have an orthologous IMPC mouse strain and display at least one phenotype (Supplement Table 2). By comparing human and mouse phenotypes, our automated pipeline identified 185 adult disease-gene associations where the IMPC mutant mouse strain modelled the human disease, with the majority (134) involving genes that have not had a mouse generated before or reported as a model of that disease from the curation efforts of MGI (Table 1, Supplement Tables 2 & 3). Each of the 889 associations had a mean of 14.7 ± 27.8 (SD) candidate genes for the disease from the algorithm, with a median rank of 3 for the true associated gene in the 185 sets of recalled associations.
Table 1. Frequency of IMPC Models that correspond to Mendelian Disease-Gene Associations in OMIM or Orphanet.
650 known rare disease-gene associations covered by OMIM and Orphanet have a phenotyped IMPC strain involving the orthologous mouse gene. The PhenoDigm automated pipeline and manual curation approaches identified matching phenotypes between mouse strains and human disease. A correspondence between the human disease and mouse model was defined when at least one of the human clinical phenotypes was recapitulated by the IMPC line. Novel models were defined when MGI contains no curated mouse line or literature asserted disease model for the gene. The manual lethality matching category corresponds to IMPC mutant strains for which homozygosity produced embryo or neonatal lethality/subviability and matched reports of human lethality/subviability in the OMIM/Orphanet summaries (see methods).
| Category | Frequency |
|---|---|
| Automated IMPC Disease Model (novel only) | 134/889 (15.1%) |
| Automated IMPC Disease Models (all) | 185/889 (20.8%) |
| Additional Manual Lethality IMPC Disease Models (all) | 175/889 (19.7%) |
| Total IMPC Disease Models (all) | 360/889 (40.5%) |
The range of human Mendelian diseases with matching mouse phenotypes was broad and included multiple biological systems (Table 2). Three examples of new mouse models first reported here (Figure 2) are for Bernard-Soulier syndrome, Type C (OMIM:231200), Bardet-Biedl syndrome-5 (OMIM:615983) and Gordon Holmes syndrome (OMIM:212840). Bernard-Soulier syndromes are bleeding disorders that result from mutations in genes encoding protein products of the glycoprotein Ib (GP Ib) complex that serves as the platelet membrane receptor for von Willebrand factor. GP Ib is composed of 4 subunits encoded by 4 separate genes: GP1BA, GP1BB, GP9, and GP5 with mutations in all these genes being associated with an autosomal recessive disorder characterised by prolonged bleeding times, enlarged platelets, an inability to clot, and incomplete penetrance of thrombocytopenia23. Gp9tm1.1(KOMP)Vlcg null homozygotes have a decreased number of platelets with a larger cell volume (Figure 2A,B), recapitulating key features of the disease and adding evidence that the point mutations associated with disease in humans lead to a functionally null complex. Bardet-Biedl syndromes (BBS) are heterogeneous autosomal recessive ciliopathies characterised by retinitis pigmentosa, obesity, kidney dysfunction, polydactyly, behavioral dysfunction, and hypogonadism. The disorder is associated with no fewer than 19 genes whose products form the BBSome, a protein complex involved in signaling receptor trafficking within cilia, which may also have functions not involving cilia24. Twenty mutations that include splice site, missense/nonsense, insertion, indels and deletion mutations within the BBS5 gene account for 4% of all BBS cases. Bbs5tm1b(EUCOMM)Wtsi null mice exhibit abnormal retina morphology resembling the retinal dystrophy observed in Bardet-Biedl syndrome patients. Other phenotypes were also observed in null mice recapitulating many hallmarks of BBS including obesity (Figure 2C,E) as well as other features such as impaired glucose homeostasis (Figure 2D). Gordon Holmes syndrome is another autosomal recessive disorder characterised by hypogonadism as well as cerebellar ataxia that has been associated with RNF21625,26. Male infertility was observed in Rnf216tm1b(EUCOMM)Wtsi homozygous null mice with histopathology identifying seminiferous tubule degeneration and atrophy characterised by diffuse absence of most or all germ cells and presence of occasional multinucleated spermatids with pyknotic nuclei within tubules that are lined by vacuolated Sertoli cells. Seminiferous changes were accompanied by diffuse interstitial cell (Leydig cell) hyperplasia. The epididymis was devoid of spermatozoa (epididymal aspermia) (Figure 2F).
Table 2.
Examples of IMPC Disease Models Across Diverse Biological Systems
| Biological system | Disease Gene | Human Mendelian disease | Relevant Human Phenotype | Overlapping Mouse phenotype |
|---|---|---|---|---|
| Bone | SCARF2 | Van Den Ende-Gupta Syndrome | Long metacarpals | increased length of long bones |
| Cardiovascular | LMNA | Cardiomyopathy Dilated 1a | Dilated cardiomyopathy | increased heart weight |
| Craniofacial | MSX1 | Orofacial Cleft 5 | Cleft palate | Cleft palate |
| Embryo | PSPH | Phosphoserine Phosphatase Deficiency | Intrauterine growth retardation | abnormal embryo size |
| Growth/Body size | GHRHR | Isolated Growth Hormone Deficiency, Type Ib | Short stature | decreased body length |
| Hearing | SLC52A2 | Brown-Vialetto-Van Laere Syndrome 2 | Sensorineural hearing impairment | increased or absent threshold for auditory brainstem response |
| Hematopoietic | GP9 | Bernard-Soulier Syndrome | Thrombocytopenia | Thrombocytopenia |
| Metabolism | KCNJ11 | Diabetes Mellitus, Noninsulin-Dependent | Type II diabetes mellitus | Impaired glucose tolerance |
| Muscle | COL6A2 | Bethlem Myopathy | Distal muscle weakness | Decreased grip strength |
| Neurological | GOSR2 | Epilepsy, Progressive Myoclonic, 6 | Difficulty walking | abnormal gait |
| Reproductive System | RNF216 | Gordon Holmes Syndrome | Infertility | male infertility |
| Retina | BBS5 | Bardet-Biedl Syndrome 5 | Rod-cone dystrophy | abnormal retina morphology |
Figure 2. New Mouse Models for Mendelian Human Disease.

Gp9- Bernard-Soulier syndromes are bleeding disorders that result from mutations in the glycoprotein Ib platelet membrane receptor complex. Gp9tm1.1(KOMP)Vlcg homozygotes have abnormal platelet development represented by an increased platelet volume (A; box plots representations throughout represent first and 3rd quartiles with the line indicating the median and whiskers representing the min and max values. Female control=479, female homozygous=8, male control=428, male homozygous=8; linear mixed-effects model without Weight; p=0) and decreased platelet numbers (B; Female control=439, female homozygous=8, male control=428, male homozygous=8; linear mixed-effects model without Weight; p= 2.31E-06). Bbs5- BBS5 is associated with Bardet-Biedl syndrome (BBS), a ciliopathy with multisystem involvement with severe and early-onset of symptoms. Bbs5tm1b(EUCOMM)Wtsi homozygotes display profoundly increased body fat percentage (C; Female control=1276, female homozygous=8, male control=1296, male homozygous=8; linear mixed-effects model without Weight; p=1.99E-11) and impaired glucose tolerance as shown by the time series box plot (D; blood glucose levels at time points after 16 hours fasting followed by intra-peritoneal (IP) glucose injection. Female control=491, female homozygous=8, male control=509, male homozygous=8; linear mixed-effects model without Weight; p= 2.85 E-07). Whole body X-ray visualization of Bbs5 homozygous and control, showing increased body fat in mutant animals (E). Rnf216 - Gordon Holmes syndrome is associated with RNF216 and is characterised by hypogonadism and cerebellar ataxia. Rnf216tm1b(EUCOMM)Wtsi homozygous null male mice are infertile. Histopathology images at 20x magnification show seminiferous tubule degeneration and atrophy with Leydig cell hyperplasia (Fa) and epididymal aspermia (Fb) in null mice compared to unaffected seminiferous tubules (Fc) and epididymis (Fd) in control mice.
From the 704/889 known associations where we did not detect an IMPC model, 48 have not yet been tested in the mouse for a phenotype that could recapitulate any of the clinical phenotypes, leaving 656 (74%) associations where our automatic algorithm did not detect a potential disease phenotype from the IMPC pipeline. To evaluate the sensitivity of the automated human-mouse phenotype matching, we manually evaluated 100 randomly chosen examples of these missed associations (Supplement Table 4), leading to 12 additional discoveries where the phenotype matches fell below the similarity threshold used in our algorithm e.g. the decreased startle reflex match for deafness. PhenoDigm is optimised to maximise precision and recall (Supplementary Figure 1) and reducing our threshold to detect such matches would introduce many false positives. Manual assessment is not feasible in the long term given the ever-increasing number of strains, and new data for existing strains, but future implementations will incorporate histopathology data to increase recall as seen by the additional model detected by manual assessment.
Human Mendelian disease is caused by a variety of complete loss, partial loss or gain-of-function mutations under various modes of inheritance. The IMPC only phenotypes the null allele in a homozygous state or, if embryonic/perinatal lethal, in the heterozygous state. Thus, IMPC mouse strains are suitable for putative disease gene, as opposed to variant identification e.g. for identifying which genes expressing variants of unknown significance are pathogenic. The 889 human diseases associated with genes orthologous to the IMPC mouse strains were inherited with roughly equal frequency by autosomal dominant (AD) or recessive (AR) genetics (n = 379 AD vs 423 AR, and 87 unknown/X-linked). The frequency of inheritance by AD and AR genetics was also equivalent for the 185 adult disease-gene associations where the IMPC mutant mouse line modelled the human disease (n = 82 AD vs 94 AR, and 7 unknown/X-linked). This indicates that the mouse models were effectively modelling human disease independent of the mode of human inheritance. Human AR disease is likely a consequence of a complete or partial loss-of-function mutation where haploinsufficiency is not adequate to produce symptoms. As would be expected, AR human disease was more frequently modelled by homozygous null mouse mutants: 65% (61/94) of the AR models were viable and phenotyped as homozygous mice, while 35% (33/94) were subviable/lethal as homozygotes and therefore heterozygous mice were phenotyped. AD inheritance can be attributed to either haploinsufficiency or gain-of-function mutations and we found that 46% of the dominant human mutations were modelled by heterozygous mutants in the mouse, consistent with a haploinsufficient mechanism for almost half of the diseases.
Interestingly, 227 of the 423 (54%) tested AR associations were homozygous lethal/subviable in the mouse leading us to consider whether early mortality occurred in these patients or would have occurred without extensive medical intervention. Lethality matches are not detectable by our automated algorithm, as human lethality is rarely recorded in the disease HPO annotations, and for 74 of the 889 associations (8%), homozygous lethality is the only mouse phenotype we have detected so far. To address this, we manually investigated whether the associations involving mouse homozygous lethal/subviable strains were associated in OMIM/Orphanet with human embryonic or early death (< 2 years) or with severe, early onset disorders in patients not likely to survive through puberty without significant medical support e.g. cleft palate is a lethal phenotype in mice but easily treatable in humans. This uncovered a further 97 new mouse/human disease associations (Supplement Table 2, column J annotated with Y-L) where human lethality was recorded and another 78 where the disease would probably have been lethal without medical intervention (Supplement Table 2, column J annotated with Y-PL). The majority of these lethality matches were inherited with AR genetics (73%, 122 of the 166 diseases with reported inheritance from OMIM/Orphanet) and modelled by homozygous mouse mutants, consistent with the conclusion that homozygous loss of function mutations in essential genes in humans produces either early death or severe congenital medical conditions requiring advanced medical support for survival. Examples of mouse and human embryo/early-onset lethality include: ventriculomegaly with cystic kidney disease that results in in utero or neonatal human fatality (OMIM:219730, gene: CRB2) and Stuve-Wiedermann Syndrome (OMIM:601559, gene: LIFR). Diseases that would probably have been lethal without medical support with a corresponding lethal/subviable mouse strain include: Coach Disease (OMIM:216360, gene: RPGIP1l), Meier Gorlin Syndrome 1 (OMIM:224690, gene: ORC1), and Human phospherine phosphatase deficiency (OMIM:614023, gene: PSPH). In the latter, a homozygous lethal mouse mutant in Psphtm1b(EUCOMM)Wtsi has structural abnormalities at Embryo day (E) 15.5 detected by micro-computed tomography (micro-CT) that closely resemble the developmental and structural defects in the human phosphoserine phosphatase deficiency patients (Figure 3).
Figure 3. Psph.

Phosphoserine phosphatase deficiency (OMIM: 614023) is an autosomal recessive disorder characterised by prenatal and postnatal growth retardation, psychomotor retardation and facial dysmorphologies with the severity of the symptoms requiring medical support for survival. Complete preweaning lethality was observed in Psphtm1.1(KOMP)Vlcg homozygous null mice. Pup number, genotypes and sex ratios of heterozygous intercrosses were set to generate cohorts for phenotyping. No homozygous pups were observed whereas respectively 66% (54/82) and 34% (28/82) were produced (A; # of pups, asterisks indicate no surviving homozygotes). LacZ reporter expression regulated by the Psph promoter in asymptomatic heterozygous E12.5 embryos shows extensive gene expression (B; bar 1mm). Gross images of E15.5 homozygous mutant embryos confirmed growth retardation, haemorrhage, and facial dysmorphologies (C; bar 5mm). Imaging of E15.5 embryos by microCT showed significant growth retardation, as well as facial dysmorphologies consistent with the human Mendelian disorder (D).
When we include these manually curated lethality matches, 40.5% (360) of the disease models have phenotype overlap with the 889 disease genes (Table 1) with the majority (78%; 279 of 360) being the first report of a candidate mouse model for these diseases. The discovery rate of disease models in our analysis is comparable to previous reports on smaller high-throughput mouse phenotyping studies that found modelling of 46% of 59 and 33% of 42 associations using manual investigation of data6,27.
Where we did not detect a model despite testing for at least one equivalent phenotype (54%; 484), explanations could range from differences in human and mouse biology, the genetic background and a null allele not being appropriate to model the disease, or differing methodologies used for annotation e.g. rarely observed phenotypes for a disease are often recorded in the HPO annotations and would likely fall below the statistical threshold if similarly, non-penetrant in mice. Finally, there is a slight possibility that some earlier alleles may have influenced disease modelling where a hypomorph rather than null is possible (90 tm1a) or a retained neomycin cassette may have altered expression of genes in cis (90 tm1a, 10 KOMP1).
New Functional Knowledge and Mendelian Disease-Gene Candidates
The second major clinical use case for the IMPC’s data is providing new data on the phenotypes and functions of genes. IMPC has prioritised genes with no known disease associations or minimal GO annotation to address this. Based on MGI’s literature curation of mutant strains involving any allele type except conditional mutations, 1830 of the 3,328 genes phenotyped in this IMPC release have never had a mouse produced before. No Gene Ontology (GO) molecular function or biological process annotations are available for 189 genes, while another 903 genes had inferred annotations from computational analysis (Figure 4A)28. The phenotypes of these mutant strains provide substantive insights into the function of a large class of genes (sometimes described as the ignorome)29 for which there is little or no existing functional information (Supplement Table S5).
Figure 4. Novel Mouse Models of Disease –

Over 40% of IMPC strains are for genes that lack experimental evidence for function according to the Gene Ontology Consortium (A in grey). Fam53b - Fam53btm1b(EUCOMM)Hmgu homozygous mutant mice had significantly decreased red blood cell counts (B; box plot representations throughout represent first and 3rd quartiles with the line indicating the median and whiskers representing the min and max values and asterisks indicating a significant difference between mutant and same sex controls using the mixed model with a p < 0.0000. Female control=597, female homozygous=8, male control=635, male homozygous=8; linear mixed-effects model without Weight; p=2.81E-11), and enlarged erythrocytes (C; Female control=598, female homozygous=8, male control=634, male homozygous=9; linear mixed-effects model without Weight; p=0), consistent with Diamond-Blackfan Anemia (DBA, OMIM: 105560). Dnajc5b - Dnajc5btm1b(EUCOMM)Hmgu homozygous mutants displayed significantly shortened QT interval as measured by electrocardiogram (D; Female control=7, female homozygous=6, male control=7, male homozygous=8; generalized least squares without weight; p=7.41E-08), supporting a role for DNAJC5b variants associated with human variability to statin effects on cardiovascular incident frequency.
Examples of candidate genes for human Mendelian disease with previously little functional information include Family with sequence similarity 53 member B (Fam53b), which had no reported phenotypic variants in human or mouse. The gene is differentially expressed in adult definitive erythrocytes compared to primitive erythrocytes with a >6-fold log2 change as shown in the Expression Atlas (see URLs)30,31. Homozygous Fam53btm1b(EUCOMM)Hmgu knockout mice showed increased mean corpuscular hemoglobin and decreased erythrocyte cell number (Figure 4B,C), suggesting the gene is involved in hematopoiesis and is a candidate for macrocytic hyperchromic anemias. PhenoDigm identified this gene as a phenocopy for Diamond-Blackfan Anemia (DBA; OMIM:105650), a group of fifteen unique anemias generally attributable to defects in ribosome synthesis but for which known mutations only account for approximately 54% of all DBA patients32. A single functional study suggested that Fam53b is required for Wnt signaling, which is a key step in determining cell fate, cell proliferation, stem cell maintenance and anterior-posterior axis formation33. The Fam53b knockouts thus implicate a new candidate pathway to be considered for the 46% of DBA patients where genetic causes are not known.
As well as providing fundamental insights into the function of genes with little or no previous functional annotations, the phenotype analyses are also identifying numerous new candidate disease models that may provide a foundation for relating gene function to disease phenotype. This new data and biological resources may be used to detect novel genotype to phenotype associations in disease where simply considering existing human data would lead to causative variants being overlooked among the overwhelmingly abundant associated variants of unknown significance, as happens in many exome sequencing studies: over half of diagnosed rare diseases still have no known causative gene and diagnostic rates in most high-throughput Mendelian disease sequencing projects are 20–30%, largely due to a lack of functional information for most genes. To remedy this and to start achieving better diagnostic rates we can utilise the data that the IMPC provides. As a demonstration of the potential of IMPC data for novel disease gene discovery, we identified candidate genes for Mendelian diseases with an unknown molecular mechanism but where a broad genetic localization was available in OMIM from previous studies. Our disease matching algorithm identified 135 associations where our predicted disease gene falls within these loci (Supplement Table S6).
For example, adult mice heterozygous for the Klhdc2tm1b(EUCOMM)Hmgu allele have a complex syndrome of abnormalities including altered ECG findings. The phenotypes match the clinical signs described for the autosomal dominant disease Arrhythmogenic Right Ventricular Dysplasia 3 (ARVD3; OMIM:602086) that presents with cardiac arrythmias caused by fibro-fatty replacement of right ventricle myocardium. Klhdc2 is syntenic with the ARVD3 locus, suggesting it as a candidate gene. Usmg5 null mice recapitulated the clinical symptoms of muscle weakness and abnormal gait seen in patients with the dominant intermediate A form of Charcot-Marie-Tooth disease. The human orthologue (USMG5) is located within a 9.8Mb critical region identified in patients with this disease. While the implicated human loci are sometimes Mbps in length and encompass hundreds of genes, these examples illustrate how IMPC phenotype data allows for the scoring of candidate genes with disease causing variants and has important implications for current rare disease genetic projects that are using next generation sequencing technologies.
DISCUSSION
By analysing phenotype similarities between IMPC’s mouse strains and human disease, we have provided new disease models and identified novel functional knowledge for a significant and growing proportion of protein coding genes. These models are made readily available and can be exploited to study disease mechanisms, develop new gene therapy and pharmacological treatments, and further our understanding of gene function. The novelty of the IMPC is both the scale of the vision to produce the first comprehensive catalogue of mammalian gene function across all genes as well as the non-hypothesis driven, standardised approach to the phenotyping facilitating novel discoveries about the function of genes and their role in diseases as highlighted above.
The potential of IMPC phenotype comparisons for prioritising candidates in human Mendelian syndromes is made accessible to clinical researchers performing next-generation sequencing based diagnostics through inclusion within the Exomiser software package that combines an assessment of variant pathogenicity with gene candidacy based on similarity of the patients’ phenotypes to known phenotypic knowledge from human, mouse and fish34. Exomiser is being applied within the NIH Undiagnosed Disease Program and Network (UDN)35 as well as the 100,000 Genomes Project that embeds genomics into a national healthcare system.
The IMPC goes beyond modelling Mendelian syndromes by leveraging its existing global infrastructure to address complex biological questions. IMPC mouse strains are widely used with over 1300 citations across every major biological system (see URLs) including SNP validation studies for complex traits in both humans and mice36–41. Such studies will be supported by ongoing work using the IMPC phenotyping pipeline to characterise the eight founder inbred mouse strains for the collaborative cross (CC) resource used in the study of complex traits and in targeted non-coding mutant mice strains (11 miRNA, 4 lincRNA in the current data release) to study regulatory activities. Other collaborative, multi-centre, efforts are using IMPC mouse strains to study gene function in hearing, vision, metabolism, and pervasive sexual dimorphism. Starting this year, a significant fraction (~15%) of mutant strains will be re-phenotyped at 12–18 months of age to identify genes involved with late-onset disease.
A major change in our strategy has been the adoption of CRISPR based methods to increase production rate and the opportunity to characterise strains containing the same single nucleotide variants or indels as human patients e.g. the MRC Genome Editing Mice for Medicine service (GEMM) is characterising patient variants identified through whole genome sequencing by the 100,000 Genomes Project to functionally validate variants of unknown significance and/or facilitate mechanistic and therapeutic studies in collaboration with the clinicians and researchers. Generation of these precision models is key to addressing the issue of new therapies for rare disease lagging behind the discovery of new disease genes. The approach will be expanded in the coming years to characterization of candidate non-coding, regulatory variants in undiagnosed 100,000 Genomes Project cases as well as other large-scale sequencing projects such as the UDN. We believe many of the lessons we have learned establishing the IMPC will also be of value to the recently launched precision medicine efforts whose goals are to improve treatment through the customisation of healthcare based on a patient’s genomic information and environmental factors. Harmonisation of phenotype traits captured in diverse formats across multiple centers will be critical to the stratification of disease populations for improved treatment as well as using model organism data to better identify causal disease gene variants.
While advances in CRISPR and induced-Pluripotent Stem cells (iPSc) technologies have now vastly expanded the researchers’ toolkit, the work of the IMPC highlights the continuing importance of mouse models to understanding disease mechanisms. Mice are vertebrate mammals with physiological characteristics that recapitulate all major human biological systems, allowing study of processes not possible with in vitro studies including the impacts of behavioral, inflammatory, endocrine, and gender-specific processes on disease. While CRISPR-based methodologies now allow for genome engineering in nearly every species, mice have other characteristics that have made them a widely used model organism for over a century. Inbred mouse strains such as the C57BL/6N strain used by the IMPC have standardized, uniform genetic backgrounds that reduce phenotypic variability, with most strains having a 2-year lifespan that allows for comprehensive studies in a timely manner.
Reproducibility of results in translational studies is a significant issue and we found the overlap of phenotypes between IMPC mouse strains with previously published mutant strains are in line with other studies investigating reproducibility. This highlights the importance of high-quality phenotype annotation of human clinical records and mouse phenotypes, and demonstrates the importance of open sharing of data. Towards this, the IMPC adheres to the ARRIVE guidelines for reproducibility of animal model experiments including making all data available and having transparent statistical analysis via free distribution of our PhenStat software12.
In conclusion, the IMPC has established an ever-expanding knowledgebase of mammalian gene function, a large resource of novel disease models and the capacity for functional validation of variants identified in disease sequencing projects that will be of great value to the human disease community.
ONLINE METHODS
Mouse Production
Targeted ES cell clones obtained from the International Knockout Mouse Consortium (IKMC) resource7,42 were injected into mouse blastocysts for chimera generation. The resulting chimeras were mated to C57BL/6N mice, and the progeny were screened to confirm germline transmission. Following the recovery of germline-transmitting progeny, the majority of strains (82%) were crossed with a coisogenic C57BL/6N transgenic strain bearing a germ-line expressing Cre recombinase to excise the floxed neomycin selection cassette (neo) and critical exon for EUCOMM alleles) and generate a true deletion. For the rest, the requirement early on for establishment and testing of the pipeline without additional breeding meant lines were characterised that contained either tm1a alleles (16%: rely on a stop codon that could potentially be spliced around and retain the neo cassette that can alter transcriptional activity of other genes in cis) or the KOMP1 allele (2%; retain neo cassette). The resulting C57BL/6N heterozygotes were intercrossed to determine viability and generate homozygous mutants. All strains are made accessible from the IMPC portal.
Mouse Phenotyping and Experimental Design
Based upon previous analysis on appropriate sample sizes to detect significant effects by our statistical framework (see below), a minimum of seven male and seven female mice were characterised from 9 weeks of age until 16 weeks of age using a broad-based phenotyping pipeline that assessed every major biological system. IMPC centers employed a common control strategy where cohorts of age-matched, wild-type C57BL6/N mice are phenotyped in a continuous manner alongside mutant C57BL6/N strains. These cohorts are used in quality control (e.g. baseline drift) and in statistical analysis of the data. A centralised database of consensus IMPC standard operating procedures (SOPs), IMPReSS, (see URLs) ensured that all phenotyping data and metadata are collected in a reproducible and standardised format. Cohorts of at least seven homozygous mice of each sex per line were generated. If no homozygotes were obtained from 28 or more offspring of heterozygote intercrosses during production, the strain was scored non-viable. Similarly, if less than 13% of the pups resulting from intercrossing were homozygous, the strain was scored subviable. For non-viable and subviable strains, heterozygous mice were committed to the phenotyping pipelines. The individual mouse was considered the experimental unit within the studies.
Data quality control (QC)
Defined criteria were established for QC failures (e.g. insufficient sample, incorrect instrument calibration) and detailed within IMPReSS to provide valid reasons for discarding data. A second QC cycle occurred when data was uploaded from the phenotyping center to the IMPC Data Coordination Centre (DCC) using an internal QC web interface. Data was only QC failed from the dataset if clear technical reasons were identified for a measurement being an outlier and this was tracked within the database.
Wild-type–knockout comparisons
Wild-type vs null comparisons, i.e. a dataset, were restricted to data collected at one centre and were assembled by selecting data from knockout and wild-type mice that had data collected from the same versioned protocols and with the same metadata parameters (e.g. instrument). As wild-type mice are measured every week, a null strain is generally compared to data from hundreds of wild-type control mice. In the case when all members of a null mouse strain are measured on the same day with an equal number of control mice, the comparison is restricted to this smaller set of data to eliminate batch effects.
A dataset consists of the collection of data values (mutant and control) for a single measured variable (parameter) with the same allele, zygosity, center, and experimental metadata. Using IMPC data release version 5.0, (2nd August 2016), IMPC has analysed 352,729 continuous datasets and 944,270 categorical datasets produced from 10 phenotyping centres. These raw data are available at the IMPC web portal with a page detailing the various methods by which data can be extracted (see URLs).
Statistical analysis
Statistical analysis was performed using PhenStat R package developed for IMPC. PhenStat is a statistical analysis tool suite that accounts for known variation in experimental workflow and design of phenotyping pipelines12. Briefly, categorical data analysis was completed using a Fisher’s Exact test. Continuous data analysis was performed using PhenStat Linear Mixed Model framework (see URLs) which uses linear mixed models that treat batch as a random effect. Through high throughput phenotyping programs, such as EUMODIC, where data was systematically collected on one genetic background, the significant sources of variation can be identified and it became obvious that batch (defined here as those readings collected on a particular day) can lead to large variation in phenotyping variables43. Linear mixed models (LMM) include a class of statistical models that are suited to modelling multiple sources of variability on a phenotype such as batch effects. Details of the implementation including decision tree model, descriptions and the lower FDR rates associated with multi-batch data are available in the PhenStat package user’s guide (see URLs), and described in the literature43. For this analysis, results from one batch, low batch (mutants measured in batches between 2–4 times, and multi-batch (5 or greater) experiments were used. For viability and fertility data, the center conducting the experiment used a statistical method appropriate for the breeding scheme utilised at that center (exact details are available on the IMPC data portal) and supplied the analysis results to the IMPC DCC. All available wild-type and mutant mice were used in the analysis with center-specific blinding strategies during group allocation, no specific inclusion/exclusion criteria, and no randomisation approach beyond relying on Mendelian inheritance to randomise as detailed in our ARRIVE guideline document (see URLs). All analysis presented in this publication is based on the binary assignment of a significant deviation (or not) from wild-type and the associated phenotype term. Detailed output of our statistical analysis for each test is presented on our portal pages (mousephenotype.org) including all raw data, the summary, visualisations, variance and calculated p-value for the genotype being associated with the phenotype.
Matching Mouse Phenotypes to OMIM and Orphanet Disease Descriptions: Automated PhenoDigm
We utilised the Human Phenotype Ontology (HPO) annotations available from the Monarch Initiative (Accessed 2nd September 2016) describing the clinical phenotype features of over 7,000 diseases reported in OMIM17 and Orphanet18. These HPO terms were semantically compared with the phenotype features (MP annotations) of IMPC mouse strains using the PhenoDigm algorithm22, developed by us and fellow members of the Monarch Initiative as reflected in authorship, to generate an overall score for how phenotypically similar a mouse strain is to a particular disease. PhenoDigm calculates the individual score for each HPO-MP phenotypic match based on the proximity of the two terms in the overall cross-species ontology (Jaccard index; simJ) and the observed frequency of the phenotype in common from the overall disease and mouse annotations (Information Content; IC) i.e. exact clinical and mouse phenotype matches involving rarely observed phenotypes score highest. The geometric mean of the IC and simJ is used to generate the HPO-MP pairwise score. The overall PhenoDigm percentage score is a comparison of the best and mean scores for all the pairwise HPO-MP comparison relative to the maximum possible scores for a perfectly matching mouse model to that disease. The disease models described in this paper were selected by applying a threshold of at least one HPO-MP match with a score greater than 1.35 which maximised precision and recall compared to other similarity thresholds of 1.0, 1.25, 1.5, or 1.75 (Supplementary Figure 1).
Known human genes and regions associated with diseases were extracted from OMIM and Orphanet and matching mouse orthologues were identified from HomoloGene44. Comparisons to previous mouse mutants from the MGI resource1 were achieved by download and processing a file named MGI_GenePheno.rpt containing literature curation of mouse lines associated with all allele types except those involving conditional mutations and ALL_OMIM.rpt which curates any literature assertions of a particular mouse line being a mouse model of a particular OMIM disease (see URLs and downloaded 2nd September 2016).
Lethality Matching
Screening for lethal or potentially lethal genes from data within the OMIM database could not be automated. For the set of mouse genes that were homozygous pre-weaning subviable or lethal, and also had OMIM records, we manually inspected the OMIM records to identify those with reported in utero or early deaths (prior to two-years of age) and coded these in Supplementary Table 1 as Yes-Lethal (Y-L) indicating that for some human cases with mutations for these genes, the phenotype of human lethality matched the phenotype of mouse sub-viability. We also screened for OMIM records with severe congenital defects and/or rapid progression of early onset severe disease in human patients requiring significant medical support for survival. Mice with similar phenotypes would not be likely to survive through weaning in the absence of medical support and therefore were scored as Yes-Probable Lethal (Y-PL) indicating a probable match of the human phenotype with the mouse subviable phenotype.
Matching Candidate Gene Phenotypes to Human Traits from OMIM Linkage and Cytogenetic Findings
Diseases with no known molecular mechanism but a narrowed down cytogenetic region containing the likely causative gene were extracted from OMIM (downloaded 2nd September 2016). Ensembl was used to identify the human genes contained within these regions and their mouse orthologues retrieved from HomoloGene. The overlap between these genes and candidates from the PhenoDigm analysis of the same disease were then flagged within our database and are highlighted on both our portal as well as the supplementary tables presented here.
Identifying Novel Gene-Phenotype Relationships from the IMPC Database
An online tool on the IMPC portal (see URLs) imports GO annotations daily from the Quick GO resource45 and categorises them based on the evidence codes assigned by GO curators. Annotations were analysed on 24th March 2017. We started with 2668 genes that had IMPC non-lethal phenotypes. Categories incorporate the following evidence codes:
Experimental: Inferred from Experiment (EXP), Inferred from Direct Assay (IDA), Inferred from Physical Interaction (IPI), Inferred from Mutant Phenotype (IMP), Inferred from Genetic Interaction (IGI), Inferred from Expression Pattern (IEP)
Curated computational: Inferred from Sequence or structural Similarity (ISS), Inferred from Sequence Orthology (ISO), Inferred from Sequence Alignment (ISA), Inferred from Sequence Model (ISM), Inferred from Genomic Context (IGC), Inferred from Biological aspect of Ancestor (IBA), Inferred from Biological aspect of Descendant (IBD), Inferred from Key Residues (IKR), Inferred from Rapid Divergence(IRD), Inferred from Reviewed Computational Analysis (RCA)
Automated electronic: Inferred from Electronic Annotation (IEA), Other: Traceable Author Statement (TAS), Non-traceable Author Statement (NAS), Inferred by Curator (IC)
No biological data available: No biological Data available (ND), Not listed as a gene in GO (no evidence code)
Ethical approval
Mouse production, breeding, and phenotyping at each center was done in compliance with each centers’ ethical animal care and use guidelines in addition to their applicable licensing and accrediting bodies, reflecting the national legislation under which they operate. Details of each centers’ ethical review organization, processes, and licenses are provided in Supplementary Table 7. All efforts were made to minimize suffering by considerate housing and husbandry. All phenotyping procedures were examined for potential refinements that were disseminated throughout the IMPC. Animal welfare was assessed routinely for all mice.
Urls
IMPC portal, http://www.mousephenotype.org; Glucose results for Gad2, http://mousephenotype.org/data/charts?accession=MGI:95634&allele_accession=MGI:5548938¶meter_stable_id=IMPC_IPG_010_001&metadata_group=297b1cf545aee8eea0113b14aca71ef1&zygosity=homozygote&phenotyping_center=HMGU; IMPC FTP site, ftp://ftp.ebi.ac.uk/pub/databases/impc/latest/; IMPC publications, http://www.mousephenotype.org/data/alleleref; IMPRESS, http://www.mousephenotype.org/impress; IMPC data access, http://www.mousephenotype.org/data/documentation/index; IMPC Arrive guidelines, http://www.mousephenotype.org/about-impc/arrive-guidelines; IMPC GO annotations, https://www.mousephenotype.org/data/gene2go; ExpressionAtlas result for Fam53b, http://www.ebi.ac.uk/gxa/genes/ENSMUSG00000030956?bs=%7B“mus+musculus”%3A%7B“ORGANISM_PART”%3Atrue%7D%7D&ds=%7B%7D - differential; GEMM, https://www.har.mrc.ac.uk/gemm-call-guidance-applicants; PhenStat, http://goo.gl/tfbA5k; MGI, http://www.informatics.jax.org/; MGI downloads, ftp://ftp.informatics.jax.org/pub/reports/; Monarch Initiative, https://monarchinitiative.org; OWLtools, https://github.com/owlcollab/owltools
DATA AVAILABILITY
All data presented here is openly available from the IMPC portal via our FTP site. We also provide regular data exports to the MGI group who provide public access to all available mouse data and the Monarch Initiative who integrate genotype-phenotype data from human and numerous other species.
CODE AVAILABILITY
The automated phenotype comparisons were performed using the open-source OWLtools package provided by the Monarch Initiative.
Supplementary Material
Acknowledgments
This work was supported by NIH grants U54 HG006370 (T.F.M., P.F., A.-M.M., H.E.P., D.S., S.D.M.B), U42 OD011185 (S.A.M.), U54 HG006332 (R.E.B., K.S.), U54 HG006348-S1 and OD011174 (A.L.B.), 1R24OD011883 (C.J.M., M.H, N.W., D.S.), HG006364-03S1, U54H G006364 (K.C.K.L., C.M.) and U42 OD011175 (C.M, K.C.K.L.), and additional support provided by the The Wellcome Trust, Medical Research Council Strategic Award 53658 (S.W., S.D.M.B.), Government of Canada through Genome Canada and Ontario Genomics (OGI-051)(C.M., S.D.M.B.), Wellcome Trust Strategic Award, National Centre for Scientific Research (CNRS), the French National Institute of Health and Medical Research (INSERM), the University of Strasbourg (UDS), the “Centre Européen de Recherche en Biologie et en Médecine”, the “Agence Nationale de la Recherche” under the frame programme “Investissements d’Avenir” labelled ANR-10-IDEX-0002-02, ANR-10-INBS- 07 PHENOMIN to (Y.H.), The German Federal Ministry of Education and Research by Infrafrontier grant 01KX1012 (S.M., V.G.D., H.F., M.H.d.A.) ‘EUCOMM: Tools for Functional Annotation of the Mouse Genome’ (EUCOMMTOOLS) project - grant agreement no [FP7-HEALTH-F4-2010-261492] (W.G.W)
Footnotes
AUTHOR CONTRIBUTIONS
T.F.M, D.B.W, N.C, D.Sm contributed to the data analysis, writing of the paper and design, execution of the work. N.H., M.H, N.W, C.J.M, P.M, J.O.J, C.K.C, I.T, H.M, M.R., N.K, J.W, H.W, J.M, D.Sn contributed to development of the software, statistical analysis, database and APIs. L.S, T.F, N.R, S.G performed quality control of the phenotype data. J.B., J.K.W, S.Y.C, G.F.C, M.E.S, C.L.R, J.G, V.G-D, T.S, G.P, L.R.B led the experimental work and data production. I.M, J.S, A.B, M.D, M.H.dA, M.M, Y.H, G.T-V, K.C.L, X.G, C.M, M.J.J, S.A.M, K.L.S, R.E.B, S.W, A-M.M, P.F, H.E.P, J.W, A.L.B, W.C.S, D.J.A, S.D.M.B, W.W, S.N, A.M.F, L.M.J.N, Y.O., J.K.S are senior PIs of the key programs that contributed to the paper, and were critical for the design, management, and execution of the study and writing and reviewing of manuscript. The additional IMPC consortium members all contributed to data acquisition and data handling.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests
References
- 1.Bello SM, Smith CL, Eppig JT. Allele, phenotype and disease data at Mouse Genome Informatics: improving access and analysis. Mamm Genome. 2015;26:285–94. doi: 10.1007/s00335-015-9582-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Begley CG, Ellis LM. Drug development: Raise standards for preclinical cancer research. Nature. 2012;483:531–3. doi: 10.1038/483531a. [DOI] [PubMed] [Google Scholar]
- 3.Fonio E, Golani I, Benjamini Y. Measuring behavior of animal models: faults and remedies. Nat Methods. 2012;9:1167–70. doi: 10.1038/nmeth.2252. [DOI] [PubMed] [Google Scholar]
- 4.Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8:e1000412. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown SDM, Moore MW. The International Mouse Phenotyping Consortium: past and future perspectives on mouse phenotyping. Mamm Genome. 2012;23:632–40. doi: 10.1007/s00335-012-9427-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hrabě de Angelis M, et al. Analysis of mammalian gene function through broad-based phenotypic screens across a consortium of mouse clinics. Nat Genet. 2015;47:969–78. doi: 10.1038/ng.3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Skarnes WC, et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 2011;474:337–42. doi: 10.1038/nature10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bradley A, et al. The mammalian gene function resource: The International Knockout Mouse Consortium. Mamm Genome. 2012;23:580–586. doi: 10.1007/s00335-012-9422-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosen B, Schick J, Wurst W. Beyond knockouts: the International Knockout Mouse Consortium delivers modular and evolving tools for investigating mammalian genes. Mammalian Genome. 2015;26:456–466. doi: 10.1007/s00335-015-9598-3. [DOI] [PubMed] [Google Scholar]
- 10.Dickinson M, Flenniken AM, Xiao J, Teboul L, Murray SA. High-throughput discovery of novel developmental phenotypes. Nature. 2016 doi: 10.1038/nature19356. [DOI] [PMC free article] [PubMed]
- 11.Adams D, et al. Bloomsbury report on mouse embryo phenotyping: recommendations from the IMPC workshop on embryonic lethal screening. Dis Model Mech. 2013;6:571–9. doi: 10.1242/dmm.011833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurbatova N, Mason JC, Morgan H, Meehan TF, Karp NA. PhenStat: A Tool Kit for Standardized Analysis of High Throughput Phenotypic Data. PLoS One. 2015;10:e0131274. doi: 10.1371/journal.pone.0131274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.West DB, et al. A lacZ reporter gene expression atlas for 313 adult KOMP mutant mouse lines. Genome Res. 2015;25:598–607. doi: 10.1101/gr.184184.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adissu HA, et al. Histopathology reveals correlative and unique phenotypes in a high-throughput mouse phenotyping screen. Dis Model Mech. 2014;7:515–24. doi: 10.1242/dmm.015263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koscielny G, et al. The International Mouse Phenotyping Consortium Web Portal, a unified point of access for knockout mice and related phenotyping data. Nucleic Acids Res. 2014;42:D802–9. doi: 10.1093/nar/gkt977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freedman LP, Cockburn IM, Simcoe TS. The Economics of Reproducibility in Preclinical Research. Plos Biology. 2015 doi: 10.1371/journal.pbio.1002165. http://dx.doi.org/10.1371/journal.pbio.1002165. [DOI] [PMC free article] [PubMed]
- 17.Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43:D789–98. doi: 10.1093/nar/gku1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rath A, et al. Representation of rare diseases in health information systems: The orphanet approach to serve a wide range of end users. Hum Mutat. 2012;33:803–808. doi: 10.1002/humu.22078. [DOI] [PubMed] [Google Scholar]
- 19.Kohler S, et al. The Human Phenotype Ontology in 2017. Nucleics Acid Res. 2017;45:D865–D876. doi: 10.1093/nar/gkw1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mungall CJ, et al. Use of model organism and disease databases to support matchmaking for human disease gene discovery. Hum Mutat. 2015;36:979–84. doi: 10.1002/humu.22857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith CL, Eppig JT. Expanding the mammalian phenotype ontology to support automated exchange of high throughput mouse phenotyping data generated by large-scale mouse knockout screens. J Biomed Semantics. 2015;6:11. doi: 10.1186/s13326-015-0009-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smedley D, et al. PhenoDigm: analyzing curated annotations to associate animal models with human diseases. Database (Oxford) 2013;2013:bat025. doi: 10.1093/database/bat025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Savoia A, et al. Spectrum of the mutations in Bernard-Soulier syndrome. Hum Mutat. 2014;35:1033–45. doi: 10.1002/humu.22607. [DOI] [PubMed] [Google Scholar]
- 24.Khan SA, et al. Genetics of human Bardet-Biedl syndrome, an updates. Clin Genet. 2016;90:3–15. doi: 10.1111/cge.12737. [DOI] [PubMed] [Google Scholar]
- 25.Margolin DH, et al. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N Engl J Med. 2013;368:1992–2003. doi: 10.1056/NEJMoa1215993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Santens P, et al. RNF216 mutations as a novel cause of autosomal recessive Huntington-like disorder. Neurology. 2015;84:1760–6. doi: 10.1212/WNL.0000000000001521. [DOI] [PubMed] [Google Scholar]
- 27.White JK, et al. Genome-wide generation and systematic phenotyping of knockout mice reveals new roles for many genes. Cell. 2013;154:452–64. doi: 10.1016/j.cell.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blake JA, et al. Gene ontology consortium: Going forward. Nucleic Acids Res. 2015;43:D1049–D1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pandey AK, Lu L, Wang X, Homayouni R, Williams RW. Functionally enigmatic genes: a case study of the brain ignorome. PLoS One. 2014;9:e88889. doi: 10.1371/journal.pone.0088889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petryszak R, et al. Expression Atlas update—an integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res. 2015;44:gkv1045. doi: 10.1093/nar/gkv1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kingsley PD, et al. Ontogeny of erythroid gene expression. Blood. 2013:121. doi: 10.1182/blood-2012-04-422394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boria I, et al. The ribosomal basis of Diamond-Blackfan Anemia: mutation and database update. Hum Mutat. 2010;31:1269–79. doi: 10.1002/humu.21383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kizil C, et al. Simplet/Fam53b is required for Wnt signal transduction by regulating β-catenin nuclear localization. Development. 2014;141:3529–39. doi: 10.1242/dev.108415. [DOI] [PubMed] [Google Scholar]
- 34.Smedley D, et al. Next-generation diagnostics and disease-gene discovery with the Exomiser. Nat Protoc. 2015;10:2004–15. doi: 10.1038/nprot.2015.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bone WP, et al. Computational evaluation of exome sequence data using human and model organism phenotypes improves diagnostic efficiency. Genet Med. 2016;18:608–17. doi: 10.1038/gim.2015.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harkness JH, et al. Trace Amine-Associated Receptor 1 Regulation of Methamphetamine Intake and Related Traits. Neuropsychopharmacology. 2015;40(9):2175–84. doi: 10.1038/npp.2015.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cade BE, et al. Obstructive Sleep Apnea Traits in Hispanic/Latino Americans. Am J Respir Crit Care Med. 2016;194(7):886–897. doi: 10.1164/rccm.201512-2431OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Knowles JW, et al. Identification and validation of N-acetyltransferase 2 as an insulin sensitivity gene. J Clin Invest. 2016;126(1):403. doi: 10.1172/JCI85921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lang B, et al. Recurrent deletions of ULK4 in schizophrenia: a gene crucial for neuritogenesis and neuronal motility. J Cell Sci. 2014;127:630–40. doi: 10.1242/jcs.137604. [DOI] [PubMed] [Google Scholar]
- 40.McIntyre RE, et al. A Genome-Wide Association Study for Regulators of Micronucleus Formation in Mice. G3 (Bethesda) 2016;6(8):2343–54. doi: 10.1534/g3.116.030767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Levy R, et al. Collaborative cross mice in a genetic association study reveal new candidate genes for bone microarchitecture. BMC Genomics. 2015;16:1013. doi: 10.1186/s12864-015-2213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ringwald M, et al. The IKMC web portal: A central point of entry to data and resources from the International Knockout Mouse Consortium. Nucleic Acids Res. 2011;39 doi: 10.1093/nar/gkq879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karp NA, Melvin D, Mott RF Sanger Mouse Genetics Project. Robust and sensitive analysis of mouse knockout phenotypes. PLoS One. 2012;7:e52410. doi: 10.1371/journal.pone.0052410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sayers EW, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2012;40 doi: 10.1093/nar/gkr1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Binns D, et al. QuickGO: A web-based tool for Gene Ontology searching. Bioinformatics. 2009;25:3045–3046. doi: 10.1093/bioinformatics/btp536. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data presented here is openly available from the IMPC portal via our FTP site. We also provide regular data exports to the MGI group who provide public access to all available mouse data and the Monarch Initiative who integrate genotype-phenotype data from human and numerous other species.