

SUMMARY
Cellular metabolite balance and mitochondrial function are under circadian control, but the pathways connecting the molecular clock to these functions are unclear. Peroxisome proliferator activated receptor delta (PPARδ) enables preferential utilization of lipids as fuel during exercise and is a major driver of exercise endurance. We show here that the circadian repressors CRY1 and CRY2 function as co-repressors for PPARδ. Cry1−/−;Cry2−/− myotubes and muscles exhibit elevated expression of PPARδ target genes, particularly in the context of exercise. Notably, CRY1/2 seem to repress a distinct subset of PPARδ target genes in muscle compared to the co-repressor NCOR1. In vivo, genetic disruption of Cry1 and Cry2 enhances sprint exercise performance in mice. Collectively, our data demonstrate that CRY1 and CRY2 modulate exercise physiology by altering the activity of several transcription factors, including CLOCK/BMAL1 and PPARδ and thereby alter energy storage and substrate selection for energy production.
eTOC blurb
Jordan et al. show that the circadian transcriptional repressors CRY1 and CRY2 repress the nuclear hormone receptor PPARδ and its target genes in mouse skeletal muscle and modulate exercise performance by altering substrate selectivity for energy production.
INTRODUCTION
Circadian clocks link behavioral and physiological processes to daily changes in the environment caused by the earth’s rotation. These clocks are based on interlocking transcription and translation feedback loops (Partch et al., 2014). In mammals, CLOCK and BMAL1 initiate the transcription of thousands of target genes including those encoding their repressors, periods (PER1, PER2 and PER3) and cryptochromes (CRY1 and CRY2). Dysregulation of circadian rhythms has been associated with metabolic disease (Maury et al., 2014). There is extensive crosstalk between circadian clocks and metabolism (Marcheva et al., 2013). CRY1 is phosphorylated by the cellular energy sensor AMPK (Lamia et al., 2009) and interacts with a subset of nuclear hormone receptors (NRs) (Lamia et al., 2011), suggesting a pivotal role for cryptochromes integrating circadian and metabolic gene expression. Furthermore, CRY1 and CRY2 bind about twice as many genomic sites as other clock components and sites uniquely bound by cryptochromes are enriched for NR response elements (Koike et al., 2012). Based on these prior findings, we hypothesized that cryptochromes are rhythmically expressed nutrient-regulated NR co-repressors.
PPARs are fatty acid-sensing NRs that regulate the expression of gene networks involved in metabolism, such as fatty acid oxidation, energetic uncoupling, and glucose metabolism (Ahmadian et al., 2013). PPARδ is an important regulator of muscle metabolism, in particular lipid oxidation (Barish et al., 2006). Moreover, PPARδ is activated by AMPK and this contributes to AMPK-driven changes in muscle gene expression that enhance exercise performance (Narkar et al., 2008). Thus, PPARδ may be directly implicated in the beneficial effects of exercise for the prevention and management of metabolic syndrome (Ehrenborg and Krook, 2009). Chronic activation of PPARδ either by transgenic expression or pharmacological treatment increases exercise capacity in mice (Fan et al., 2017; Narkar et al., 2008; Wang et al., 2004).
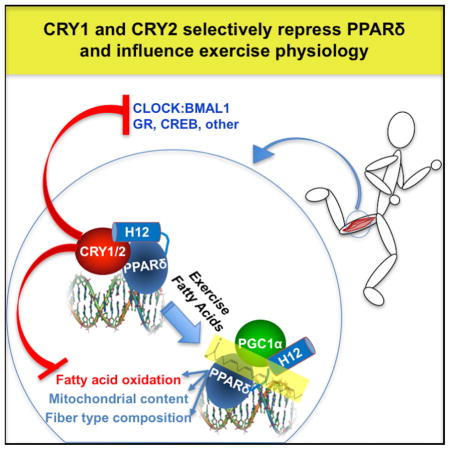
Here, we demonstrate that cryptochromes interact with PPARδ and suppress its activity. Cryptochrome-deficient mice display increased exercise capacity in the absence of major alterations in muscle fiber types. Since CRY1 and CRY2 are deleted in all tissues throughout development in these mice, their altered exercise performance may reflect a combination of physiological changes in muscle, cardiovascular, and other organ systems. We measured enhanced spare respiratory capacity and increased fatty acid oxidation in CRY-deficient myotubes, suggesting that cryptochromes modulate metabolic flexibility in muscles to coordinate with daily fluctuations in metabolic supply and demand.
RESULTS
Muscles contain AMPK-modulated circadian clocks
AMPK can alter the stability of CRY1 and PER2, and activation of AMPK increases circadian period length in fibroblasts (Lamia et al., 2009; Um et al., 2007). Given the critical role of AMPK in muscle physiology and response to exercise (Mounier et al., 2015), and that stimulation of AMPK in vivo modulates clock gene expression in muscle (Vieira et al., 2008), we investigated whether circadian clocks in myotubes can be regulated by AMPK in a cell-autonomous manner. We isolated myoblasts from mice expressing a PER2-Luciferase fusion protein (PER2-LUC) (Yoo et al., 2004) and differentiated them into myotubes. Successful isolation of myoblasts and differentiation into myotubes was confirmed by immunostaining of DESMIN and expected morphology (Figure 1A). We synchronized circadian rhythms with stimuli reported in other cell types (Balsalobre et al., 2000; Nagoshi et al., 2004; Ramanathan et al., 2014), demonstrating that these primary mouse myotubes harbor robust circadian clocks (Figures 1B–C). As previously shown in fibroblasts (Lamia et al., 2009), the AMP analog AICAR, which activates AMPK, lengthened the period and shifted the phase of circadian rhythms in myotubes (Figures 1D–E). Similar to (Wolff and Esser, 2012), we also observed rhythmicity synchronized by serum shock in muscle explants (Figure S1). In vivo, we measured rhythmic expression of clock genes in quadriceps as reported by others (Hodge et al., 2015). We also confirmed (McCarthy et al., 2007) diurnal expression of PPARδ target genes pyruvate dehydrogenase kinase 4 (Pdk4) and uncoupling protein 3 (Ucp3) (Figure 1F). Together, these data support the hypothesis that myotubes contain circadian clocks that are responsive to AMPK and could play a role in the adaptive response to exercise.
Figure 1. Muscle cell clocks are influenced by AMPK activation. See also Figure S1.

(A) Representative images of myoblasts (MBs) and myotubes (MTs). (B,D) Luciferase activity in Per2Luc MTs stimulated with horse serum (HS, grey), dexamethasone (Dex, blue) or forskolin (Fsk, green) (B) or in the absence (black) or presence (red) of AICAR after dexamethasone (D). (C,E) Average period of rhythms measured as in (B,D). (F) Expression of the indicated transcripts measured by quantitative PCR (qPCR) in quadriceps collected at the indicated zeitgeber times (time after lights on, ZT), normalized to Hprt. (C,E) show mean ± s.e.m. for 4–8 samples per group. *p ≤ 0.05, ** p ≤ 0.01. (F) shows mean ± s.e.m. for 3 samples per condition, each measured in triplicate.
Cryptochromes exhibit co-repressor-like interactions with PPARδ
We previously showed that cryptochromes interact with the glucocorticoid receptor and mediate rhythmic repression of its activity (Lamia et al., 2011). A broader survey of NR interactions revealed that CRY1 and CRY2 also interact with PPARs (Figure 2A). PPARs, like other NRs, are regulated by ligand-dependent association and dissociation of co-activators and co-repressors. We found that agonist ligand binding to PPARs reduces the association of CRY1 and CRY2 (Figure 2B), suggesting that they may behave like co-repressors for NRs, including PPARs. The effect of ligand on cryptochrome association varied between the three PPAR isoforms, and was most dramatic for PPARδ.
Figure 2. Cryptochromes exhibit co-repressor-like interactions with PPARδ.

Proteins detected by immunoblot (IB) following FLAG IP from lysates of 293T cells expressing the indicated plasmids and treated with PPAR ligands or vehicle (-). WCL whole cell lysate, FL full length PPARδ, DBD DNA binding domain, LBD ligand binding domain, ΔH12 helix 12 deleted.
Typically, NRs contain an N-terminal DNA-binding domain (DBD) and a C-terminal ligand-binding domain (LBD) that includes the ligand-dependent activation function (AF-2), also known as AF-2 helix or helix-12 (H12). Agonist ligand binding changes the LBD conformation, releasing co-repressors and allowing co-activators to bind (Renaud et al., 1995; Shiau et al., 1998). Deletion of H12 results in constitutive co-repressor binding (Dowell et al., 1999). Consistent with their hypothesized role as co-repressors, CRY1 and CRY2 preferentially bind the LBD of PPARδ (Figure 2C) and deletion of H12 prevents the dissociation of CRY1 and CRY2 in response to ligand (Figure 2D).
Cryptochromes suppress PPARδ activity in a cell-intrinsic manner
To determine whether the interaction of cryptochromes and PPARδ is relevant in the context of muscle physiology, we isolated myoblasts from wildtype and Cry1−/−;Cry2−/− double knockout (dKO) mice and differentiated them into myotubes. The absence of cryptochromes had no effect on differentiation, as evidenced by the indistinguishable morphology (Figure 3A) and expression of myogenic marker genes (Figures 3B and S2A). We also found that PPARδ protein levels in myotube nuclei are unaltered by deletion of CRY1/2 (Figure 3C). Since dKO myoblasts differentiate normally and express normal levels of PPARδ, we used them to examine the effect of cryptochrome deficiency on the expression of PPARδ target genes. The agonist ligand GW1516 activates PPARδ and thereby induces the transcription of Pdk4 and Ucp3, among other target genes (Narkar et al., 2008). Pdk4 and Ucp3 were significantly increased in dKO compared to wildtype cells and were further increased by prolonged ligand stimulation (Figure 3D). Several other transcripts involved in fatty acid transport and metabolism that are activated by PPARδ in other systems (Ehrenborg and Krook, 2009) did not respond to PPARδ ligand under the conditions tested, but carnitine palmityoyl transferase 1b (Cpt1b), fatty acid binding protein 3 (Fabp3) and lipoprotein lipase (Lpl) were highly elevated in dKO myotubes compared to wildtype (Figures 3D and S2B-F). These data support the hypothesis that CRY1 and CRY2 repress PPARδ activity in myotubes and suggest that they modulate additional pathways critical for muscle metabolism as well. Since PPARδ can be activated by AMPK, we considered the hypothesis that AMPK-mediated activation of PPARδ requires degradation of CRYs. However, pharmacological activation of AMPK increased PPARδ target gene expression in dKO myotubes to a similar, if not greater, degree than in wildtype myotubes (Figures 3E, S2G and S2H).
Figure 3. Cryptochromes suppress PPARδ in a cell-intrinsic manner. See also Figure S2.

(A) Representative images of MBs differentiated into MTs. (B,D,E) Relative expression of indicated transcripts in WT (open symbols) or dKO (filled symbols) cells during differentiation (B) or after treatment with vehicle (DMSO, black) or 100nM GW1516 (GW, red) (D) or at the indicated times, 3 hours after treatment with vehicle (DMSO, black) or GW (red) following three days of stimulation with AICAR at 9am (E). Data represent mean ± s.e.m of experimental triplicates. (C) Proteins detected by IB in nuclear extracts from MTs of indicated genotypes. (F) Oxygen consumption rate (OCR) in MTs treated sequentially with oligomycin (oligo), FCCP, and rotenone+antimycin A (R/AA). (G) Spare respiratory capacity calculated from (F). (H) OCR in WT (left) and dKO (right) MTs in the presence or absence of exogenous palmitate (16:0) and etomoxir (Eto.). (I) Fatty acid oxidation (FAO) calculated from (H). Data represent mean ± s.e.m of three experiments each done in triplicate (D), or 3, 15 or 6–9 replicates in a representative experiment (E, F–G, or H–I). *p ≤ 0.05, ** p ≤ 0.01, ***p ≤ 0.001 by 2-way (D) or 3-way (E) ANOVA or t-test (G, I). In (D,E) black and red asterisks represent effects of genotype or treatment respectively.
By increasing the expression of genes involved in fuel utilization, PPARδ activation enhances the ability of myotubes to oxidize fatty acids (Tanaka et al., 2003). Analysis of cellular respiration in dKO primary myotubes revealed increased mitochondrial reserve capacity compared to wildtype cells (Figures 3F, 3G). We also observed greater oxidation of exogenously supplied palmitate (Figure 3H, 3I), consistent with increased PPARδ activity and enhanced expression of lipid metabolism genes, including Pdk4, Cpt1b and Fabp3. Together, these results indicate that cryptochromes regulate skeletal muscle metabolism, at least in part by repressing fatty acid transport and oxidation, via CRY1/2-mediated inhibition of PPARδ and other transcription factors.
Deletion of Cry1 and Cry2 alters exercise physiology in mice
Increased PPARδ activity in muscle improves exercise endurance in mice (Fan et al., 2017; Wang et al., 2004). Though improved physiological performance is rare in knockout models, muscle-specific deletion of the NR co-repressor NCOR1 results in ~30% increased endurance, attributed to increased muscle mass and mitochondrial number driven by activation of PPARδ and other transcription factors (Yamamoto et al., 2011). Given that CRY1 and CRY2 also repress PPARδ and likely additional transcription factors, we analyzed running performance in wildtype mice and mice deficient for either Cry1 (1KO) or Cry2 (2KO) or both (dKO). While 1KO and 2KO mice performed like wildtype littermates, sedentary dKO mice ran faster and further than wildtype littermates in every test that included an endpoint based on speed (Figures 4A protocols I-III, S3A and S3B). In contrast, they maintained a similar duration of low intensity exercise when the endpoint was defined as exhaustion connected with blood glucose depletion (Figure 4A protocol IV). We also measured enhanced fitness in dKO mice after prolonged training by either supervised treadmill running (Figure S3A and S3C) or continuous access to a running wheel (Figure S3A and S3D). Notably, dKO mice ran significantly less than wildtype mice when given voluntary access to a running wheel (Figure S3E), though they have more similar levels of ambulatory activity (Figure S3F). Therefore, the enhanced performance in dKO mice reflects neither increased behavioral motivation nor increased training. These data suggest that dKO mice have improved capacity for higher power output exercise that typically utilizes stored intramuscular substrates. They show less marked differences in prolonged, lower intensity exercise, which normally relies more on plasma substrate turnover. Additionally, sedentary state whole body energy production in dKO and wildtype mice is derived from similar relative utilization of carbohydrate and lipid fuels (Figure S3G). Moreover, both genotypes exhibit similar daily loss of heat as estimated by calorimetry (Figure S3H), though the diurnal rhythms are somewhat blunted in CRY dKO mice.
Figure 4. Deletion of Cry1 and Cry2 increases exercise capacity. See also Figures S3 and S4.

(A) Maximum speed or initial and final blood glucose (top) and total distance (bottom) in male mice. (B) Glycogen content in gastrocnemius and liver (N=5 each). (C) Representative plantaris (pl) and soleus (sol) muscle staining. (D) Quantification of fiber types in plantaris (N=7 each). (E) Representative vasculature stains. Scale bars, 50 μm. (F,G) Quantification of fiber cross-sectional area (CSA, F) and capillary density (G) (N=3 mice each, five sections per mouse). (H) Relative amount of mitochondrial DNA in the indicated muscles (N=7–18). (I) Protein content in isolated mitochondria relative to tissue weight in quadriceps (N=3 each). Data represent mean ± s.e.m. * P < 0.05, *** P < 0.001 vs. WT by t-test.
Since CRY1 and CRY2 modulate several transcription factors (Huber et al., 2016; Lamia et al., 2011; Zhang et al., 2010) and we are studying mice in which they have been deleted in all tissues, we examined several possible contributions to the enhanced exercise performance in CRY-deficient animals. Glycogen is a major source of carbohydrate-derived energy during exercise and we measured increased glycogen content in both skeletal muscle and liver of CRY dKO mice compared to wildtype littermates (Figure 4B). Interestingly, CRY1/2-deficient mice seem to deplete their glycogen stores more rapidly during intense exercise (Figure S3I), which may reflect more efficient glycolysis and could contribute to their greater power output.
Enhanced exercise capacity in transgenic animals is often associated with an increase in muscle mitochondrial content and/or changes in fiber type composition. However, it remains unclear whether structural alterations are required for enhanced fitness. For example, eight weeks of chronic pharmacological activation of PPARδ enhances exercise endurance without altering muscle fiber type composition or mitochondrial number and this requires PPARδ in muscle (Fan et al., 2017). To examine whether CRY1/2 deletion affects fiber type composition, we analyzed the expression of myosin heavy chain (Myh) genes classifying the four main fiber types in murine skeletal muscle. In dKO mice, fiber type marker gene expression was similar to wildtype in both predominantly fast-twitch (quadriceps, gastrocnemius and plantaris) and slow-twitch (soleus) muscles (Figure S3J). To obtain more precise information about fiber type composition, we performed multicolor immunofluorescence of myosin heavy chain (MHC) proteins in cross-sections of the gastrocnemius-plantaris-soleus muscle compartment (Figures 4C, S3K and S3L). Though MHC-I positive oxidative slow-twitch (type I) fibers represented a tiny fraction of the fibers in plantaris muscle in both genotypes, they were significantly increased in dKO mice (Figure 4C and 4D). This may contribute to the observed enhancement of exercise capacity, but seems unlikely to fully explain the phenotype, especially with respect to enhanced sprint capacity, which would more likely be favored by glycolytic fiber types. Other MHC fiber type markers were unaffected by Cry1 and Cry2 deletion.
Exercise capacity is greatly influenced by the cardiovascular and circulatory systems; enhanced muscle vascularization improves performance (Fitts, 1994). We examined fiber and capillary density in gastrocnemius, plantaris, soleus, and quadriceps muscles by immunostaining frozen sections for LAMININ, and platelet and endothelial cell adhesion molecule (PECAM) (Figure 4E). The fiber cross-sectional area was reduced in dKO muscles (Figure 4F); the number of fibers per unit area was thus increased. Vascular staining indicated that while the number of capillaries per fiber was reduced in dKO mice compared to wildtype, there was no alteration in capillary density per unit area of muscle tissue (Figure 4G). These data reinforce earlier morphometric observations in the dKO mouse (Lamia et al., 2011) and suggest that altered muscle vascularization does not contribute to their enhanced exercised capacity.
Enhanced exercise capacity has also been linked to increased skeletal muscle mitochondrial biogenesis and/or activity (Narkar et al., 2008; Wang et al., 2004). Histochemical analysis of succinate dehydrogenase (SDH) activity and Western blot analysis of mitochondrial complex composition looked similar in wildtype and dKO muscles (Figures S4A and S4B). Enhanced exercise capacity in dKO mice also could not be attributed to increased mitochondrial number as mitochondrial DNA and protein content were either reduced or unchanged (Figures 4H and 4I).
To gain insight into how CRY1 and CRY2 alter muscle-intrinsic properties in muscle groups of relatively homogeneous fiber types, we analyzed isolated soleus (oxidative) and extensor digitorum longus (EDL, glycolytic) muscles. Soleus muscles from dKO mice have the same length as those from wildtype littermates, but have less mass and a smaller cross-sectional area (Figures S4C-E), consistent with our fiber observations. Their ability to generate force ex vivo was indistinguishable but the dKO muscles showed a tendency toward reduced fatiguability (Figures S4F-K). The lower muscle weights and reduced cross-sectional area are correlated with lower body weight (Lamia et al., 2011) and reduced lean mass (Figure S4L–N) of dKO mice.
Taken together, these data indicate that cryptochrome deficiency improves exercise performance without causing major fiber type changes or increasing mitochondrial mass or vascularization. These findings suggest that CRY1 and CRY2 influence exercise physiology primarily by altering metabolic substrate storage and utilization rather than altering structural development of the musculoskeletal or circulatory systems. Additonal muscle-extrinsic physiological changes may also contribute to enhanced performance in mice ubiquitously lacking CRY1 and CRY2.
Cryptochromes limit exercise-induced activation of PPARδ target genes
To examine whether cryptochromes modulate PPARδ-driven gene expression in response to exercise in vivo, we analyzed Pdk4 expression in muscles of untrained (Figures 5A and 5B) or running-wheel trained (Figures 5A and 5C) mice after an acute exercise bout. As seen in myotubes, Pdk4 expression was higher in dKO quadriceps of exercised mice compared to wildtype littermates. Notably, Pdk4 expression was unaffected by ablation of Cry1 and Cry2 in soleus muscle (Figure 5C), where basal expression of Pdk4 is much higher than in the quadriceps. PPAR gamma co-activator 1 alpha (PGC1α) is a major activator of PPARδ activity and Pgc1α mRNA is induced by exercise (Goto et al., 2000). We measured Pgc1α and found that it was robustly enhanced by the acute treadmill exercise protocol that we employed. Furthermore, the exercise-induced increase in Pgc1α was not affected by deletion of Cry1 and Cry2 (Figure S5A). Recently, alternative isoforms of Pgc1α that are differentially affected by exercise have been described, which may contribute to exercise-driven enhancement of PPARδ activity (Martinez-Redondo et al., 2015). We examined the expression of several Pgc1α variants and found them to be unaffected by CRY1/2 ablation (Figure S5B).
Figure 5. Cryptochromes limit exercise-induced activation of PPARδ. See also Figure S5.

(A) Experimental flow chart. (B,C,F) Expression of indicated transcripts in quadriceps of untrained sedentary or exercised mice (N=7–8 each). n.s. not significant, *p ≤ 0.05, ***p ≤ 0.001 vs sedentary control or as indicated. (C) Pdk4 expression in the indicated muscle groups of trained mice after exercise. Data represent mean ± s.e.m of 7–8 mice per genotype. **p ≤ 0.01 vs WT. (D–E) Heat maps showing relative expression (red high, blue low) detected by deep sequencing of RNA isolated from mice as described in (B). (F) Relative expression in quadriceps of trained mice after exercise. Data represent mean ± s.e.m of 7–8 mice per genotype. *p ≤ 0.05 vs WT.
To investigate the role of CRY1 and CRY2 in transcriptional regulation during exercise in an unbiased manner, we sequenced RNA from quadriceps of untrained sedentary and acutely exercised wildtype and dKO mice. Using gene set enrichment analysis (GSEA) (Mootha et al., 2003; Subramanian et al., 2007) to search the resulting data for regulatory sequences near transcription start sites, we found that “V$HNF4_Q6” (Genes with promoter regions ±2kb around transcription start site containing the motif AARGTCCAN, matching annotation for HNF4A: hepatocyte nuclear factor 4, alpha) is overrepresented (FDR = 0.05) in promoters of transcripts with increased expression in sedentary dKO, compared to sedentary wildtype mice. HNF4α is a NR that is not expressed in muscle (Bookout et al., 2006) so this probably does not reflect increased HNF4α activity. This motif has sufficient homology to the PPRE optimal half-site sequence (AGGTCA) that it is likely to interact with PPARδ and other closely related NRs. Notably, this analysis did not reveal overrepresentation of E-boxes which are preferentially bound by CLOCK and BMAL1; other targeted pathways may also be missed.
We also used GSEA to search for pathway-specific gene networks overrepresented among genes that were differentially expressed between wildtype and dKO muscles. In the sedentary state, several gene sets related to mitochondria exhibit decreased expression in CRY dKO mice (Supplemental Table S1). The expression levels for one such gene set (the so-called “Hallmark” pathway (Liberzon et al., 2015) for Oxidative Phosphorylation) is shown in Figure 5D. Notably, the PPARδ target Pdk4 is located near the bottom of the ranked gene list because it is increased in dKO mice. Using GSEA to compare sedentary and exercised gene expression in wildtype mice, we identified hypoxia-related gene sets as those most significantly altered by exercise. One such gene set (Weinmann et al., 2005) was enhanced in dKO mice compared to wildtype specifically in the exercised state (Figure 5E).
To examine the effect of CRY1/2 deficiency specifically on PPARδ target gene expression, we used gene expression data from mice exposed to chronic treatment with the PPARδ agonist GW1516 (Fan et al., 2017), to generate a defined set containing all genes significantly increased by activation of PPARδ (GW_UP, Table S2). These genes exhibited a bimodal distribution in response to the genetic deletion of Cry1 and Cry2 such that they were enriched among transcripts altered by the loss of cryptochromes but the direction of the change in their expression can be either positive or negative (Figure S5C). When we limited our analysis to the 15 genes with the greatest fold induction by GW1516, we found that these transcripts were more highly activated in dKO mice, especially in the context of acute exercise (not shown). Among those, nine genes are predicted to impact exercise physiology. Four of them were expressed at detectable levels in myotubes, including stearoyl-coenzyme A desaturase 1 (Scd1) and solute carrier family 27 member 1 (Slc27a1, encoding fatty acid transport protein 1 FATP1), which were significantly increased in dKO cells (Figure S5D). All nine transcripts were elevated in quadriceps of running-wheel trained dKO mice compared to trained wildtype littermates, after an acute exercise bout (Figure 5F). In untrained mice, these transcripts were much less affected by the loss of CRY1 and CRY2 (Figure S5E). Together these data indicate that the circadian transcriptional repressors CRY1 and CRY2 limit exercise-induced activation of PPARδ in vivo, which likely contributes to altered substrate utilization and exercise capacity.
DISCUSSION
Detailed investigation of skeletal muscle circadian clocks has only recently begun (Harfmann et al., 2015). Accumulating evidence suggests that muscle clocks play a major role in coordinating substrate metabolism with daily cycles of activity and feeding (Dyar et al., 2014; Hodge et al., 2015). Transitioning between these two phases is not only associated with fluctuations in fuel supply and demand but also with dynamic flux between fuel sources. Circadian regulation of mitochondrial substrate utilization has also been observed in other cell types (Peek et al., 2013). Recently, it was shown that circadian clocks modulate mitochondrial morphology (Jacobi et al., 2015) and the composition of the mitochondrial proteome (Neufeld-Cohen et al., 2016), each of which may influence fuel selection (Mishra and Chan, 2016; Stanley et al., 2014). Metabolic flexibility is key to muscles’ adaptation to variable energy demands during exercise. Metabolic sensors, such as AMPK, are crucial in these adaptive responses. We previously demonstrated that AMPK phosphorylates the circadian clock protein CRY1 and enhances its degradation (Lamia et al., 2009), suggesting that cryptochromes may connect energy sensing with daily fluctuations in mitochondrial fuel utilization.
AMPK can activate PPARδ, leading to increased expression of PPARδ target genes and enhanced exercise endurance (Narkar et al., 2008). AMPK-dependent phosphorylation of PGC1α contributes to AMPK activation of some PPARδ target genes in skeletal muscle. However, AMPK-driven activation of other PPARδ targets, including Pdk4, does not require PGC1α (Jager et al., 2007). We found that the circadian repressors CRY1 and CRY2 interact with PPARδ. These interactions are increased by deletion of PPARδ H12 and disrupted by agonist ligands, as expected for an NR:co-repressor interaction. Deletion of Cry1 and Cry2 increases exercise activation of PPARδ target genes, suggesting that AMPK-dependent phosphorylation of CRY1 may contribute to this process in vivo. However, degradation of CRY1 and/or CRY2 are not required for AMPK dependent activation of PPARδ in myotubes. Intriguingly, the activation of PPARδ targets either by acute exercise in muscles in vivo or by AMPK activation in primary myotubes seems to require repeated daily exposure to the AMPK-activating stimulus (running wheel access in mice or daily AICAR stimulation in cell culture, Figs. 3E, S2B-H, 5F, and S5E). Since AMPK activation can act as a circadian timing cue (Lamia et al., 2009), the cumulative effect of daily AMPK activation may involve integration with clocks. The relatively low amplitude of skeletal muscle clocks that we and others have observed may reflect a lack in sedentary mice of the exercise stimulus, which has been shown to influence clock time and amplitude (Wolff and Esser, 2012). It will be interesting to investigate whether time of day profoundly impacts exercise physiology in mice exposed to voluntary running wheel training. Further investigation is required to determine whether and to what extent PGC1α and CRY1 cooperate to transduce signals from exercise and other AMPK-activating stimuli to PPARs, other NRs, and additional downstream pathways.
We found that cryptochromes suppress PPARδ activity in primary cultured myotubes and limit activation of the PPARδ transcriptional network in response to exercise in vivo. Interestingly, we observed a bimodal distribution of expression changes in PPARδ-regulated genes in response to genetic disruption of Cry1 and Cry2. Since PPARδ interacts with many co-regulators including PGC1α, SRC1/2/3, NCOR1 and SMRT/NCOR2, the absence of CRY1 and CRY2 may alter the formation of PPARδ complexes with these or other transcriptional regulators. In addition, PPARδ-coregulator complexes modulate enhancer activity in the context of three-dimensional chromatin structures (Fang et al., 2014; Zhu et al., 2015). Thus, it is not surprising that the directionality of altered transcriptional regulation upon loss of Cry1 and Cry2 depends on the target genes examined. Agonist ligand stimulation increases Pdk4 and Ucp3 levels in dKO myotubes, indicating that the absence of cryptochromes alone does not lead to maximal PPARδ activation of these transcripts. NCOR1 has been shown to bind Pdk4 and Ucp3 promoter elements (Yamamoto et al., 2011), and release of NCOR1 and/or other repressors may enhance their transcription by PPARδ in response to agonist ligands in Cry dKO cells.
Unbiased analysis of gene expression patterns in response to exercise revealed that the transcriptional network most responsive to acute exercise in muscle is that stimulated by hypoxia, and that this response is enhanced in CRY dKO mice. Hypoxia and the hypoxia inducible factors (HIF1α, HIF1β) can enhance glycolysis in response to high intensity training (Abe et al., 2015) or chronic hypoxia (for example high altitude) (Favier et al., 2015), which could contribute to increased power output in the context of exercise. Since we observed both increased liver glycogen depletion and enhanced transcriptional response to hypoxia during exercise in dKO mice, chronic enhancement of the hypoxia response may play a role in the altered exercise physiology of CRY-deficient animals. This is especially intriguing given the recent demonstrations that HIF1α both modulates circadian clock function and is altered by circadian disruption in several systems (Adamovich et al., 2017; Peek et al., 2017; Wu et al., 2017). Further investigation will be required to understand the mechanism(s) by which the transcriptional response to exercise-induced hypoxia is enhanced in CRY dKO mice.
We and others previously showed that mice lacking CRY1 and CRY2 exhibit elevated gluconeogenesis and corticosterone production (Lamia et al., 2011; Zhang et al., 2010). Acute increases in circulating corticosteroids in response to high intensity exercise in rodents and in humans (Davies and Few, 1973; Sasaki et al., 2016) may enhance rapid energy production. This heightened adrenal output would increase gluconeogenesis and more rapidly restore carbohydrates as they are burned. Although it is unclear whether or how these global phenotypes alter performance in the running protocols tested here, it is possible that constitutively elevated corticosterone and gluconeogenesis in CRY dKO mice impacts their exercise performance.
PPARδ is a master regulator of muscle mitochondrial function and adaptation to exercise training. Together with the prior demonstration that muscle-specific loss of NCOR1 increases exercise capacity (Yamamoto et al., 2011), our findings support the notion that increased PPARδ activity in skeletal muscles enhances exercise capacity in vivo. Interestingly, NCOR1 and CRY1/2 seem to regulate unique aspects of muscle physiology via distinct subsets of genes within the PPARδ transcriptional network, which ultimately affect athletic performance. In NCOR1-deficient mouse muscle, key transcripts involved in oxidative phosphorylation were preferentially enhanced compared to those required for fatty acid oxidation (Perez-Schindler et al., 2012). The authors of that study argued that in sedentary animals, NCOR1 predominantly inhibits ERRα-mediated transcription of oxidative phosphorylation genes in muscle. Hence, the reduced expression of the oxidative phosphorylation gene set in the absence of cryptochromes may indicate increased NCOR1-mediated repression of ERRα. Though we have observed interactions between CRY1/2 and several NRs, they do not seem to bind ERRα (A. Kriebs and K. Lamia, unpublished). In addition to their apparent division of labor in repressing a subset of PPARδ-driven transcripts, NCOR1 and CRY1/2 each primarily suppresses a fraction of the PPARδ effects on muscle physiology. Though loss of either NCOR1 or CRY1/2 enhances exercise capacity in mice, NCOR1 deficiency increases muscle mass and enhances the activity of the mitochondrial electron transport chain (Yamamoto et al., 2011), while myotubes lacking CRY1 and CRY2 exhibit increased fatty acid oxidation and CRY dKO mice have decreased muscle mass without major alterations in muscle fiber type distribution. The distinct physiological effects on muscle structure and metabolism caused by genetic disruption of Ncor1 or Cry1/2 may be explained by the specific branches of the PPARδ transcriptional network that are altered by the loss of each co-repressor, as well as overlapping but distinct patterns of suppressing additional transcription factors. Exercise is a complex physiological process and depends on a multitude of muscle-intrinsic and extrinsic factors (Fitts, 1994). While enhanced PPARδ-driven fatty acid oxidation in muscles likely contributes to enhanced exercise performance in dKO mice, other effects of global CRY1 and CRY2 deficiency throughout development may also contribute to this improvement in fitness. Further investigation, including examination of animals lacking CRY1 and CRY2 in specific anatomic locations, is needed to determine the relative contributions of various underlying mechanisms to modulation of exercise physiology by cryptochromes.
The increased exercise capacity of dKO mice is consistent with higher PPARδ activity, but enhanced performance in a model of circadian disruption was unexpected. Mice harboring mutation or deletion of Clock, Bmal1, or Per2 exhibit mild or severe muscle pathologies (Andrews et al., 2010; Bae et al., 2006; Dyar et al., 2014). This discrepancy highlights the important distinction between measuring the effect of circadian disruption and understanding the influence of specific gene products. It is clear that circadian repressors, including PER1 and PER2 in addition to CRY1 and CRY2, can modulate transcription independent of their feedback repression of CLOCK and BMAL1 by independently binding to thousands of additional chromatin loci (Koike et al., 2012). Accumulating evidence suggests that circadian repression of NRs by PERs and CRYs likely plays a pivotal role in these non-clock circadian output pathways (Grimaldi et al., 2010; Koike et al., 2012; Lamia et al., 2011; Schmutz et al., 2010; Yang et al., 2006; Yang et al., 2007). Our finding that CRY1 and CRY2 modulate PPARδ in skeletal muscles supports this emerging paradigm.
STAR METHODS
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Katja Lamia (klamia@scripps.edu).
Experimental model and subject details
Mouse models
Cry1−/−;Cry2−/− mice were from Dr. Aziz Sancar (Thresher et al., 1998). They were reportedly backcrossed ≥10 generations to c57Bl6/J prior to transfer to us and we backcrossed them an additional 3 generations to c57Bl6/J mice from the TSRI breeding colony; we recently performed PCR analysis of some marker genes and realized that the backcrossing was not complete as they are heterogeneous at the Nnt locus. Mice were maintained in standard 12:12 light:dark conditions unless otherwise indicated and were group housed except when given voluntary access to running wheels in which case they were singly housed in running wheel cages. They were given ad libitum access to normal mouse chow and water. All animal care and treatments were in accordance with The Scripps Research Institute guidelines for the care and use of animals. Per2::Luciferase mice (Yoo et al., 2004) were purchased from Jackson Laboratories (B6.129S6-Per2tm1Jt/J; strain 006852; Bar Harbor, ME). Male mice 3–5 months of age were used in all experiments. Littermates were used as controls and were randomly assigned to experimental groups. Mice were group housed except during voluntary running wheel training when they were single housed.
Cell lines
Primary myoblasts were isolated from six-week-old male Cry1−/−;Cry2−/− mice and wildtype littermates. Three sets of littermate wildtype and dKO myoblasts were isolated from three independent breedings of Cry1+/−;Cry2+/− parents. Culture conditions for myoblasts and mytobues are provided in method details. 293T (ATCC® CRL3216™) cells were purchased from the American Type Culture Collection (ATCC) and are derived from female human embryonic kidney cells. HEK 293T cells were grown in complete Dulbecco’s Modified Eagle’s Medium (DMEM) (Invitrogen #10569) supplemented with 10% fetal bovine serum, and 1% penicillin and streptomycin. Cells were grown in a 37°C incubator maintained at 5% CO2.
Method details
Myoblast isolation and culture
Quadriceps muscles were isolated from 6-week-old male mice within 2 minutes of CO2 euthanasia. Muscle tissue was rinsed in sterile PBS containing 40 μg/ml gentamycin and transferred to a sterile 10-cm plate in a biosafety cabinet. 1 ml of plating media (1:1 solution of DMEM:HAMS F12 media + 40% heat inactivated fetal bovine serum and 10% AmnioMax) was added to the muscle tissue and the tissue was gently cut into small (1–3 mm3) fragments with a sterile scalpel. The fragments were transferred to a 6-cm dish in which the surface was pre-coated with coating solution [1:1 solution of DMEM:HAMS F12 media containing 0.17 mg/ml Collagen I (Gibco #A10644-01) and 2% BD Matrigel™ Basement Membrane Matrix (BD Biosciences #356234], keeping them well separated from each other. An additional 0.8 ml plating media was gently added to the transferred tissue fragments. Up to three 6-cm plates each containing tissue fragments from a single quadriceps muscle were placed in a sterile moist chamber consisting of a 15-cm dish containing Whatman paper cut to fit the dish and dampened with sterile deionized water. This chamber was placed inside a sterile plastic bag with one end left open and transferred to a 37°C incubator with 5% CO2 for 48 hours. After 48 hours without disturbance, when the fragments were sufficiently attached to the coated plates, an additional 2 ml of plating media were gently added. After an additional 2–5 days of growth, myoblasts were visibly growing out of the tissue explants and were harvested three times at 2 day intervals. To harvest the outgrowing myoblasts, the 6-cm plates containing tissue explants were washed once gently and quickly (~ 10 seconds) with PBS containing gentamycin. An additional 1 ml PBS + gentamycin was added to the plate and left to sit for 1 minute, then collected and replaced with 1 ml TrypLE (Gibco). The plate containing TrypLE was placed in a 37°C incubator for 3 minutes and then tapped very gently to dislodge myoblasts without detaching the tissue explants. Then the TrypLE containing myoblasts was collected and combined with the myoblasts collected in PBS and 8 ml of myoblast media [1:1 solution of DMEM:HAMS F12 media + 20% heat inactivated fetal bovine serum (Gibco #10437-028) + 10% AmnioMax-C100 (Gibco #12556-023)] were added. 2ml of warm plating media were added to the remaining tissue explants which were returned to the moist chamber and maintained in a 37°C incubator with 5% CO2. The combined collected myoblasts were collected by centrifugation at 200 × g for 3 minutes, resuspended in warm myoblast media, and transferred to T25 or T75 flasks pre-coated with coating solution. These cells were enriched for myoblasts by passaging three times in PBS + gentamycin at 2 day intervals: each flask was washed quickly (~10 seconds) and gently with PBS, and then incubated with warm PBS at 37°C, 5% CO2 for three minutes. Flasks were tapped to dislodge myoblasts while other cell types remain attached in PBS. Myoblasts were then collected by centrifugation at 200 × g for 3 minutes, resuspended in warm myoblast media, and transferred to a new coated flask. After three such passages in warm PBS, the cultures typically contained >95% myoblasts.
Desmin Staining
For immunofluorescence cells were rinsed twice with PBS before fixation with 2% paraformaldehyde in PBS for 5 minutes at room temperature (RT), then rinsed three times with PBS before adding 100% ice-cold methanol for permeabilization. After 5 minutes at -20°C cells were rinsed again three times with PBS. Cells were blocked in PBS with 10% donkey serum (blocking solution) for 30 minutes at RT. Monoclonal anti-mouse Desmin antibody (Sigma #D1033 clone DE-U-10) was diluted 1/400 in blocking solution and cells were incubated with primary antibody overnight at 4°C. Cells were washed three times for 5 minutes with blocking solution and incubated with secondary antibody Alexa Fluor 488 donkey anti-mouse IgG (Life Technologies #R37114 Ready-to-use; 2drops/ml) for 1 hour at RT protected from light. Cells were then washed three times for 5 minutes with blocking solution.
Oxygen consumption rate measurements
For Seahorse experiments (XF96, Seahorse Biosciences), XF96 plates were coated with Coating Solution (1:1 solution of DMEM:HAMS F12 media supplemented with 0.17 mg/ml Collagen I and 2% BD Matrigel™ Basement Membrane Matrix) by adding 25 μl per well and centrifuging the plate for 2 minutes at 200 × g, then placing on a flat surface at 4°C. After 1–4 hours, the coating solution was removed by aspiration and the wells were washed three times in cold PBS to remove excess coating solution. 40 μl of sterile differentiation media [DMEM/HAMS F12 1:1 media supplemented with 3% horse serum (Gibco #26050-088), 1% Insulin-Selenium- Transferrin (Invitrogen #41400-045) and 50μg/ml gentamicin] were added to each well of the coated plate and centrifuged for 2 minutes at 200 × g to remove bubbles and achieve a uniform base layer of media. 2×104 primary myoblasts in an additional 40 μl of differentiation were seeded in each well of XF96 plates prepared in this manner. The plated cells were maintained in a moist chamber as described above at 37°C, 5% CO2. During 5 days of differentiation, 80% of the differentiation media was replaced every day without exposing the cells to air. OCR and ECAR were measured on day 6 following manufacturer’s instructions with the injection of palmitate-BSA, oligomycin, FCCP, and rotenone/antimycin A. For the mitochondrial stress test, we used 1 μM oligomycin, 1 μM FCCP and 2 μM each rotenone and antimycin A. For measurement of fatty acid oxidation, we used 2 μM oligomycin, 2 and 4 μM FCCP, 4 μM each rotenone and antimycin A, 40 μM etomoxir, 667 μM 16:0 palmitate and 167 μM bovine serum albumin (BSA). We performed fatty acid oxidation assays after incubating myotubes in substrate-limited media [glucose- and glutamine-free DMEM base media (Sigma #D5030) supplemented with 0.5 mM glucose, 1 mM glutamine, 0.5 mM carnitine and 1% heatin-activated horse serum] for 3–8 hours; it was difficult to achieve appropriate substrate-limited conditions such that fatty acid oxidation derived primarily from exogenous palmitate without impacting cell viability and this was variable between experiments; data shown represent one of six experiments performed with similar differences in overall palmitate oxidation between genotypes in which we achieved the best utilization of exogenous palmitate in all cells (e.g. very little utilization of endogenous palmitate) such that reliable calculations could be performed. The plate setup was varied between replicate experiments and wells on the perimeter of the plate were excluded from all analyses.
Lumicycle analysis
For analysis of circadian rhythmicity in primary myotubes, myoblasts were isolated from transgenic mice in which luciferase coding sequence is fused to the C-terminus of PER2 expressed from the endogenous Per2 locus (B6.129S6-Per2tm1Jt/J; Jackson Labs strain # 006852) (Yoo et al., 2004). Per2Luc myoblasts were plated at 1×106 cells per plate in 3.5-cm dishes in DMEM/HAMS F12 1:1 media supplemented with 3% horse serum (Gibco #26050-088), 1% Insulin-Selenium-Transferrin (Invitrogen #41400-045) and 50μg/ml gentamicin. Two days after plating, the differentiating myotubes were treated with 50% horse serum, 1 μM dexamethasone, or 10 μM forskolin in differentiation media containing 100 μM D-luciferin to synchronize circadian rhythms. After 1 hour (dexamethasone or forskolin) or two hours (horse serum), the media was replaced with DMEM/HAMS F12 1:1 media supplemented with 1% horse serum, 1% Insulin-Selenium-Transferrin, 50μg/ml gentamicin and 100 μM D-luciferin. Plates were sealed with vacuum grease (Dow Corning high vacuum grease; VWR cat #59344-055) and glass cover slips (40CIR-1, Fisher Scientific cat #22038999) and placed into the Actimetrics Lumicycle 32. Data was recorded using Actimetrics Lumicycle Data Collection software and analyzed using Actimetrics Lumicycle Analysis program. Background subtraction of the recorded data was performed with Running Average setting, and fit by least mean squares calculation to a damped sine wave to calculate the period, amplitude, and phase. Only data with a goodness of fit percentage of 80 or above was included in the analysis. Analysis of rhythmicity in muscle explants was performed similarly except that the tissue explants were cultured in DMEM supplemented with 5% fetal bovine serum and penicillin-streptomycin. Following synchronization of circadian rhythms, they were transferred to DMEM supplemented with 1% FBS, penicillin-streptomycin and 100 μM D-luciferin.
Cell culture and transfection
Transfections in HEK 293T cells were carried out using calcium phosphate or polyethylenimine (PEI; Polysciences Inc #23966-2) following standard protocols. pcDNA3-2xFlag-mCRY1 and pcDNA3-2xFlag-mCRY2 are described in (Lamia et al., 2009). pcDNA3-Myc-mCRY1 and pcDNA3- Myc-mCRY2 are described in (Huber et. al., 2016). pcDNA3.2-mPPARα-V5, pcDNA3.2-mPPARδ-V5, pcDNA3.2-mPPARγ-V5 and pcDNA3-2xFlag-PPARδ-FL, pcDNA3-2xFlag-PPARδ-DBD, pcDNA3- 2xFlag-PPARδ-LBD are described in (Lamia et al., 2011; Narkar et al., 2008). pcDNA3.2- mPPARδΔH12-V5 was generated using Q5 Site-Directed Mutagenesis kit and protocol (NEB #E0554S).
PPAR ligand treatments
In experiments shown in Figure 2, HEK 293T cells were co-treated with 10μM MG-132 (Millipore #474790-10MG) and isoform-specific PPAR ligand for 6–8 hours using the following concentrations: GW1516 (Enzo #ALX-420-032-M001) 0.1μM and 1μM for PPARδ, WY-14643 (Enzo #GR-200) 1 μM and 10μM for PPARα, and Rosiglitazone (Cayman #71742) 10μM and 100μM for PPARγ.
For gene expression analysis in myotubes, GW1516 (Enzo #ALX-420-032-M001) was used at a final concentration of 100 nM for the indicated times. Ligand treatment experiments were performed in triplicate and repeated at least 3 times.
Co-immunoprecipitation and Western blotting
HEK 293T whole cell extracts were prepared using lysis buffer containing 1%TX-100 as previously described (Lamia et al., 2004). Immunoprecipitation was performed using anti-Flag M2 agarose beads (Sigma #A2220). Antibodies for Western Blots were anti-Flag polyclonal (Sigma cat #F7425), anti-V5 polyclonal (Bethyl Labs cat# A190-120A), anti-βActin (Sigma cat #A1978), anti-Cry1-CT and anti-Cry2-CT as described (Lamia et al., 2011). Anti-PPARδ antibody was raised in guinea pig using a peptide containing the first twenty amino acids of mouse PPARδ (Fan et al., 2017).
Treadmill Exercise Testing
To analyze treadmill running performance in sedentary mice, 11–12-week old male mice were habituated to a motorized treadmill (Exer3/6M with 10° grade, Columbus Instruments) in the week prior to experimentation with three 10-minute sessions of walking/running at a slow speed (first session up to 10 m/min; second session up to 12.5 m/min; third session up to 15 m/min). On the day of experimentation for Protocol I, the treadmill was started at a speed of 6 m/min for a period of 3 minutes. Subsequently, the speed was continuously increased every minute by 1 m/min, until the mouse could no longer sustain that current sprint speed. During the experimental sessions for Protocols II and III, the treadmill was started at a speed of 12.5 m/min and after 5 minutes the speed was increased to 15 m/min, for either 5 minutes (Protocol II) or 25 minutes (Protocol III). Next the speed was increased by 2.5 m/min every 3 minutes until each mouse was no longer able to maintain running. For each animal, dropping off the treadmill more than 3 times during a period of 15 seconds was considered the endpoint of the experiment and maximum speed and accumulated running time was noted. In the experimental test for Protocol IV, the treadmill was started at a speed of 15 m/min for a period of 2 minutes. Subsequently, the treadmill was gradually increased by 1 m/min every 2 minutes until a speed of 20 m/min was reached. The speed then remained constant at 20 m/min for the rest of the experiment, until mice became exhausted and could no longer endure running. Blood glucose levels were assessed via the tail vein (Aviva Accu-chek, Roche Diagnostics) immediately postendurance run to confirm that mice had run to true exhaustion. Mice that did not exhaust blood glucose to <100 mg/dL were excluded from analyses. To calculate the total distance covered by each mouse, the speed and time spent at this speed level were multiplied. Statistical analyses were done using 2-way ANOVA.
Supervised treadmill training
13-week-old male mice were trained 5 days a week for 3 weeks by alternating supervised speed and endurance challenge runs using a motorized treadmill (Exer3/6M with 10° grade, Columbus Instruments). Speed challenge runs were performed twice a week starting at 17.5m/min with 2.5m/min increments for 10 minutes. Endurance challenge runs were performed three times a week with 15 minutes at 17.5 m/min in week 1, 30 minutes at 17.5 m/min in week 2 and 15 minutes at 20m/min in week 3.
Running wheel training
13-week-old male mice were single housed and given access to running wheels for 3 weeks in standard 12h light-12h dark cycle with ad libitum access to food and water. Voluntary running wheel activity was analyzed using digital recordings of wheel rotations.
Treadmill exercise in running-wheel trained mice
Treadmill running performance in running-wheel trained mice was analyzed in 16-week-old male mice directly after the 3-week training period using a motorized treadmill (Exer3/6M with 10° grade, Columbus Instruments). During the experimental sessions, the treadmill was started at a speed of 15 m/min after 5 minutes the speed was increased to 17.5 m/min for either 5 minutes (Protocol 1) or 25 minutes (Protocol 2). Next the speed was increased 2.5 m/min every 3 minutes until each mouse was no longer able to maintain running. For each animal, dropping off the treadmill more than 3 times during a period of 15 seconds was considered the endpoint of the experiment and maximum speed and accumulated running time was noted. To calculate the total distance covered by each mouse speed and time spent at this speed level were multiplied. Statistical analyses were done using two-tailed student’s t-test.
Body Composition Analysis by EchoMRI
Following calibration, un-anaesthetized male mice were placed in a glass cylinder one at a time, and a weighted adjustable plastic restrainer was placed on top of the mouse to prevent movement during imaging. Each mouse was restrained for approximately two minutes while measuring whole body fat, lean, free water, and total water masses using an EchoMRI 3-in-1 instrument (EchoMRI LLC, Houston, TX).
Immunohistochemistry and histochemistry
Gastrocnemius, soleus, and plantaris muscles were dissected and freshly frozen in liquid N2- cooled isopentane. 10μm cryo-sections were prepared using a cryostat (Zeiss). Neighboring sections were stained for fiber-typing using monoclonal antibodies BA-F8 (type I), SC-71 (IIa), and BF-F3 (IIb), obtained from DSHB at the University of Iowa, and mitochondrial complex I/IV activities with NADH tetrazolium reductase and cytochrome C oxidase stainings. All stainings were performed following a previously published protocol (De Paepe et al., 2009). For fiber type quantification, a total of 2–4 tissue sections of 7 mice per genotype were scanned using an automated slide scanner (Zeiss Axio Scan.Z1) and analyzed by either quantitative fluorescence (gastrocnemius) or manual count (plantaris) using Image J. Statistical analyses were done using two-tailed student’s t-test.
Muscle Isolation and Staining for Fiber Size and Vascularization
Immediately following euthanasia, skinned, intact legs were isolated from mice, rinsed with PBS and then quickly placed into PBS on ice. Following all isolations (n = 3 per group), legs were pinned onto cork dissection boards so that the gastrocnemius/plantaris/soleus complex and quadriceps muscles were stretched, thereby enabling the preservation of tissue integrity during fixation. Legs were fixed overnight in 4% paraformaldehyde at 4°C. Following fixation, the abovementioned muscles were individually dissected and cryopreserved using a sucrose gradient. Muscles were placed sequentially into 1%, 5% and 20% sucrose solutions (w/v in PBS) until equilibration. Muscle samples were then placed into OCT (Tissue-Tek) and flash-frozen using a dry ice/ethanol slurry, after which samples were maintained frozen at -80°C. For cryosectioning, muscle samples were cut into 12μm transverse sections, captured onto slides and air dried. Sections were then rehydrated with PBS containing 0.1% Triton for 15 minutes and permeabilized for 15 minutes with PBS containing 0.3% Triton. Samples were blocked with 4% BSA/1% goat serum (in PBST) for 1–2 hours at room temperature, followed by overnight incubation with primary antibodies in blocking buffer (rabbit anti-laminin, 1:200, Sigma and mouse anti-CD31, 1:200, BD Biosciences), at 4°C in a humidifying chamber. The next day, sections were washed 3 times (20 minutes each) in PBST (0.1%) and incubated at room temperature for 1.5 hours with secondary antibodies (Alexa-Fluor conjugated 594 goat anti-rabbit, 1:200 and 488 goat anti-rat, 1:200; Jackson Immunoresearch), as well as conjugated dyes for F-actin and nuclei (Alexa Fluor conjugated 647 phalloidin, 1:200; Thermo Fisher and Hoechst, 1:1000; Thermo Fisher). Sections were again washed 3 times in PBST (0.1%) and mounted with ProLong Gold mounting reagent (Thermo Fisher). Images were acquired using a digital BZ-X700 Keyance microscope and imaging system (Keyence Corporation, Japan). Five digital images were taken from non-overlapping fields (at 40X) for each muscle section, per mouse; totaling ~15 fields for each group. Muscle fiber cross-sectional area, as well as fiber and capillary density were quantified using Keyence BZ-X analysis software. Staining, imaging and analysis were performed by a single experimenter blinded to mouse genotype.
Mechanical testing of EDL and soleus muscles
Dissection was performed in mammalian Ringer’s solution containing (in mM): NaCl (137), KCl (5), NaH2PO4 (1), NaHCO3 (24), CaCl2 (2), MgSO4 (1) and glucose (11) containing 10 mg/l curare. Proximal and distal tendons of the muscles were tied off as close to the fibers as possible to minimize tendon-series compliance using 6 – 0 silk suture (Harvard Apparatus, Holliston, MA). The proximal and distal tendons were then transected, and the muscle was placed into a custom testing chamber filled with Ringer’s solution, the distal tendon was secured to the arm of a dual-mode ergometer (model 300B; Aurora Scientific, Richmond Hill, ON, Canada), and the proximal tendon was secured to a fixed stainless steel pin. Muscle slack was taken up, and muscle length was measured. The muscle was then adjusted to a sarcomere length of 3 μm confirmed using laser diffraction, then muscle fiber length was measured. To determine the voltage that would yield maximal force production, the muscle was stimulated with an electrical stimulator (Pulsar 6bp, FHC, Inc., Bowdoin, ME) via platinum plate electrodes that extended the length of the muscle with a pulse duration of 0.3 ms. Muscle twitches were administered beginning with a twitch voltage of 4 V, and twitch voltage was increased by 1 V until the output by the muscle no longer increased in response. This peak twitch voltage was doubled to ensure supramaximal stimulation of the muscles. Two twitches were recorded thirty seconds apart, and analyzed for time to peak force, net peak force, half relaxation time and full width at half maximum. Force frequency characteristics of the muscles were then determined by taking a series of tetanic contractions (400 ms train duration) with a two minute rest between each contraction while increasing the stimulation frequency. The stimulation frequencies used for the soleus were 5 Hz, 10 Hz, 30 Hz, 50 Hz, 70 Hz, 90 Hz, and 120 Hz. The stimulation frequencies used for the EDL were 5 Hz, 10 Hz, 20 Hz, 40 Hz, 60 Hz, 80 Hz, and 100 Hz. The force frequency data was analyzed for peak stress, half fusion frequency and fusion frequency. The muscle then underwent a fatigue protocol consisting of 300 tetanic contractions delivered every three seconds using a stimulation frequency corresponding to the half fusion frequency and a 330 ms train duration.
Mitochondrial DNA quantification
Genomic DNA was isolated using Trizol (Life Technologies #15596018) following a standard protocol for DNA recovery from the organic phase using back extraction buffer (4M guanidine thiocyanate, 50mM sodium citrate, 1M Tris). 10ng of gDNA were used to determine the relative amount of mitochondrial DNA by quantitative real-time PCR using SYBR Green (Life Technologies #4367659) and primers detecting the mitochondrial Cox I gene and the nuclear RNaseP gene for normalization. All samples were measured in technical triplicates using QuantStudio 6 Flex Real Time PCR system (Applied Biosystems). Statistical analyses were done using two-tailed student’s t-test.
Mitochondrial protein quantification
Isolated mouse quadriceps muscles were weighed before mincing on ice for 2–3 minutes and then placed in a glass mortar containing 10mL of HM Buffer (70mM sucrose, 110mM KCL, 1mM EGTA, 20mM MOPS, pH 7.5). Minced quadriceps were subjected to homogenization using a fitted Teflon pestle attached to a Potter homogenizer. Tissue homogenates were centrifuged at 4°C for 10 minutes at 800g. The resulting supernatant was collected and the centrifugation step was repeated 1–2 more times, until the supernatant appeared clear. Clean supernatants were centrifuged at 4°C for 10 minutes at 8000 × g. The resulting pellets were resuspended in 100–300 μl of H Buffer (210mM mannitol, 70mM sucrose, 1mM EGTA, 0.2% fatty acid free BSA, 5mM HEPES, pH 7.2). An aliquot of isolated mitochondria was analyzed for protein content and compared to initial tissue weight to determine relative amount of mitochondrial protein. Isolated mitochondria were stored on ice for 4–6 hours prior to Seahorse assay in which 2–4 μg isolated mitochondria were used in each well of a 96-well Seahorse plate.
Analysis of mitochondrial OXPHOS complexes
Muscle tissue was prepared as previously described (Philp et al., 2011). Equal amounts of protein (20μg) were separated on 10% Bis-Tris gels and immunoblotting was performed following standard protocols. Mitochondrial OXPHOS subunits were detected using Mitoprofile total OXPHOS antibody mixture (MS604) from Mitosciences according to the manufacturer’s instructions.
Timing of experiments and tissue collections
Exercise assessment protocols were started at ZT8 (protocols I, II, III) or ZT6 (protocol IV). Mice from which quadriceps were isolated for RNA isolation and deep sequencing were sacrificed at ZT9; for those subjected to acute exercise prior to sacrifice, it was initiated at ZT8.
Quantitative PCR
RNA was extracted using Trizol (Life Technologies #15596018) and RNeasy Mini Kit (Qiagen #74106). Muscle tissue samples were homogenized in 1ml Trizol using a Precellys homogenizer with attached Cryolys for 30–90s at 5000rpm at 4°C prior to RNA extraction. cDNA was prepared using QuantiTect Reverse Transcription Kit (Qiagen #205314). Quantitative real-time PCR used SYBR Green (Life Technologies #4367659) and QuantStudio 6 Flex Real Time PCR system (Applied Biosystems). See Table S2 for primer sequences.
RNA sequencing
Total RNAs were extracted from the glycolytic white quadriceps muscle with Trizol (Life Technologies #15596018) and the RNeasy Mini kit with on-column DNase treatment (Qiagen). RNA quality was confirmed using the Agilent 2100 Bioanalyzer and RNA-Seq libraries prepared using the TruSeq RNA Sample Preparation Kit v2 according to Illumina protocol. Multiplexed libraries were validated using the Agilent BioAnalyzer, normalized and pooled for sequencing. High-throughput sequencing was performed on the HiSeq 2500 system (Illumina) with a 100-bp read length.
Quantification and statistical analysis
Detailed descriptions of sample numbers and statistical tests can be found in Figure Legends and Method Details. In general, ANOVA analyses were used for data sets with more than two groups and student’s t-tests were used for datasets with only two groups. Statistics were calculated in either Microsoft Excel (t-tests) or GraphPad Prism (ANOVA and t-tests). For RNA-Seq, image analysis and base calling were done with Illumina CASAVA-1.8.2. Short read sequences were mapped to a UCSC mm9 reference sequence using the RNA-Seq aligner STAR (Dobin et al., 2013). Known splice junctions from mm9 were supplied to the aligner and de novo junction discovery was also permitted. Transcript expression was calculated as gene-level relative abundance in fragments per kilobase of exon model per million mapped fragments and employed correction for transcript abundance bias (Roberts et al., 2011). Differential gene expression analysis, statistical testing and annotation were performed using Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2007).
Data and software availability
RNA-Seq data reported in this paper have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database, Accession # SRP090320.
Supplementary Material
Highlights.
CRY1 and CRY2 interact with PPARδ in a ligand-dependent manner.
CRY1/2 repress PPARδ target gene expression in myotubes.
Genetic ablation of CRY1 and CRY2 increases exercise capacity in mice.
Exercise-induced muscle gene signature is altered in CRY−/− mice.
Acknowledgments
This work was funded by NIH grants K01 DK090188 and R01 DK097164 (to K.A.L.), R37 DK057978, R01 HL105278, and CCSG P30 CA014195 (to R.M.E.), R01 DK105126 (to A.Kralli), 1S10OD16357 which funded the Seahorse Instrument at The Scripps Research Institute, P30 AR061303 San Diego Muscle Research Center (SDMRC), R24 HD050837 National Skeletal Muscle Resource Center (NSMRC), a Searle Scholars award to K.A.L. from the Kinship Foundation, by the Helmsley Charitable Trust to R.M.E., by fellowships from Deutsche Forschungsgemeinschaft to S.D.J., from the American Heart Association (15POST22510020 to S.D.J. and 16PRE3041001 to S.J.P.) and from the Swedish Research Council to E.H. R.M.E is an investigator of the Howard Hughes Medical Institute and March of Dimes Chair in Molecular and Developmental Biology at the Salk Institute. We thank the Cancer Research Laboratory Molecular Imaging Center at UC Berkeley and the Biology Faculty Research Fund for use of a Slide Scanner. We thank Enrique Saez, Simon Schenk, Richard Lieber, Velia Fowler, Jamie Williamson, Shannon Bremmner, Carsten Merkwirth and Andrew Dillin for helpful discussions, sharing of technical expertise, equipment and reagents and/or critical reading of the manuscript, and J. Valecko for administrative assistance.
Footnotes
AUTHOR CONTRIBUTIONS
S.D.J., A. Kriebs, D.D., W.F., M.V., E.H., A.-L. H., S.J.P., M.N., M.A., and K.A.L. performed experiments; S.D.J., A. Kriebs, D.D., M.V., M.A., W.F., M.D., R.Y., and K.A.L. analyzed data; S.D.J. and K.A.L. made figures and wrote the paper with input from the other authors; A. Kralli developed methods for isolating and differentiating primary mouse myoblasts and helped design and interpret the experiments using primary myotubes and examining treadmill exercise performance; R.M.E. and K.A.L. conceived and supervised the study. All authors edited and approved the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe T, Kitaoka Y, Kikuchi DM, Takeda K, Numata O, Takemasa T. High-intensity interval training-induced metabolic adaptation coupled with an increase in Hif-1alpha and glycolytic protein expression. Journal of applied physiology. 2015;119:1297–1302. doi: 10.1152/japplphysiol.00499.2015. [DOI] [PubMed] [Google Scholar]
- Adamovich Y, Ladeuix B, Golik M, Koeners MP, Asher G. Rhythmic Oxygen Levels Reset Circadian Clocks through HIF1alpha. Cell metabolism. 2017;25:93–101. doi: 10.1016/j.cmet.2016.09.014. [DOI] [PubMed] [Google Scholar]
- Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, Evans RM. PPARgamma signaling and metabolism: the good, the bad and the future. Nature medicine. 2013;19:557–566. doi: 10.1038/nm.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews JL, Zhang X, McCarthy JJ, McDearmon EL, Hornberger TA, Russell B, Campbell KS, Arbogast S, Reid MB, Walker JR, et al. CLOCK and BMAL1 regulate MyoD and are necessary for maintenance of skeletal muscle phenotype and function. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:19090–19095. doi: 10.1073/pnas.1014523107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae K, Lee K, Seo Y, Lee H, Kim D, Choi I. Differential effects of two period genes on the physiology and proteomic profiles of mouse anterior tibialis muscles. Molecules and cells. 2006;22:275–284. [PubMed] [Google Scholar]
- Balsalobre A, Marcacci L, Schibler U. Multiple signaling pathways elicit circadian gene expression in cultured Rat-1 fibroblasts. Current biology: CB. 2000;10:1291–1294. doi: 10.1016/s0960-9822(00)00758-2. [DOI] [PubMed] [Google Scholar]
- Barish GD, Narkar VA, Evans RM. PPAR delta: a dagger in the heart of the metabolic syndrome. The Journal of clinical investigation. 2006;116:590–597. doi: 10.1172/JCI27955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126:789–799. doi: 10.1016/j.cell.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies CT, Few JD. Effects of exercise on adrenocortical function. J Appl Physiol. 1973;35:887–891. doi: 10.1152/jappl.1973.35.6.887. [DOI] [PubMed] [Google Scholar]
- De Paepe B, De Bleecker JL, Van Coster R. Histochemical methods for the diagnosis of mitochondrial diseases. Current protocols in human genetics/ editorial board, Jonathan L. Haines et al. 2009;Chapter 19(Unit19):12. doi: 10.1002/0471142905.hg1902s63. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowell P, Ishmael JE, Avram D, Peterson VJ, Nevrivy DJ, Leid M. Identification of nuclear receptor corepressor as a peroxisome proliferator-activated receptor alpha interacting protein. The Journal of biological chemistry. 1999;274:15901–15907. doi: 10.1074/jbc.274.22.15901. [DOI] [PubMed] [Google Scholar]
- Dyar KA, Ciciliot S, Wright LE, Bienso RS, Tagliazucchi GM, Patel VR, Forcato M, Paz MI, Gudiksen A, Solagna F, et al. Muscle insulin sensitivity and glucose metabolism are controlled by the intrinsic muscle clock. Molecular metabolism. 2014;3:29–41. doi: 10.1016/j.molmet.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenborg E, Krook A. Regulation of skeletal muscle physiology and metabolism by peroxisome proliferator-activated receptor delta. Pharmacological reviews. 2009;61:373–393. doi: 10.1124/pr.109.001560. [DOI] [PubMed] [Google Scholar]
- Fan W, Waizenegger W, Lin CS, Sorrentino V, He MX, Wall CE, Li H, Liddle C, Yu RT, Atkins AR, et al. PPARdelta Promotes Running Endurance by Preserving Glucose. Cell metabolism. 2017;25:1186–1193. e1184. doi: 10.1016/j.cmet.2017.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang B, Everett LJ, Jager J, Briggs E, Armour SM, Feng D, Roy A, Gerhart-Hines Z, Sun Z, Lazar MA. Circadian enhancers coordinate multiple phases of rhythmic gene transcription in vivo. Cell. 2014;159:1140–1152. doi: 10.1016/j.cell.2014.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favier FB, Britto FA, Freyssenet DG, Bigard XA, Benoit H. HIF-1-driven skeletal muscle adaptations to chronic hypoxia: molecular insights into muscle physiology. Cellular and molecular life sciences: CMLS. 2015;72:4681–4696. doi: 10.1007/s00018-015-2025-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitts RH. Cellular mechanisms of muscle fatigue. Physiological reviews. 1994;74:49–94. doi: 10.1152/physrev.1994.74.1.49. [DOI] [PubMed] [Google Scholar]
- Goto M, Terada S, Kato M, Katoh M, Yokozeki T, Tabata I, Shimokawa T. cDNA Cloning and mRNA analysis of PGC-1 in epitrochlearis muscle in swimming-exercised rats. Biochemical and biophysical research communications. 2000;274:350–354. doi: 10.1006/bbrc.2000.3134. [DOI] [PubMed] [Google Scholar]
- Grimaldi B, Bellet MM, Katada S, Astarita G, Hirayama J, Amin RH, Granneman JG, Piomelli D, Leff T, Sassone-Corsi P. PER2 controls lipid metabolism by direct regulation of PPARgamma. Cell metabolism. 2010;12:509–520. doi: 10.1016/j.cmet.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfmann BD, Schroder EA, Esser KA. Circadian rhythms, the molecular clock, and skeletal muscle. Journal of biological rhythms. 2015;30:84–94. doi: 10.1177/0748730414561638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge BA, Wen Y, Riley LA, Zhang X, England JH, Harfmann BD, Schroder EA, Esser KA. The endogenous molecular clock orchestrates the temporal separation of substrate metabolism in skeletal muscle. Skeletal muscle. 2015;5:17. doi: 10.1186/s13395-015-0039-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber AL, Papp SJ, Chan AB, Henriksson E, Jordan SD, Kriebs A, Nguyen M, Wallace M, Li Z, Metallo CM, et al. CRY2 and FBXL3 Cooperatively Degrade c-MYC. Molecular cell. 2016;64:774–789. doi: 10.1016/j.molcel.2016.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobi D, Liu S, Burkewitz K, Kory N, Knudsen NH, Alexander RK, Unluturk U, Li X, Kong X, Hyde AL, et al. Hepatic Bmal1 Regulates Rhythmic Mitochondrial Dynamics and Promotes Metabolic Fitness. Cell metabolism. 2015;22:709–720. doi: 10.1016/j.cmet.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, Takahashi JS. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science. 2012;338:349–354. doi: 10.1126/science.1226339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamia KA, Papp SJ, Yu RT, Barish GD, Uhlenhaut NH, Jonker JW, Downes M, Evans RM. Cryptochromes mediate rhythmic repression of the glucocorticoid receptor. Nature. 2011;480:552–556. doi: 10.1038/nature10700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamia KA, Peroni OD, Kim YB, Rameh LE, Kahn BB, Cantley LC. Increased insulin sensitivity and reduced adiposity in phosphatidylinositol 5-phosphate 4-kinase beta−/− mice. Molecular and cellular biology. 2004;24:5080–5087. doi: 10.1128/MCB.24.11.5080-5087.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamia KA, Sachdeva UM, DiTacchio L, Williams EC, Alvarez JG, Egan DF, Vasquez DS, Juguilon H, Panda S, Shaw RJ, et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science. 2009;326:437–440. doi: 10.1126/science.1172156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell systems. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcheva B, Ramsey KM, Peek CB, Affinati A, Maury E, Bass J. Circadian clocks and metabolism. Handbook of experimental pharmacology. 2013:127–155. doi: 10.1007/978-3-642-25950-0_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Redondo V, Pettersson AT, Ruas JL. The hitchhiker’s guide to PGC-1alpha isoform structure and biological functions. Diabetologia. 2015;58:1969–1977. doi: 10.1007/s00125-015-3671-z. [DOI] [PubMed] [Google Scholar]
- Maury E, Hong HK, Bass J. Circadian disruption in the pathogenesis of metabolic syndrome. Diabetes & metabolism. 2014;40:338–346. doi: 10.1016/j.diabet.2013.12.005. [DOI] [PubMed] [Google Scholar]
- McCarthy JJ, Andrews JL, McDearmon EL, Campbell KS, Barber BK, Miller BH, Walker JR, Hogenesch JB, Takahashi JS, Esser KA. Identification of the circadian transcriptome in adult mouse skeletal muscle. Physiological genomics. 2007;31:86–95. doi: 10.1152/physiolgenomics.00066.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. The Journal of cell biology. 2016;212:379–387. doi: 10.1083/jcb.201511036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature genetics. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Mounier R, Theret M, Lantier L, Foretz M, Viollet B. Expanding roles for AMPK in skeletal muscle plasticity. Trends in endocrinology and metabolism: TEM. 2015;26:275–286. doi: 10.1016/j.tem.2015.02.009. [DOI] [PubMed] [Google Scholar]
- Nagoshi E, Saini C, Bauer C, Laroche T, Naef F, Schibler U. Circadian gene expression in individual fibroblasts: cell-autonomous and self-sustained oscillators pass time to daughter cells. Cell. 2004;119:693–705. doi: 10.1016/j.cell.2004.11.015. [DOI] [PubMed] [Google Scholar]
- Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, Mihaylova MM, Nelson MC, Zou Y, Juguilon H, et al. AMPK and PPARdelta agonists are exercise mimetics. Cell. 2008;134:405–415. doi: 10.1016/j.cell.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld-Cohen A, Robles MS, Aviram R, Manella G, Adamovich Y, Ladeuix B, Nir D, Rousso-Noori L, Kuperman Y, Golik M, et al. Circadian control of oscillations in mitochondrial rate-limiting enzymes and nutrient utilization by PERIOD proteins. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:E1673–1682. doi: 10.1073/pnas.1519650113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partch CL, Green CB, Takahashi JS. Molecular architecture of the mammalian circadian clock. Trends in cell biology. 2014;24:90–99. doi: 10.1016/j.tcb.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peek CB, Affinati AH, Ramsey KM, Kuo HY, Yu W, Sena LA, Ilkayeva O, Marcheva B, Kobayashi Y, Omura C, et al. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science. 2013;342:1243417. doi: 10.1126/science.1243417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peek CB, Levine DC, Cedernaes J, Taguchi A, Kobayashi Y, Tsai SJ, Bonar NA, McNulty MR, Ramsey KM, Bass J. Circadian Clock Interaction with HIF1alpha Mediates Oxygenic Metabolism and Anaerobic Glycolysis in Skeletal Muscle. Cell metabolism. 2017;25:86–92. doi: 10.1016/j.cmet.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Schindler J, Summermatter S, Salatino S, Zorzato F, Beer M, Balwierz PJ, van Nimwegen E, Feige JN, Auwerx J, Handschin C. The corepressor NCoR1 antagonizes PGC-1alpha and estrogen-related receptor alpha in the regulation of skeletal muscle function and oxidative metabolism. Molecular and cellular biology. 2012;32:4913–4924. doi: 10.1128/MCB.00877-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philp A, Chen A, Lan D, Meyer GA, Murphy AN, Knapp AE, Olfert IM, McCurdy CE, Marcotte GR, Hogan MC, et al. Sirtuin 1 (SIRT1) deacetylase activity is not required for mitochondrial biogenesis or peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) deacetylation following endurance exercise. The Journal of biological chemistry. 2011;286:30561–30570. doi: 10.1074/jbc.M111.261685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan C, Xu H, Khan SK, Shen Y, Gitis PJ, Welsh DK, Hogenesch JB, Liu AC. Cell type-specific functions of period genes revealed by novel adipocyte and hepatocyte circadian clock models. PLoS genetics. 2014;10:e1004244. doi: 10.1371/journal.pgen.1004244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renaud JP, Rochel N, Ruff M, Vivat V, Chambon P, Gronemeyer H, Moras D. Crystal structure of the RAR-gamma ligand-binding domain bound to all-trans retinoic acid. Nature. 1995;378:681–689. doi: 10.1038/378681a0. [DOI] [PubMed] [Google Scholar]
- Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome biology. 2011;12:R22. doi: 10.1186/gb-2011-12-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H, Hattori Y, Ikeda Y, Kamagata M, Iwami S, Yasuda S, Tahara Y, Shibata S. Forced rather than voluntary exercise entrains peripheral clocks via a corticosterone/noradrenaline increase in PER2::LUC mice. Scientific reports. 2016;6:27607. doi: 10.1038/srep27607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmutz I, Ripperger JA, Baeriswyl-Aebischer S, Albrecht U. The mammalian clock component PERIOD2 coordinates circadian output by interaction with nuclear receptors. Genes & development. 2010;24:345–357. doi: 10.1101/gad.564110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- Stanley IA, Ribeiro SM, Gimenez-Cassina A, Norberg E, Danial NN. Changing appetites: the adaptive advantages of fuel choice. Trends in cell biology. 2014;24:118–127. doi: 10.1016/j.tcb.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP. GSEA-P: a desktop application for Gene Set Enrichment Analysis. Bioinformatics. 2007;23:3251–3253. doi: 10.1093/bioinformatics/btm369. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Yamamoto J, Iwasaki S, Asaba H, Hamura H, Ikeda Y, Watanabe M, Magoori K, Ioka RX, Tachibana K, et al. Activation of peroxisome proliferator-activated receptor delta induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:15924–15929. doi: 10.1073/pnas.0306981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thresher RJ, Vitaterna MH, Miyamoto Y, Kazantsev A, Hsu DS, Petit C, Selby CP, Dawut L, Smithies O, Takahashi JS, et al. Role of mouse cryptochrome blue-light photoreceptor in circadian photoresponses. Science. 1998;282:1490–1494. doi: 10.1126/science.282.5393.1490. [DOI] [PubMed] [Google Scholar]
- Um JH, Yang S, Yamazaki S, Kang H, Viollet B, Foretz M, Chung JH. Activation of 5′-AMP-activated kinase with diabetes drug metformin induces casein kinase Iepsilon (CKIepsilon)-dependent degradation of clock protein mPer2. The Journal of biological chemistry. 2007;282:20794–20798. doi: 10.1074/jbc.C700070200. [DOI] [PubMed] [Google Scholar]
- Vieira E, Nilsson EC, Nerstedt A, Ormestad M, Long YC, Garcia-Roves PM, Zierath JR, Mahlapuu M. Relationship between AMPK and the transcriptional balance of clockrelated genes in skeletal muscle. American journal of physiology. Endocrinology and metabolism. 2008;295:E1032–1037. doi: 10.1152/ajpendo.90510.2008. [DOI] [PubMed] [Google Scholar]
- Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR, Ham J, Kang H, Evans RM. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS biology. 2004;2:e294. doi: 10.1371/journal.pbio.0020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinmann M, Belka C, Guner D, Goecke B, Muller I, Bamberg M, Jendrossek V. Array-based comparative gene expression analysis of tumor cells with increased apoptosis resistance after hypoxic selection. Oncogene. 2005;24:5914–5922. doi: 10.1038/sj.onc.1208748. [DOI] [PubMed] [Google Scholar]
- Wolff G, Esser KA. Scheduled exercise phase shifts the circadian clock in skeletal muscle. Medicine and science in sports and exercise. 2012;44:1663–1670. doi: 10.1249/MSS.0b013e318255cf4c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Tang D, Liu N, Xiong W, Huang H, Li Y, Ma Z, Zhao H, Chen P, Qi X, et al. Reciprocal Regulation between the Circadian Clock and Hypoxia Signaling at the Genome Level in Mammals. Cell metabolism. 2017;25:73–85. doi: 10.1016/j.cmet.2016.09.009. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Williams EG, Mouchiroud L, Canto C, Fan W, Downes M, Heligon C, Barish GD, Desvergne B, Evans RM, et al. NCoR1 is a conserved physiological modulator of muscle mass and oxidative function. Cell. 2011;147:827–839. doi: 10.1016/j.cell.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Downes M, Yu RT, Bookout AL, He W, Straume M, Mangelsdorf DJ, Evans RM. Nuclear receptor expression links the circadian clock to metabolism. Cell. 2006;126:801–810. doi: 10.1016/j.cell.2006.06.050. [DOI] [PubMed] [Google Scholar]
- Yang X, Lamia KA, Evans RM. Nuclear receptors, metabolism, and the circadian clock. Cold Spring Harbor symposia on quantitative biology. 2007;72:387–394. doi: 10.1101/sqb.2007.72.058. [DOI] [PubMed] [Google Scholar]
- Yoo SH, Yamazaki S, Lowrey PL, Shimomura K, Ko CH, Buhr ED, Siepka SM, Hong HK, Oh WJ, Yoo OJ, et al. PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:5339–5346. doi: 10.1073/pnas.0308709101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang EE, Liu Y, Dentin R, Pongsawakul PY, Liu AC, Hirota T, Nusinow DA, Sun X, Landais S, Kodama Y, et al. Cryptochrome mediates circadian regulation of cAMP signaling and hepatic gluconeogenesis. Nature medicine. 2010;16:1152–1156. doi: 10.1038/nm.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B, Gates LA, Stashi E, Dasgupta S, Gonzales N, Dean A, Dacso CC, York B, O’Malley BW. Coactivator-Dependent Oscillation of Chromatin Accessibility Dictates Circadian Gene Amplitude via REV-ERB Loading. Molecular cell. 2015;60:769–783. doi: 10.1016/j.molcel.2015.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.