

Abstract
Objective
Sepsis remains an unresolved clinical problem. Therapeutic strategies focusing on inhibition of neutrophils (PMNs) have failed, which indicates that a more detailed understanding of the underlying pathophysiology of sepsis is required. PMN activation and chemotaxis require cellular ATP release via pannexin-1 (pnx1) channels that fuel autocrine feedback via purinergic receptors. In the current study, we examined the roles of endogenous and systemic ATP on PMN activation and host defense in sepsis.
Design
Prospective randomized animal investigation and in vitro studies.
Setting
Preclinical academic research laboratory.
Subjects
Wild-type (WT) C57BL/6 mice, pnx1 knockout (KO) mice, and healthy human subjects used to obtain PMNs for in vitro studies.
Interventions
WT and pnx1 KO mice were treated with suramin or apyrase to block the endogenous or systemic effects of ATP. Mice were subjected to cecal ligation and puncture (CLP) and PMN activation (CD11b expression), organ (liver) injury (plasma aspartate aminotransferase; AST), bacterial spread, and survival were monitored. Human PMNs were used to study the effect of systemic ATP and apyrase on chemotaxis.
Measurements and Main Results
Targeting endogenous ATP reduced PMN activation and organ injury, but increased the spread of bacteria and mortality in sepsis. By contrast, targeting of systemic ATP improved bacterial clearance and survival in sepsis by improving PMN chemotaxis.
Conclusions
Systemic ATP impairs PMN functions by disrupting the endogenous purinergic signaling mechanisms that regulate cell activation and chemotaxis. Targeting systemic ATP improves PMN function and host defenses, making this a promising new treatment strategy for sepsis.
Keywords: Sepsis, cecal ligation and puncture, systemic inflammatory response syndrome (SIRS), multiple organ dysfunction syndrome (MODS), autocrine purinergic signaling, neutrophil chemotaxis
INTRODUCTION
Sepsis is a leading cause of death worldwide (1). Despite intensive research, there are still no effective treatments available today (1–2). Much emphasis has been placed on strategies to block the inflammatory response that causes secondary host organ damage. However, the disappointing results of several recent clinical trials have raised doubts about the feasibility of this approach and renewed interest has focused on immunosuppression in sepsis (3–5).
The inflammatory response to sepsis involves the activation of polymorphonuclear neutrophils (PMNs) that cause collateral damage to host organs such as the lung, liver, and kidney. In the past, we studied the activation processes that regulate PMN functions with the hope of finding novel targets that allow modulation of PMNs in critical care patients. We found that cellular ATP release via pannexin-1 channels (pnx1) and autocrine stimulation of purinergic receptors are essential steps in PMN activation (6). ATP release and purinergic signaling regulate complex cell functions such as chemotaxis (6–12). Chemotactic stimuli like the bacterial peptide N-formyl-met-leu-phe (fMLP) trigger ATP release from the PMN surface closest to the chemotactic source (7). This initial ATP release stimulates P2Y2 receptors that define the leading edge and guide PMN chemotaxis. Ectonucleotidases (e.g., CD39) and A2a adenosine receptors also contribute to chemotaxis (8–10). Together, P2Y2, A2a, and other purinergic receptors elicit excitatory and inhibitory responses at the front and back of cells that resulting in a push-pull mechanism that guides the movement of PMNs to sites of infection (10–12). Inhibition or genetic ablation of pnx1, P2Y2, or A2a receptors impairs chemotaxis (6,7). However, these endogenous purinergic signaling mechanisms of PMNs can be impaired by external, systemic ATP that is released in response to inflammatory processes or host tissue damage (13–15). Sepsis causes the release of large amounts of ATP that promotes PMN activation (15).
In the current study, we examined how endogenous ATP release and systemic ATP affect PMN functions in sepsis. We found that systemic ATP in sepsis promotes PMN activation but that it also impairs PMN chemotaxis and the ability of PMNs to locate and eliminate invading bacteria. We could show that removal of systemic ATP restores PMN chemotaxis, improves bacterial clearance, and reduced mortality in a mouse model of sepsis.
MATERIALS AND METHODS
Mouse model of sepsis
All animal experiments were approved by the Institutional Animal Care and Use Committee of the Beth Israel Deaconess Medical Center (BIDMC) and performed according to the guidelines of the National Institutes of Health. C57BL/6 wild-type (WT) mice were obtained from Charles River Laboratories (Wilmington, MA). Pannexin-1 knock-out (pnx1 KO) mice were from The Knockout Mouse Project Center for Comparative Medicine (University of California, Davis, CA) and backcrossed into the C57BL/6 background. For all experiments, male mice, aged 8 to 10 weeks and weighing 20–25 g were used.
Polymicrobial sepsis was induced by cecal ligation and puncture (CLP) as described previously (15). Briefly, mice were anesthetized with isoflurane. Half of the cecum was ligated with 3–0 silk suture and punctured with a 22-G needle. Sham animals were subjected to the identical surgical procedure without cecal ligation and puncture. Mice were resuscitated by subcutaneous (s.c.) injection of 1 ml of warmed 0.9% saline solution (Baxter, Deerfield, IL). Buprenorphine (0.1 mg/kg s.c.; Hospira, Lake Forest, IL) was administered for pain control immediately after surgery and every 12 h until euthanasia.
Animal treatment
Suramin (Calbiochem EMD Merck, Darmstadt, Germany) was dissolved in 0.9% saline at a concentration of 4 mM, and mice were treated by dorsal s.c. injection with 4 μl/g body weight (BW) of this solution (16 nmol/g) 10 min before or 60 or 180 min after CLP. Apyrase (Sigma Chemical Co, MO) was administered intraperitoneally (i.p.) at a dose of 0.6 U/g BW (6 μl/g of a 100-IU/ml apyrase solution) 30 min before CLP.
Bacterial counts, white blood cell counts, and AST measurement
Mice were anesthetized and sacrificed by cardiac puncture and heparinized blood was collected. Bronchoalveolar lavage fluid (BALF) was obtained by flushing the lungs twice with 1 ml ice-cold 0.9% saline. The BALF was centrifuged at 2,000 rpm for 10 min at 4°C and the supernatant was collected and stored at −80°C until use. Peritoneal lavage fluid was obtained by injection of 3 ml ice-cold 0.9% saline containing 1% fetal calf serum using a 26-G needle; the abdomen was gently massaged and the peritoneal fluid collected through the same needle after 5 min. Blood, bronchoalveolar and peritoneal lavages were collected and spleens, livers, and lungs homogenized in 1 ml normal saline. Bacterial counts were assessed as previously described (6, 7, 15). Colony forming units (CFUs) were counted after 24 h. White blood cell counts were determined with a hemocytometer and aspartate aminotransferase (AST) activity in plasma was assessed with a commercially available assay kit (Biotron Diagnostics, Inc., Hemet, CA).
Survival
Animals were subjected to CLP and randomly assigned to 3 different treatment groups: control group (0.9% saline i.p. 10 min prior to CLP), suramin group (suramin, 16 nmol/g BW s.c. 10 min prior to CLP), and apyrase group (apyrase, 0.6 IU/g BW i.p. 30 min before CLP). Animals were monitored in 8-h intervals. Vital signs, distress, and discomfort were scored and moribund animals sacrificed as needed. After 72 h, all remaining mice were sacrificed.
In vivo PMN activation
Neutrophil activation was assessed by measuring CD11b on the surface of Ly-6G positive leukocytes using flow cytometry as previously described (15). Blood samples were subjected to red blood cells (RBCs) lysis, washed, and incubated with allophycocyanin (APC)-conjugated anti-mouse CD11b antibodies (eBioscience; 1:100) and phycoerythrin (PE)-conjugated anti-mouse Ly-6G antibodies (eBioscience; 1:200). Then samples were washed, fixed, and analyzed with a BD FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA). For gating, isotype controls were used and only debris and platelets were exclude from analysis. In some experiments, blood samples were stimulated in vitro with 100 μM W-peptide for 15 min at 37°C.
In vitro PMN chemotaxis
The Institutional Review Board of BIDMC approved all studies involving human subjects. PMNs were isolated from healthy volunteers and chemotaxis was assessed using a live cell imaging system as described previously (7). Briefly, freshly isolated PMNs plated onto 25-mm glass coverslips coated with fibronectin were placed under a microscope (Leica DMI6000B, Leica, Wetzlar, Germany) and the movement of cells in a chemoattractant gradient field generated with a micropipette loaded with 100 nM fMLP was recorded by time-lapse video microscopy (20 serial images at 20-s intervals). From these images, migration speeds and directionality of individual cells were calculated (10).
Statistical analysis
Data are expressed as mean ± SEM (standard error of the mean) unless stated otherwise. Statistical analyses were done using 2-tailed, unpaired Student’s t-tests or Mann-Whitney U test when comparing two groups, or one-way analysis of variance (ANOVA) and Fisher’s post-hoc least significant difference test for multiple comparisons. The survival data were analyzed by the Kaplan-Meier test. Differences between groups were considered statistically significant at p<0.05. All statistical analyses were performed using Statistical Package for the Social Sciences (SPSS; IBM, Chicago, IL).
RESULTS
Pannexin-dependent and independent mechanisms contribute to PMN activation in sepsis
Sepsis increased the number of CD11b-positive PMNs (suppl. Fig. 1A–C) and the abundance of CD11b molecules on the surface of PMNs (suppl. Fig. 1D). PMN activation peaked between 3 and 12 h after CLP and was paralleled by an increase in WBC counts (suppl. Fig. 2A) and plasma AST levels indicating liver damage (suppl. Fig. 2B). Previously, we have shown that sepsis-induced ATP release in the plasma correlates with PMN activation (15). In response to cell activation, human PMNs release ATP via pannexin 1 (pnx1) channels (6, 8). In order to study the role pnx1 channels in mouse PMNs, we used cells from pnx1 knockout (KO) mice. In contrast to wild-type controls, PMNs from healthy pnx1 KO mice were unresponsive to in vitro stimulation with the mouse formyl peptide receptor (FPR) agonist W-peptide (Fig. 1A). These findings confirm previous work demonstrating that pnx1 channels deliver the ATP that fuels the autocrine purinergic signaling that is needed for PMN activation (6). However, in response to sepsis, PMN of pnx1 KO mice were clearly activated (Fig. 1B–D). Taken together, these findings indicate that pnx1 is required for the endogenous signaling that activate PMNs, but that additional pnx1-independent mechanisms contribute to PMN activation in sepsis.
Figure 1. Pnx1-dependent and independent mechanisms contribute to PMN activation in sepsis.
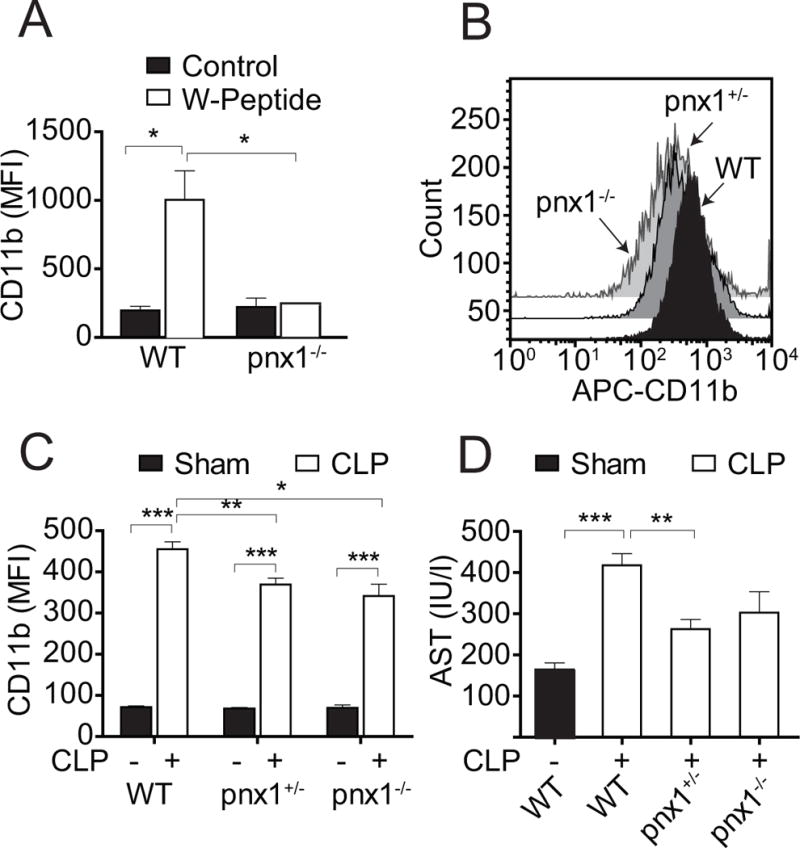
(A) Blood samples of wild-type (WT) and homozygous pnx1 KO mice (pnx1−/−) were stimulated in vitro with W-peptide (100 μM, 15 min at 37°C) and PMN activation was assessed. (B-C) Heterozygous (pnx1+/−) or homozygous (pnx1−/−) pnx1 KO and WT mice were subjected to CLP and PMN activation was determined after 4 h. Panel B shows representative flow cytometer results. Panel C shows cumulative data from mice subjected to CLP or sham surgery. (D) Plasma AST levels of pnx1 KO and WT mice subjected to sham surgery or CLP were assessed 4 h after CLP. Data are expressed as mean ± SEM of 5 animals in each group (ANOVA; *p<0.05, **p<0.01, ***p<0.001).
Pnx1 contributes to bacterial clearance in sepsis
The findings above suggest that pnx1-independent mechanisms deliver most of the ATP that promotes PMN activation in sepsis. In WT mice with CLP, bacteria rapidly spread from the peritoneum to the peripheral blood and the lungs, suggesting inadequate host defense in sepsis (Fig. 2A). In order to study the role of pnx1 channels in host defense, we compared the spread of bacteria in WT and pnx1 KO mice. Mice were subjected to CLP and bacteria were counted in the blood, bronchoalveolar lavage, spleen, liver, lungs, and in the peritoneum 4 h later. With the exception of the liver, the numbers of bacteria were significantly higher in all compartments of homozygous pnx1 KO mice compared to WT mice (Fig. 2B). These findings demonstrate that pnx1-induced ATP release contributes to the defense against invading bacteria. The importance of pnx1 in host defense was further supported by the finding that the mortality rate was higher in pnx1 KO mice than in WT mice (Fig. 2C). Taken together, these results indicate that pnx1 channels contribute to the host defense in sepsis.
Figure 2. Pnx1-dependent mechanisms contribute to host immune defense in sepsis.
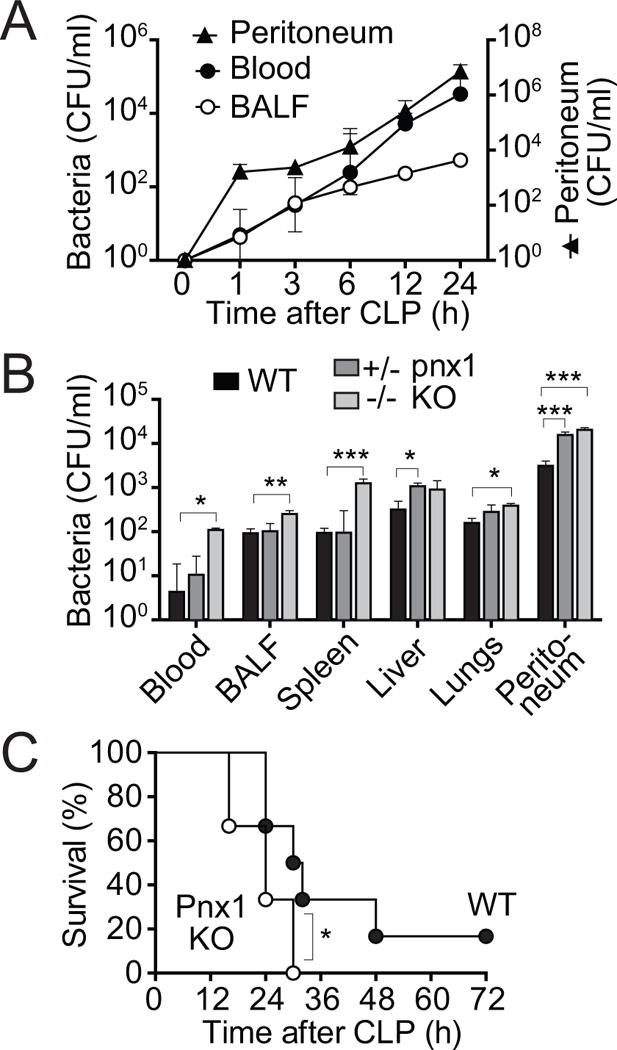
(A) WT mice were subjected to sham surgery (t=0) or CLP for the indicated times. Then bacterial counts (colony forming units; CFU/ml) were determined in peritoneal lavage, blood, and bronchoalveolar lavage (BALF) samples. (B) Heterozygous (pnx1+/−) and homozygous (pnx1−/−) pnx1 KO and WT mice were subjected to CLP for 4 h, sacrificed, and bacterial counts determined in blood, BALF, spleen, liver, lungs, and peritoneal lavage. Data shown represent the mean and SEM of 3–5 mice in each group. Statistical comparisons between groups were made with ANOVA, *p<0.05, **p<0.01, ***p<0.001. (C) WT mice and homozygous (pnx1−/−) pnx1 KO mice were subjected to CLP and survival rates were recorded for a period of 72 h following CLP. Each group consisted of 11 animals. Survival statistics were assessed using the Kaplan-Meier test, *p<0.05.
Blocking purinergic signaling reduces host tissue damage but also host immune defense
The findings above demonstrate that ATP release has dual roles in sepsis, namely by facilitating PMN activation that can either promote host organ damage or facilitate host defense. We have previously shown that inhibition of purinergic signaling with the P2 receptor antagonist suramin reduces PMN activation in sepsis (15). Therefore, we wondered how suramin affects the protective and destructive effects of PMNs in sepsis. We tested various treatment strategies with suramin in our CLP model (Fig. 3A). In a pilot study, we established the dose of suramin that reduced PMN activation in response to sepsis by 50%. At that dose (16 nmol/g BW), suramin had no noticeable effect on CD11b expression or AST levels in sham animals (Fig. 3B–D). When administered 10 min prior to CLP, this dose PMN activation and liver damage were reduced (Fig. 3B–D), but the numbers of bacteria in the peripheral blood was significantly higher than in untreated CLP mice (Fig. 3E). These findings suggest that inhibition of PMNs with suramin at the onset of sepsis can decrease PMN activation and organ damage but that this treatment also impairs the protective role of PMNs. Next, we tested whether delayed treatment with suramin would allow PMNs to mount an adequate host defense response, but prevent subsequent activation that may contribute to organ damage. For that purpose, we administered suramin either 60 min or 180 min after CLP. Delayed suramin treatment at 60 min was still able to reduce organ damage (Fig. 3D) but delayed suramin treatment also reduced host defense and promoted the spread of bacteria in sepsis (Fig. 3E). Taken together, these findings suggest that blocking PMN activation in sepsis inevitably impairs the ability of PMNs to defend the host from infection.
Figure 3. Inhibition of P2 receptor signaling reduces host organ damage but also host immune defense in sepsis.
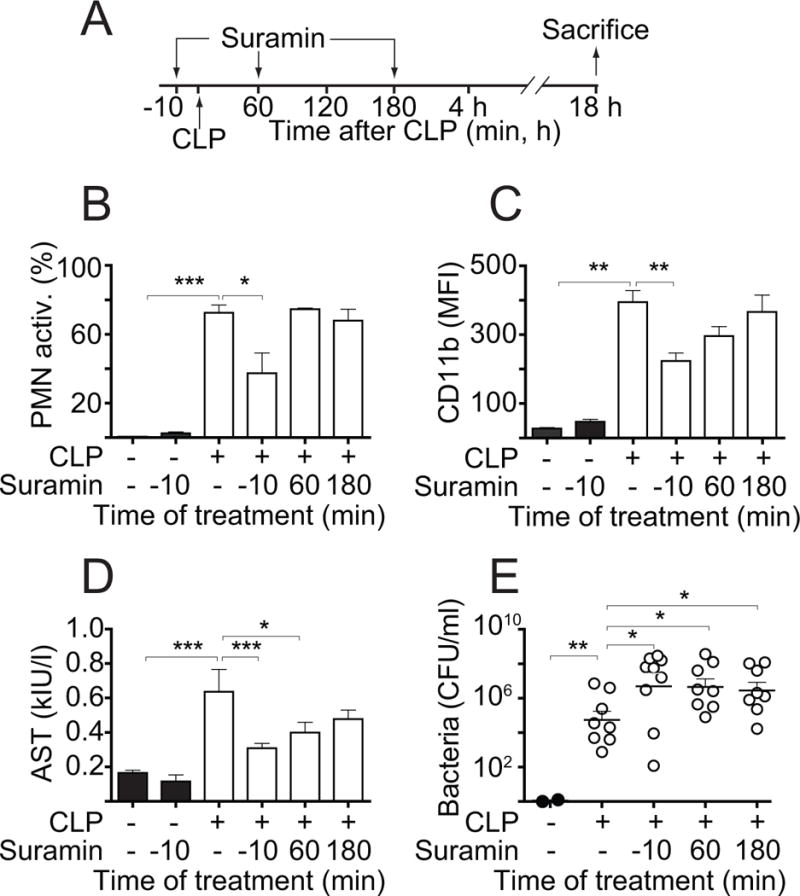
(A) Three different strategies with the aim to reduce PMN activation and preserving host defense were employed, administering suramin subcutaneously at a dose of 4 μl/g BW of a 4-mM solution either 10 min before or 1 h or 3 h after CLP. (B–C) Animals were sacrificed 18 h after CLP and PMN activation was assessed. Panel B shows the percentage of activated (CD11b+) PMNs and panel C shows the abundance of CD11b on the surface of PMNs. (D–E) AST levels or bacteria counts in the circulation of mice treated as described above were determined as readouts of host organ damage or host immune defense, respectively. Data shown are expressed as mean ± SEM of 8–12 animals per group (ANOVA; *p<0.05, **p<0.01, ***p<0.001).
Removal of systemic ATP improves survival and host immune defense in sepsis
Our findings indicate that inhibition of PMNs with suramin reduces host organ damage but also host immune defense. In order to overcome this dilemma, we test other strategies to modulate purinergic signaling. Apyrase is an ATPase that promotes the enzymatic breakdown of extracellular ATP. We administered apyrase i.p. with the goal of reducing extracellular ATP levels in the peritoneal cavity, where excessive ATP release may be released from damaged tissues at the source of infection. Apyrase or suramin was administered, mice were subjected to CLP, and PMN activation, organ damage, and the spread of bacteria were assessed (Fig. 4). While apyrase treatment had little effect on PMN activation and organ damage (Fig. 4A), it markedly reduced mortality (Fig. 4B–C). These effects were paralleled by a significant decrease in the number of bacteria in the peritoneum and the peripheral blood (Fig. 4D). We found that apyrase treatment diminished the concentration of extracellular ATP in peritoneal lavages, but that PMN counts in the peritoneum were unchanged (Fig. 4E). These findings demonstrate that apyrase treatment significantly improves outcome in sepsis by promoting host immune defense and bacterial clearance in sepsis.
Figure 4. Removing systemic ATP improves host immune defense and survival in sepsis.
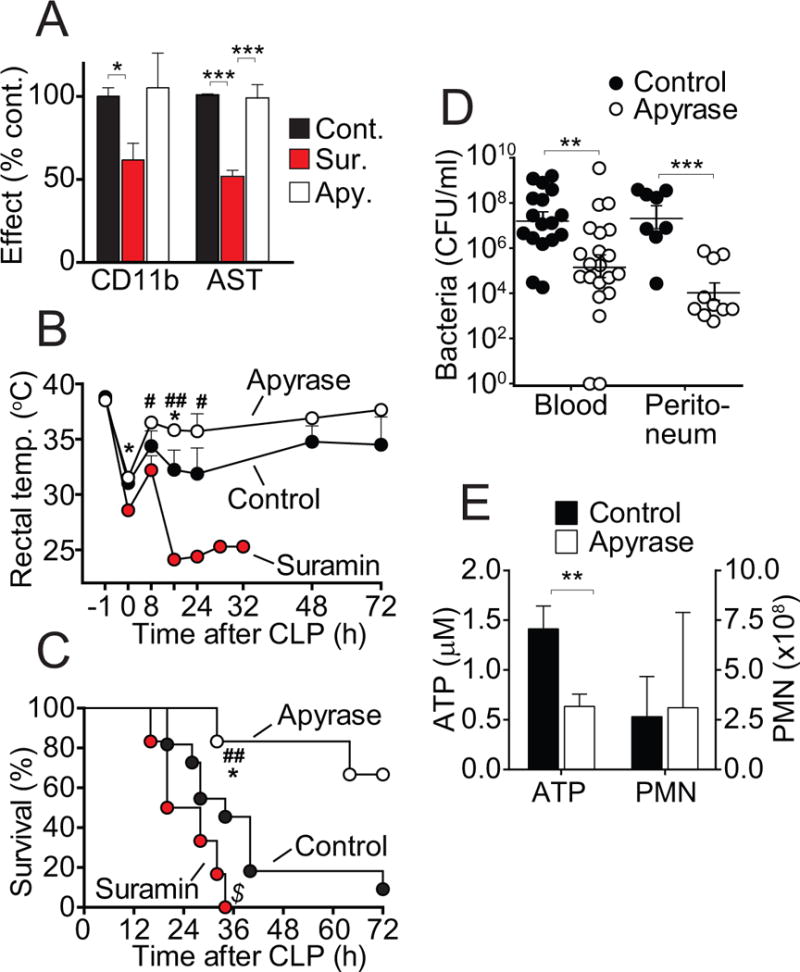
(A) WT mice were randomly divided into three groups. Control mice received normal saline vehicle prior to CLP; suramin was administered subcutaneously as a bolus (16 nmol/g BW) 10 min prior to CLP; and apyrase was administered intraperitoneally as a bolus (0.6 IU/g BW) 30 min prior to CLP. Animals were sacrificed after 18 h and PMN activation and plasma AST levels were determined. Data are expressed as percent change compared to untreated controls and represent the means ± SEM of 8–12 animals per group (ANOVA; *p<0.05, **p<0.01, ***p<0.001). (B) Body temperature was recorded with a rectal temperature probe before surgery (t=−1 h), at the end of surgery (t=0 h), and throughout the 72-h observation period following CLP. Each data point represents the mean ± SEM of 6 animals in each group. Differences among the three groups were analyzed with ANOVA (*p<0.05 control vs. apyrase; #p<0.05; ##p<0.01 apyrase vs. suramin). (C) Survival rates in the three treatment groups were recorded for a period of 72 h after CLP. Each group consisted of 11 animals. Survival statistics were assessed using the Kaplan-Meier test (*p<0.02 control vs. apyrase; $p<0.05 control vs. suramin; ##p<0.01 apyrase vs. suramin). (D) Numbers of bacteria in the blood and the peritoneum of mice with or without apyrase treatment were assessed 18 h after CLP. Data are expressed as means ± SEM of 17–21 animals per group (blood) and 6–10 animals per group (peritoneum). Comparisons between groups were made with ANOVA (**p<0.005, ***p<0.0001 control vs. apyrase). (E) ATP concentrations and total PMN counts in the peritoneal lavage of CLP mice without (control) and with apyrase treatment were determined (n=3 per group). Comparisons between groups were made with ANOVA (**p<0.005, ***p<0.0001 control vs. apyrase).
Eliminating systemic ATP with apyrase restores PMN chemotaxis
PMNs form the first line of defense against invading bacteria. Chemotaxis allows PMNs to locate and eliminate invading bacteria. Our previous work has shown that endogenous purinergic signaling regulates PMN chemotaxis through a delicate endogenous purinergic guidance system that directs migration to sites of infection (6–10). We hypothesized that the beneficial effects of apyrase in our CLP model may be due to improved PMN chemotaxis. To test this possibility, we used a live-cell imaging system for real-time monitoring of PMN chemotaxis in a chemotactic gradient field. Freshly isolated PMNs from healthy human donors were exposed to a chemotactic gradient field generated with a micropipette loaded with the bacterial product fMLP (Fig. 5A). Addition of ATP at concentrations similar to the systemic ATP levels found in sepsis (1–10 μM; ref. 15) dose-dependently impaired chemotaxis (Fig. 5B; video 1). Analysis of the chemotactic behavior of individual cells revealed that systemic ATP impaired the ability of PMNs to properly orient in a chemotactic gradient field, significantly reducing the percentage of PMNs that migrate in the correct direction to the micropipette tip simulating the site of an infection (Fig. 5C). Addition of apyrase at a concentration of 1 mU/ml did not affect PMN chemotaxis under control conditions, because this concentration of apyrase is not sufficiently high to interfere with the endogenous ATP signaling events that regulate PMN chemotaxis (Fig. 5A–C; video 1). However, this concentration of apyrase completely restored chemotaxis of PMNs in the presence of systemic ATP. These findings indicate that systemic ATP in sepsis can impair PMN chemotaxis by obscuring the endogenous purinergic guidance system these cells need for proper chemotaxis and their ability to find and kill invading bacteria.
Figure 5. Removing systemic ATP improves PMN chemotaxis.
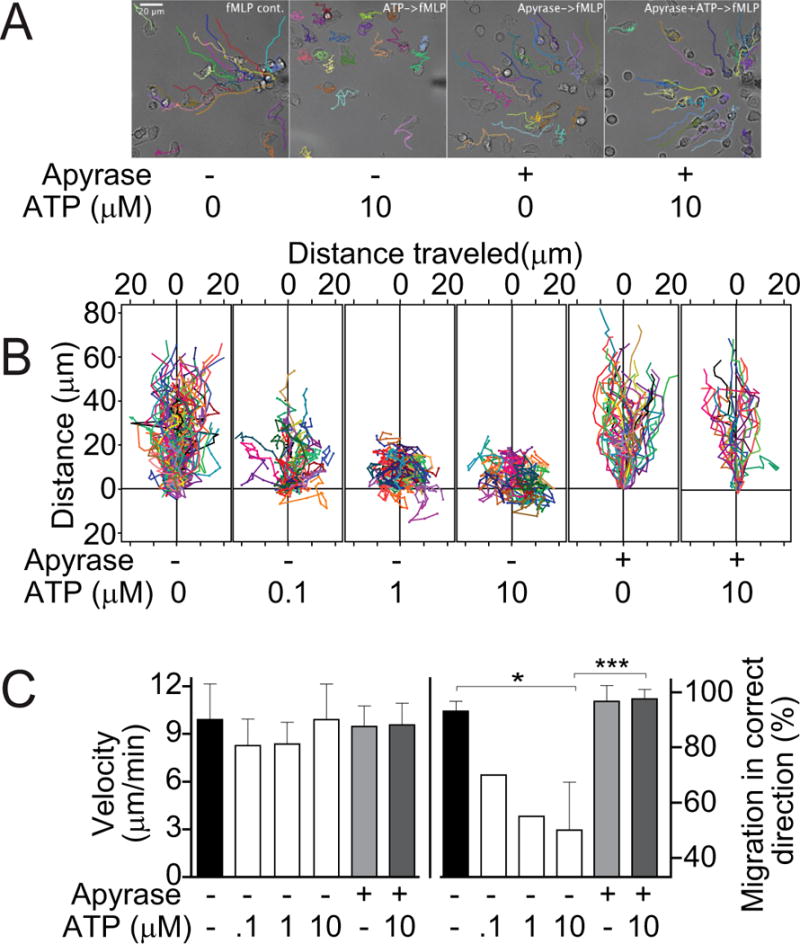
Freshly isolated primary human PMNs were exposed to a chemotactic gradient using a micropipette loaded with fMLP (100 nM). Individual cell traces were recorded with time lapse imaging and analyzed with image processing software. (A) Representative images of cell traces showing chemotaxis of PMNs to the chemotactic source (micropipette tip at right edge of each image); 20x objective. (B) Increasing concentrations of exogenous ATP were added in the presence or absence of apyrase and PMN chemotaxis was assessed (see also video 1). Individual cell traces were aligned with their starting points at coordinate x=y=0 μm and the micropipette tip at coordinate x=0, y=200 μm. (C) The velocity of migration was calculated from the total distance each cell traveled divided by the observation time. Gradient sensing ability of cells was estimated by determining the percentage of PMNs that migrated in the correct direction, i.e., on a migration path that did not deviate by more than 60° from a straight line between their starting points and the micropipette tip. The experiments shown were performed with cells isolated from at least three different healthy individuals. Data are expressed as means ± S.D. of n=20–40 cells from each donor.
DISCUSSION
Sepsis is accompanied by a systemic inflammatory response syndrome (SIRS) that promotes multi-organ failure and is a leading cause of death in trauma and critical care patients (1–5). Most therapeutic strategies designed to treat sepsis have focused on blocking SIRS. However, virtually all clinical trials that tested this approach have yielded disappointing results and no effective treatments for sepsis are available to this day (1,2). These findings have cast serious doubt over the feasibility of current treatment strategies and reemphasized the need for a deeper understanding of the pathophysiological mechanisms that are involved in sepsis (16–18). The findings of our current study support the notion that strategies aimed at the inflammatory process in sepsis are not suitable to improve clinical outcome. We found that blocking of the endogenous purinergic signaling mechanisms that activate PMNs can inhibit cell activation and PMN-induced organ damage but that this approach does not improve outcome in sepsis. Instead, we found that this approach impairs immune defenses because PMNs are no longer capable of locating and eliminating bacteria that spread throughout the host and propagate the process that leads to full-blown sepsis.
Recent work has revealed that ATP release via pnx1 and autocrine purinergic signaling regulate the activation and function of PMNs and of other immune and non-immune cell typs (12, 19–24). We demonstrated that these purinergic signaling mechanisms have a central role in the regulation of PMN chemotaxis (7–11). However, elevated systemic ATP levels in the plasma and at the site of infection in trauma and critical care patients defeat the endogenous purinergic signaling mechanisms that regulate PMN chemotaxis. Stimulation of P2Y2 receptors by systemic ATP levels can enhance PMN degranulation that is involved in host tissue damage (6, 15, 25, 26). However, our current findings indicate that excessive P2Y2 receptor stimulation by systemic ATP can also impair PMN chemotaxis, which is needed to protect the host from infection. Our findings suggest that systemic ATP interferes with the endogenous purinergic signaling mechanisms that regulate PMN chemotaxis. Thus, strategies that inhibit PMNs with purinergic drugs or other therapeutic agents studied in the past have two opposing effects: they reduce host organ damage but also impair the host immune defenses.
This dilemma is probably a major reason for that lack of progress in the development of effective therapies for sepsis. We could overcome this problem with the use of apyrase that allows the removal of systemic ATP without depleting the endogenous purinergic signaling mechanisms that are required for PMN chemotaxis. This endogenous purinergic signaling mechanism allow PMNs to determine the direction to which they must migrate in order to locate sites of infection and involves P2Y2 receptors and pnx1-mediated ATP release from the cell surface closest to the source of chemoattractants such as fMLP (7–8; see suppl. Fig. 3). Under normal circumstances, this purinergic signaling process results in the efficient migration of PMNs upstream of chemotactic gradient fields following a localized ATP signal in close proximity to the cell surface (11–12). The presence of systemic ATP interferes with this gradient sensing mechanism by obscuring the endogenous ATP signal generated by localized ATP release and by indiscriminately stimulating P2Y2 receptors across the surface of PMNs (suppl. Fig. 3). As a consequence, PMNs lose their ability to locate and kill bacteria. Removal of systemic ATP can restore PMN chemotaxis and significantly improve bacterial clearance and survival in the CLP model we used. These observations are in line with recent reports by others who demonstrated that apyrase treatment improves survival in a mouse sepsis model (27, 28x). We conclude that systemic ATP is a major culprit responsible for the pathophysiological processes involved in sepsis. Removal of systemic ATP is a promising novel treatment strategy to treat sepsis.
Supplementary Material
Video 1: Chemotaxis in the presence of the indicated concentrations of additional (systemic) ATP and apyrase.
Supplemental Figure 1. Sepsis induces PMN activation. Wild-type mice were subjected to cecal ligation and puncture (CLP) and PMN activation was determined by flow cytometry and anti-Ly-6G antibodies to differentiate PMNs and anti-CD11b to assess PMN activation. (A–B) Representative flow cytometry data at different times after CLP are shown. (C–D) The percentage of CD11b+ PMNs and the abundance of CD11b on the cell surface of Ly-6G+ cells (MFI, mean fluorescence intensity) at the indicated times after CLP is shown. Each data point represents the mean ± standard error of the mean (SEM) of 3–5 mice in each group. Statistical comparisons between groups were done with ANOVA, *p<0.05, **p<0.01 compared to sham controls (t=0 h).
Supplemental Figure 2. Sepsis causes leukocytosis and liver damage. (A) Wild-type mice were subjected to CLP or sham surgery for the indicated times and total leukocyte counts in peripheral blood were determined by enumeration with a hemocytometer. (B) Plasma samples of mice subjected to CLP as described above were used to determine the activity of aspartate aminotransferase (AST) as a marker of liver damage. Each data point shown represents the mean ± SEM of 3–5 mice in each group. Statistical comparisons between groups were done with ANOVA, *p<0.05.
Supplemental Figure 3. Systemic ATP impairs chemotaxis by obscuring the purinergic guidance system of PMNs. Autocrine purinergic signaling regulates PMN chemotaxis by localized pnx1-induced ATP release and P2Y2 receptor stimulation at the front of cells. This endogenous purinergic signaling mechanism is required for the directed migration to and the killing of bacteria at sites of infection. (A) Systemic ATP disrupts these endogenous purinergic signaling systems, resulting in impaired PMN chemotaxis and host immune defenses against invading bacteria. (B–C) Removal of systemic ATP with apyrase restores chemotaxis and the ability of PMNs to find and kill invading bacteria at sites of infection.
Acknowledgments
This work was funded in part through National Institutes of Health grants GM-51477, GM-60475, AI-080582, and T32 GM103702 (W.G.J.), through National Natural Science Foundation of China grants 8123001 and 81470236 (X.L.), and through a German Research Foundation (DFG) grant, LE-3209/1-1 (C.L.). We thank Dr. Mahtab Fakhari for her valuable comments and for carefully revising our manuscript.
Funding sources: This work was funded in part through National Institutes of Health grants GM-51477, GM-60475, AI-080582, and T32GM103702 (W.G.J.), National Natural Science Foundation of China grants 8123001 and 81470236 (X.L.), a German Research Foundation (DFG) grant, LE-3209/1-1 (C.L.), and a grant from Nakatomi Foundation, Japan (Y.K.).
Footnotes
Copyright form disclosures: Dr. Junger received support for article research from the National Institutes of Health (NIH); DFG; Germany National Science Foundation; and the China Nakatomi Foundation, Japan and disclosed other support (Various types of remuneration for grant review activities from the NIH, DOD, and other foreign funding agencies). His institution received funding from the NIH, DFG, and the National Natural Science Foundation of China. Dr. Li received support for article research from the NIH and disclosed that this work was funded in part through National Institutes of Health grants GM-51477, GM-60475, AI-080582, and T32GM103702 (W.G.J.); through National Natural Science Foundation of China grants 8123001 and 81470236 (X.L.); and through a German Research Foundation (DFG) grant, LE-3209/1-1 (C.L.). Dr. Lee received support for article research from the NIH. Dr. Ledderose received support for article research from the German Research Foundation (DFG). The remaining authors have disclosed that they do not have any potential conflicts of interest.
References
- 1.Angus DC. The search for effective therapy for sepsis: back to the drawing board? JAMA. 2011;306:2614–2615. doi: 10.1001/jama.2011.1853. [DOI] [PubMed] [Google Scholar]
- 2.Wenzel RP, Edmond MB. Septic shock–evaluating another failed treatment. New Engl J Med. 2012;366:2122–2124. doi: 10.1056/NEJMe1203412. [DOI] [PubMed] [Google Scholar]
- 3.Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, Bricker TL, Jarman SD, Kreisel D, Krupnick AS, Srivastava A, Swanson PE, Green JM, Hotchkiss RS. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA. 2011;306:2594–2605. doi: 10.1001/jama.2011.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hotchkiss R, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunction to immunotherapy. Nat Rev Immunol. 2013;13:862–874. doi: 10.1038/nri3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. 2013;13:260–268. doi: 10.1016/S1473-3099(13)70001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Y, Yao Y, Sumi Y, Li A, To UK, Elkhal A, Inoue Y, Woehrle T, Zhang Q, Hauser C, Junger WG. Purinergic signaling: a fundamental mechanism in neutrophil activation. Sci Signal. 2010;3(125):ra45. doi: 10.1126/scisignal.2000549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, Nizet V, Insel PA, Junger WG. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science. 2006;314:1792–1795. doi: 10.1126/science.1132559. [DOI] [PubMed] [Google Scholar]
- 8.Bao Y, Chen Y, Ledderose C, Li L, Junger WG. Pannexin 1 channels link chemoattractant receptor signaling to local excitation and global inhibition responses at the front and back of polarized neutrophils. J Biol Chem. 2013;288:22650–22657. doi: 10.1074/jbc.M113.476283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bao Y, Ledderose C, Seier T, Graf AF, Brix B, Chong E, Junger WG. Mitochondria regulate neutrophil activation by generating ATP for autocrine purinergic signaling. J Biol Chem. 2014;289:26794–26803. doi: 10.1074/jbc.M114.572495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bao Y, Ledderose C, Graf AF, Brix B, Birsak T, Lee A, Zhang J, Junger WG. mTOR and differential activation of mitochondria orchestrate neutrophil chemotaxis. J Cell Biol. 2015;210:1153–1164. doi: 10.1083/jcb.201503066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Junger WG. Purinergic regulation of neutrophil chemotaxis. Cell Mol Life Sci. 2008;65:2528–2540. doi: 10.1007/s00018-008-8095-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol. 2011;11:201–212. doi: 10.1038/nri2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma P, Lysiak JJ, Harden TK. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461:282–286. doi: 10.1038/nature08296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, Isakson BE, Bayliss DA, Ravichandran KS. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 2010;467:863–867. doi: 10.1038/nature09413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sumi Y, Woehrle T, Chen Y, Bao Y, Li X, Yao Y, Inoue Y, Tanaka H, Junger WG. Plasma ATP is required for neutrophil activation in a mouse sepsis model. Shock. 2014;42:142–147. doi: 10.1097/SHK.0000000000000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stearns-Kurosawa DJ, Osuchowski MF, Valentine C, Kurosawa S, Remick DG. The pathogenesis of sepsis. Annu Rev Pathol. 2011;6:19–48. doi: 10.1146/annurev-pathol-011110-130327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fink MP, Warren HS. Strategies to improve drug development for sepsis. Nat Rev Drug Discov. 2014;13:741–758. doi: 10.1038/nrd4368. [DOI] [PubMed] [Google Scholar]
- 18.Hutchins NA, Unsinger J, Hotchkiss RS, Ayala A. The new normal: immunomodulatory agents against sepsis immune suppression. Trends Mol Med. 2014;20:224–233. doi: 10.1016/j.molmed.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burnstock G. Purinergic signalling: from discovery to current developments. Exp Physiol. 2014;99:16–34. doi: 10.1113/expphysiol.2013.071951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bours MJ, Swennen EL, Di Virgilio F, Cronstein BN, Dagnelie PC. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther. 2006;112:358–404. doi: 10.1016/j.pharmthera.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 21.Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med. 2012;367:2322–2333. doi: 10.1056/NEJMra1205750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Penuela S, Harland L, Simek J, Laird DW. Pannexin channels and their links to human disease. Biochem J. 2014;461:371–381. doi: 10.1042/BJ20140447. [DOI] [PubMed] [Google Scholar]
- 23.Antonioli L, Pacher P, Vizi ES, Haskó G. CD39 and CD73 in immunity and inflammation. Trends Mol Med. 2013;19:355–367. doi: 10.1016/j.molmed.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adamson SE, Leitinger N. The role of pannexin1 in the induction and resolution of inflammation. FEBS Lett. 2014;588:1416–1422. doi: 10.1016/j.febslet.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y, Shukla A, Namiki S, Insel PA, Junger WG. A putative osmoreceptor system that controls neutrophil function through the release of ATP, its conversion to adenosine, and activation of A2 adenosine and P2 receptors. J Leukoc Biol. 2004;76:245–253. doi: 10.1189/jlb.0204066. [DOI] [PubMed] [Google Scholar]
- 26.Chen Y, Hashiguchi N, Yip L, Junger WG. Hypertonic saline enhances neutrophil elastase release through activation of P2 and A3 receptors. Am J Physiol Cell Physiol. 2006;290:C1051–C1059. doi: 10.1152/ajpcell.00216.2005. [DOI] [PubMed] [Google Scholar]
- 27.Cauwels A, Rogge E, Vandendriessche B, Shiva S, Brouckaert P. Extracellular ATP drives systemic inflammation, tissue damage and mortality. Cell Death Dis. 2014;5:e1102. doi: 10.1038/cddis.2014.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Csóka B, Németh ZH, Törő G, Koscsó B, Kókai E, Robson SC, Enjyoji K, Rolandelli RH, Erdélyi K, Pacher P, Haskó G. CD39 improves survival in microbial sepsis by attenuating systemic inflammation. FASEB J. 2015;29:25–36. doi: 10.1096/fj.14-253567. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video 1: Chemotaxis in the presence of the indicated concentrations of additional (systemic) ATP and apyrase.
Supplemental Figure 1. Sepsis induces PMN activation. Wild-type mice were subjected to cecal ligation and puncture (CLP) and PMN activation was determined by flow cytometry and anti-Ly-6G antibodies to differentiate PMNs and anti-CD11b to assess PMN activation. (A–B) Representative flow cytometry data at different times after CLP are shown. (C–D) The percentage of CD11b+ PMNs and the abundance of CD11b on the cell surface of Ly-6G+ cells (MFI, mean fluorescence intensity) at the indicated times after CLP is shown. Each data point represents the mean ± standard error of the mean (SEM) of 3–5 mice in each group. Statistical comparisons between groups were done with ANOVA, *p<0.05, **p<0.01 compared to sham controls (t=0 h).
Supplemental Figure 2. Sepsis causes leukocytosis and liver damage. (A) Wild-type mice were subjected to CLP or sham surgery for the indicated times and total leukocyte counts in peripheral blood were determined by enumeration with a hemocytometer. (B) Plasma samples of mice subjected to CLP as described above were used to determine the activity of aspartate aminotransferase (AST) as a marker of liver damage. Each data point shown represents the mean ± SEM of 3–5 mice in each group. Statistical comparisons between groups were done with ANOVA, *p<0.05.
Supplemental Figure 3. Systemic ATP impairs chemotaxis by obscuring the purinergic guidance system of PMNs. Autocrine purinergic signaling regulates PMN chemotaxis by localized pnx1-induced ATP release and P2Y2 receptor stimulation at the front of cells. This endogenous purinergic signaling mechanism is required for the directed migration to and the killing of bacteria at sites of infection. (A) Systemic ATP disrupts these endogenous purinergic signaling systems, resulting in impaired PMN chemotaxis and host immune defenses against invading bacteria. (B–C) Removal of systemic ATP with apyrase restores chemotaxis and the ability of PMNs to find and kill invading bacteria at sites of infection.