

Abstract
Objectives
Hydrogen peroxide (H2O2) contributes to flow-induced dilation (FID) of human arterioles. This study is designed to examine the roles of mitochondria and NADPH oxidase in modulating the release of ROS and in mediating FID. We tested whether NADPH oxidase contributes to mitochondrial ROS generation in arterioles during coronary artery disease (CAD).
Methods
Visceral adipose arterioles obtained from patients with or without CAD were cannulated and pressurized for videomicroscopic measurement of arteriolar diameters. Dilator responses and ROS production during flow were determined in the presence and absence of the NADPH oxidase inhibitor gp91ds-tat and the mitochondrial electron transport inhibitor rotenone.
Results
Both dilation and H2O2 generation during flow were reduced in the presence of rotenone (13.5±8% vs. 97±17% without rotenone) or gp91ds-tat in patients with CAD, while patients without CAD exhibited H2O2-independent dilations. Mitochondrial superoxide production during flow was attenuated by gp91ds-tat in arterioles from CAD patients.
Conclusions
These findings indicate that ROS produced by NADPH oxidase are an upstream component of the mitochondria-dependent pathway contributing to flow-dependent H2O2 generation and dilation in peripheral microvessels from patients with CAD. We conclude that in CAD, both mitochondria and NADPH oxidase contribute to FID through a redox mechanism in visceral arterioles.
Keywords: Adipose, Microcirculation, Flow Mediated Dilation, and Mitochondria
Introduction
Vasodilation to an increase in blood flow is an important physiologic stimulus for regulating peripheral vascular tone and homeostasis. Several mediators of flow induced dilation (FID) have been described, including nitric oxide (NO) (1), prostacyclin (PGI2) (2), and cytochrome P450 metabolites of arachidonic acid (3). Previous data shows a complex role for reactive oxygen species (ROS) in regulating vascular tone in cardiovascular disease. On one hand cardiovascular disease is associated with reduced flow-induced NO release due to the increased levels of superoxide. However, we and others have shown that an increase in the ROS hydrogen peroxide (H2O2), is responsible for endothelium- dependent vasodilation in animals (4) and in coronary (5) and visceral (6) arterioles from patients with coronary artery disease (CAD). When compared to subcutaneous fat storage depots, visceral fat accumulation is more strongly associated with cardiovascular risk factors such as hyperlipidemia, insulin resistance, and endothelial dysfunction (7). Since human adipose is a source of paracrine modulation of vasomotion and vascular inflammation (8), alterations in the mechanisms mediating FID in arterioles embedded in adipose tissue may closely reflect this pro-inflammatory environment and may contribute to the development of cardiovascular disease.
In resistance arteries evidence supports H2O2 as the endothelial derived hyperpolarizing factor (EDHF) in the mesenteric and coronary circulations of mice (9) and humans (10). As a component of FID, EDHF comprises only a portion of the dilatory response to flow with the well-established endothelium-derived relaxing factor, NO, contributing substantially in patients without CAD. Importantly, we previously showed that flow-stimulated H2O2 production occurs in place of NO-dependent FID in CAD patients (11). The mechanosensitive transducer sensitive to shear and responsible for mitochondrial ROS production in adipose vessels is not known, however, the NADPH oxidase is a mechanosensitive enzyme complex located on the endothelial cell surface that produces superoxide (O2−) and may contribute to vascular H2O2 generation (12). Communication between NADPH oxidase and mitochondria has been described as a mechanism leading to a redox amplification process termed ROS-induced ROS release (RIRR) (13). We previously reported the critical involvement of mitochondrial ROS in FID (14). Furthermore it has been shown that NADPH oxidase is responsible for some increase in ROS during mechanical shear stress (15, 16). Therefore, we tested the hypothesis that the FID in visceral adipose microvessels from patients with CAD is regulated by NADPH oxidase generation of O2− which acts on mitochondria to promote redox mediated vasodilation through an RIRR-dependent mechanism.
Materials and Methods
Tissue acquisition
Discarded and de-identified visceral adipose tissue was obtained at the time of abdominal or thoracic surgery. Patients with coronary artery disease (CAD) were identified by chart documented diagnosis. Control subjects had at least one cardiovascular risk factor (e.g. hypercholesterolemia, smoking, hypertension, diabetes, etc.) but no prior diagnosis of CAD. Tissues were placed into iced HEPES buffer. All studies were approved by the Medical College of Wisconsin/Froedtert Hospital Institutional Review Board.
Videomicroscopic studies
Arterioles were dissected from the tissue of CAD and non-CAD patients as close to the time of retrieval as possible but within 24hrs following surgery and cleaned to remove adjacent connective tissue and fat. In an organ chamber, vessels were cannulated with glass micropipettes filled with Krebs solution, secured with monofilament suture, and the vessel was maintained at an intraluminal pressure of 20 mmHg for 30 minutes. Each preparation was transferred to the stage of an inverted microscope (magnification 200x) attached to a video camera, video monitor, and a video measuring device (Boeckeler VIA-100) (14). The external bathing medium was made fresh daily, continuously superfused with heated buffer solution aerated with a gas mixture of 21% O2, 5% CO2 and 74% N2 and maintained at 37°C. After pressure was slowly increased to 60 mmHg and maintained for 30 minutes, vessels were constricted 30–50% with endothelin-1 (ET-1). Vessels that do not constrict more than 30% are excluded from analysis. Flow was produced by simultaneously changing the heights of the reservoirs in equal and opposite directions to generate a pressure gradient as described previously (5).
Experimental Protocols
Steady-state internal arterial diameter was measured before and during intraluminal flow corresponding to pressure gradients of 10, 20, and 100 cmH2O in the absence or presence of rotenone (10−6 M) or the NADPH oxidase inhibitor gp91ds-tat (5×10−5M) or a scrambled gp91 sequence (scramb-tat; 5×10−5M). gp91ds-tat inhibits p47phox association with gp91phox, a key assembly unit of the NADPH oxidase complex that is required for O2− generation in vascular cells as previously described (17). In separate experiments, vessels were superfused and perfused with the superoxide dismutase mimetic manganese tetrakis (4-benzoic acid) porphyrin chloride (MnTBAP; 10−4 M) or the hydrogen peroxide scavenger polyethylene glycol (PEG-CAT; 500 U/ml) prior to flow. Inhibitors were added to the superfusion and/or perfusion PSS 60 minutes prior to constriction with ET-1 to 30–50% of resting diameter. The passive diameter (maximal diameter) of every vessel was determined in the presence of papaverine (10−4 M) and the diameter in response to flow at a gradient of 100 cmH2O was measured in the presence of papaverine first in one direction, then with the gradient reversed to verify matched pipette resistance. Arterioles were dissected from the tissue within 24hrs following surgery and cleaned to remove adjacent connective tissue and fat cells. Microvascular segments were prepared for assessment of internal diameter changes over a range of physiological intraluminal flows using incremental pressure gradients of 5, 10, 20, and 100cm H2O with and without inhibitors, as previously described (6).
Fluorescence Measurement of Vascular ROS Production
Production of vascular cytosolic peroxide and mitochondrial O2− in adipose arterioles was assessed by fluorescence microscopy using 2′, 7′-dichlorodihydrofluorescein (DCFH; 5× 10−6 M, Invitrogen), and the specific mitochondrial O2− indicator (MitoSOX Red; 5×10−6 M, Invitrogen), respectively. Polythylene glycol-catalase (PEG-CAT; 500U/ml) was used to confirm the specificity of DCFH fluorescence for H2O2. Briefly, two vessels dissected from each adipose tissue segment were used per experiment, one superfused with a vehicle and another with an inhibitor for 30 minutes. Vessels were then incubated with either DCFH or MitoSOX for additional 30 min at 37°C. Next intraluminal flow (pressure gradient of 100cm H2O) was initiated for 15 minutes and fluorescent images were taken at an excitation/emission frequency of 488/526 nm for DCF and 510/580 nm for MitoSox. The recorded images were analyzed with MetaMorph software to calculate the changes in fluorescence intensity before and after the flow.
Materials
Gp91ds-tat was purchased from AnaSpec Inc. All other chemicals were obtained from Sigma. Endothelin-1 was prepared in saline with 1% bovine serum albumin. MnTBAP was dissolved in ethanol. Rotenone, DCFH, and MitoSOX stocks were prepared in DMSO. Similar concentrations of ethanol and DMSO were used for vehicles. All other agents were prepared in distilled water. Final molar concentrations of agents in the organ chambers are indicated.
Statistical Analysis
All data are expressed as mean ± SEM. Percent dilation was calculated as the percent change from constricted diameter to the diameter after flow with 100% representing maximal diameter usually in the presence of papaverine (10−4M). To determine the effect of treatment on the dilatory response to flow, data from vessels exposed to flow before and after treatment were compared using a two-way repeated measures ANOVA using SigmaStat version 3.5. To determine the effect of a treatment on DCFH or MitoSOX fluorescence intensity either a one-way ANOVA with Bonferroni correction or a t-test was used where appropriate. All fluorescence responses were compared to no flow conditions. P<0.05 was considered statistically significant. Only one vessel was used per protocol from each patient.
Results
Patient characteristics
Visceral adipose arterioles from forty-five patients were utilized for this study. Patient demographics including diagnoses are summarized in Table 1. Vascular characteristics are presented in Table 2. No statistical significance was found between CAD and non-CAD subjects for baseline internal diameter, maximal diameter, or percent tone.
Table 1.
Patient Demographics
| Characteristic | Non-CAD (n) | CAD (n) |
|---|---|---|
| Sex, M/F | 10/18 | 15/2 |
| Age, years | 55±3 | 62±2 |
| * BMI | 26±1 | 29±1 |
| Race/Ethnicity | ||
| Caucasion | 24 | 14 |
| African American | 4 | 2 |
| Hispanic | 0 | 1 |
| Diseases | ||
| DM | 2 | 6 |
| HTN | 12 | 15 |
| Hl | 1 | 8 |
| MI | 0 | 4 |
| CHF | 0 | 3 |
| Tobacco | 3 | 5 |
| Other | 2 | 5 |
| No. of risk factors | ||
| 1 | 10 | 1 |
| 2 | 3 | 2 |
| 3 or more | 1 | 14 |
Data shown as mean±SE; n indicates the number of patients studied. M indicates male; F, female; CAD, coronary artery disease; DM, diabetes mellitus; HTN, hypertension; HL, hyperlipidemia; MI, myocardial infarction; and CHF, congestive heart failure
Data collected from a subset of patients; non-CAD (24/28), CAD (40/45)
Table 2.
Vascular Characteristics
| Characteristic | Non-CAD (μm) | CAD (μm) |
|---|---|---|
| Baseline IDs | 158.9±11.8 | 170.9±13.7 |
| Maximal Diameters | 166.0±12.4 | 174.5±13.6 |
| % Tone | 3.1±0.1% | 2.5±1.3% |
Data shown as mean±se. ID = internal diameter. % Tone represents the developed tone during pressurizing at 60 mm Hg prior to preconstriction with ET-1.
Role of mitochondria in FID and ROS generation
The mitochondrial electron transport chain (ETC) is a major source of cellular ROS, specifically at complex I. We therefore used rotenone, an inhibitor of complex I, to test whether reductions in mitochondria-derived ROS reduced dilation to flow in patients with CAD. In pressurized arterioles isolated from visceral adipose of subjects undergoing thoracic or abdominal surgery, rotenone decreased dilations to flow in subjects with CAD (maximum dilation (MD): 72.5 ± 10.6% no rotenone vs. 32.6 ± 9.9% rotenone, P < 0.05; Figure 1A) but had no effect in subjects without CAD (Figure 1B) implicating a specific role for mitochondrial complex I in patients afflicted with CAD. Furthermore, MnTBAP, an SOD mimetic, reduced FID in subjects with CAD (MD: 76.6 ± 3.0% no MnTBAP vs. 7.4 ± 7.9% MnTBAP, P < 0.05) indicating a prominent role for ROS in FID. Rotenone also prevented the increase in DCFH fluorescence during flow (Figure 2A, fluorescence intensity (FI) increase: 96.9 ± 16.9% vehicle vs. 13.5 ± 8.0% rotenone, P < 0.05; Figure 2B) thereby linking the inhibition of complex I to mitochondrial ROS-induced FID in patients with CAD. This finding also supports our previous report where rotenone prevents mitochondrial production of H2O2 in human coronary resistance arteries (14). Summary data from five experiments show a catalase-sensitive increase in FI due to peroxide production during shear (Figure 2B). These findings support our hypotheses that mitochondrial-derived H2O2 is the major 1) ROS produced in response to flow in patients with CAD and 2) endothelium derived vasodilator in patients with CAD.
Figure 1. Inhibition of mitochondrial complex I reduces FID in CAD subjects.

Rotenone (1 μM) reduces FID in adipose microvessels from subjects with CAD (A) but has no effect on FID in adipose microvessels from subjects without CAD (B). N=5–6. *P<0.05 vs. vehicle.
Figure 2. Inhibition of mitochondrial complex I prevents the flow-induced increase in H2O2 in CAD subjects.

Panel A: Representative images (20x) of H2O2 production in visceral adipose arterioles from CAD patients as determined by DCF fluorescence in the presence of flow (100 cm) before (top) and after (bottom) a 30 min exposure to rotenone (1uM). Panel B: Summary data showing an increase in DCF fluorescence after 15 min of flow in adipose arterioles from CAD patients. The increase in fluorescence was reduced by rotenone and Peg-Cat (500U/ml) in independent treatments. *P<0.05 vs. vehicle.
Role of NADPH oxidase in FID and ROS generation
For the mitochondrial ETC to be involved in dilations to flow, mechanosensitve, membrane-bound receptors of the vascular endothelium would have to translate mechanical shear stimuli. To determine whether NADPH oxidase is involved in the dilator response to flow we used the selective inhibitor, gp91ds-tat in arterioles from patients with and without CAD. In CAD patients, gp91ds-tat abolished arteriolar FID (MD: 61.2 ± 2.8% control vs. 5.1 ± 4.4% gp91ds-tat, P < 0.05; Figure 3A) but there was no effect of gp91ds-tat on vessels from non-CAD patients (Figure 3B). The scrambled peptide had no effect on FID in CAD patients (Figure 3C). This finding suggests that NADPH oxidase is a critical component of the pathway mediating flow-induced dilation in human visceral adipose arterioles specifically from patients with CAD. To test the possibility that NADPH-oxidase is coupled to mitochondrial ROS formation via a RIRR mechanism, we assessed mitochondrial superoxide production using the dye MitoSox. MitoSox fluorescence increased over time in response to flow in arterioles form CAD patients (top panel, Figure 4A). Inhibiting NADPH-oxidase with the gp91-ds-tat peptide abrogated the increase in MitoSox fluorescence (bottom panel, Figure 4A. FI increase: 218 ± 16.2% vehicle vs. 11.3 ± 5.8% gp91-ds-tat, P < 0.05; Figure 4B). Together, these data suggest that the flow-induced elevation in mitochondrial ROS requires NADPH oxidase in subjects afflicted with CAD.
Figure 3. NADPH-oxidase is a source of flow-induced ROS in CAD subjects.

Inhibiting NADPH-oxidase with the peptide gp91ds-tat (50 μM) reduces FID in adipose microvessels exclusively in subjects with CAD (A) while the scrambled peptide control (50 μM) has no effect on this population (C). The peptide has no effect on FID in adipose microvessels from subjects without CAD (B). *P<0.05 vs. vehicle.
Figure 4. NADPH-oxidase is upstream of mitochondria-derived ROS in CAD subjects.

Panel A: Representative images (20x) of mitochondrial superoxide production in visceral adipose arterioles from CAD patients as determined by MitoSox fluorescence in the presence of 100cm of flow before (top) and after (bottom) exposure to gp91ds-tat (50uM) for 30 min. Panel B: Summary data showing an increase in MitoSox fluorescence after 15 min of flow in adipose arterioles from CAD patients (n=5). The increase in fluorescence was significantly reduced with gp91ds-tat (50uM). * P<0.05 vs. vehicle.
Discussion
The endothelium plays a key role in vascular homeostasis by synthesizing and releasing vasorelaxing factors including NO, PGI2, and EDHFs. Atherosclerosis and its risk factors can reduce the ambient levels of NO and any associated dilation (18). We previously established a compensatory switch in the flow-induced dilator molecule in arterioles of patients with CAD from NO to H2O2 (11). Identifying the underlying mechanisms leading to the flow-induced production of H2O2 and subsequent vasodilation in arterioles of patients afflicted with CAD is the main goal of this study.
Mitochondrial production of O2− is a major source of cellular ROS and oxidative damage contributing to cardiovascular dysfunction in many disease states (19). In this study, we confirm the crucial role of mitochondrial ROS production in flow-mediated dilation in arterioles of CAD patients by blocking the mitochondrial complex I, the predominant source of O2−, with rotenone. Inhibition of complex I drastically reduced dilations to flow (Fig 1) as well as the production of flow-induced H2O2 as detected by PEG-catalase-quenchable DCF fluorescence (Fig 2) in CAD patients; effects not observed in patients without CAD. To verify that blockade of complex I prevents the production of O2−, the mitochondrial specific ROS scavenger MnTBAP was used on arterioles isolated from CAD patients. Scavenging flow-induced mitochondrial ROS inhibits dilations to flow to a similar extent as complex I inhibition. Together, these data show that mitochondrial specific production of ROS, most likely generated by complex I, are essential for maximal dilations to flow in the visceral arterioles of CAD patients. Importantly, these data also support the foundation of previous studies by our group and others that the dilator to flow ‘switches’ from NO to ROS in distinct arteriolar beds (5, 6, 20).
While we are aware that rotenone is classically considered to inhibit complex I and thereby promote mitochondrial ROS production in healthy cells (21), our data supports studies indicating that it alternatively inhibits O2− production (22). In order for an increase in flow to stimulate endothelial cell ROS production, membrane-bound receptors most likely translate mechanical forces into appropriate cellular signals. NADPH oxidase, a mechanosensitive membrane-bound protein, has been linked to increased superoxide production with pathological (oscillatory) shear stress (23, 24). In the present study NADPH oxidase is shown to be a necessary upstream mediator of RIRR-dependent dilations in arterioles isolated from CAD patients. Blockade of NADPH oxidase with gp91-ds-tat: 1) nearly abolishes dilations to flow (Fig. 3) and 2) prevents flow-induced mitochondrial ROS production (Fig. 4). These findings implicate a link between flow-stimulated NADPH oxidase O2− production and H2O2 generation from increased mitochondrial O2−. The mechanical or chemical link between these two sources of ROS production is not known but could involve the endothelial cell cytoskeleton, the integrity of which is essential for shear-induced production of H2O2 and for FMD in human arterioles (14).
In the presence of CAD NADPH oxidase expression and activity is increased and promotes the generation of ROS (25). Although we have previously shown that mitochondrial ROS contribute to FID in coronary and adipose circulations (22), this is the first demonstration that NADPH oxidase plays a critical role in FID of human adipose vessels. While the exact mechanism linking NADPH oxidase to mitochondrial ROS generation during CAD has not been determined, the phenomenon of RIRR has been reported by others (26). Figure 5 schematically represents our hypothesis that ROS induced dilation in arterioles from patients with CAD is driven by mechanical stimulation of endothelial NADPH oxidase and subsequent RIRR mechanisms resulting in mitochondrial production of superoxide which is converted to H2O2, presumably through superoxide dismutase. In addition, mitochondrial ROS can activate NADPH oxidase supporting the theory that there is positive feedback which accelerates RIRR between mitochondria and NADPH oxidase during cardiovascular pathology (13). Importantly, inhibiting NADPH oxidase had no effect on FID in vessels from patients without CAD (Figure 3B). This indicates that the ROS-generating systems are functionally coupled to shear-induced mechano-signaling systems only in the presence of disease. We speculate that under normal conditions NO production inhibits the NADPH-oxidase-mitochondrial ROS activation pathway. However, in the presence of CAD, ambient ROS elevation quenches NOS-derived NO and releases this inhibition. Additionally, elevated ROS is also known to uncouple eNOS through oxidation of essential cofactors, specifically tetrahydrobiopterin (BH4), which when supplemented experimentally recovers endothelial function in coronary arteries of hypercholesteremic humans (27, 28). It is possible that increased activity of NADPH oxidase elevates ROS leading to an initial uncoupling of eNOS via loss of BH4 which then contributes to RIRR signaling.
Figure 5. Schematic illustrating the proposed mechanism demonstrated by the current study.
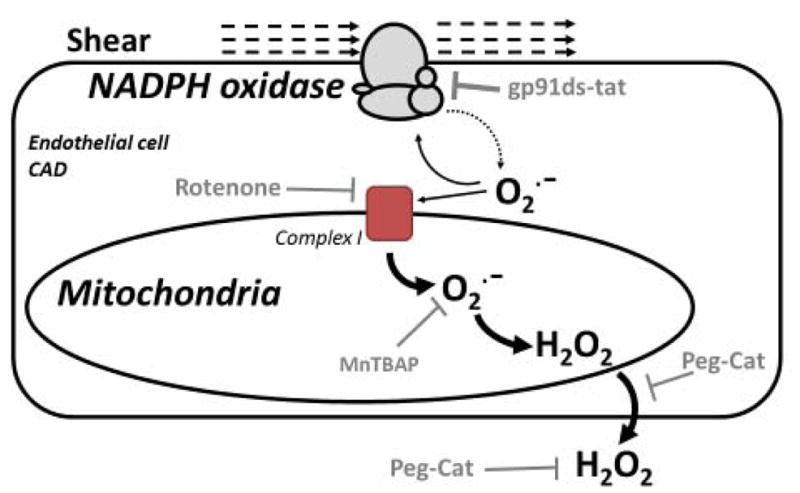
Shear stress implemented on the vascular wall activates endothelial NADPH-oxidase in subjects afflicted with CAD leading to a RIRR cascade. The small amount of O2− produced by mechanically stimulated NADPH-oxidase (dotted arrow) can feed back and continue activation of NADPH-oxidase but also promote additional O2− production from mitochondrial electron transport complexes, specifically Complex I. O2− production from mitochondrial Complex I (large solid arrow) is most likely the major source of ROS in this system which ultimately results in the conversion of O2− to H2O2 probably through SOD (not shown). The pharmacological tools we used to test our hypotheses are shown.
Further evidence is required to establish the mode of action of O2− generated by NADPH oxidase on mitochondria and how this results in increased mitochondria O2− production. Furthermore, a role for endothelial superoxide dismutase is postulated in this study as the enzyme responsible for conversion of mitochondrial O2− to H2O2 but we have not yet confirmed this assumption.
Study limitations
A limitation to the present study is the small quantity of tissue in each vascular sample which does not allow for the direct quantitative measurement of superoxide from NADPH oxidase in live vessels. However relative fluorescence provides accurate assessment of directional change in ROS production. Methodological limitations inherent in the study design of discarded human tissue include the absence of a healthy control group, since all the subjects had underlying health problems that required surgical intervention. However, in most circumstances each vessel served as its own control where flow-response curves were performed sequentially on the same vessel. The use of discarded tissue samples precluded us, due to IRB requirements, from obtaining comprehensive clinical information about human subjects; therefore we cannot take into consideration the extent to which multiple factors such as severity of CAD, disease progression, and medical treatment had on FID. Non-specificity may be a concern for mitochondrial O2− and H2O2 probes. For MitoSOX, the concern lies in the detection of the intended O2− and hydroethidine byproduct, 2-hydroxyethidium, over nonspecific detection of ethidium (29). Importantly, our data reflect that of a study conducted by Robinson et al. where two separate excitation wavelengths for 2-hydroxyethidium (385 and 405 nm) were used to avoid detecting the formation of ethidium (30). Concerns regarding H2O2 detection are tempered by the use of PEG-CAT; a specific scavenger of H2O2 which reduces the DCF signal in microvessels and is consistent with findings in the functional studies supporting the findings of H2O2 generation.
Clinical implications
Flow-induced dilation of adipose arterioles from patients with CAD is mediated by ROS that are produced by endothelium. Given that endothelial cells are constantly exposed to shear stress, early redox signaling events are likely to involve membrane-bound enzymes such as NADPH oxidase. Superoxide produced by NADPH oxidase is an important second messenger regulating expression of redox-sensitive genes (12). The release of superoxide from NADPH oxidase results in NO scavenging and peroxynitrite production. Nitric oxide donors suppress NADPH oxidase activation via S-nitrosylation of the crucial organizer subunit p47phox, thus preventing atherogenesis (31). NO also inhibits the mitochondrial electron transport chain and mitochondrial ROS production. In the absence of NO, activation of ROS generation from mitochondria by NADPH oxidase may be an initiating event in the signaling cascade that preserves vasodilation in patients with CAD when nitric oxide is no longer present. This conservation of vascular function may be a short term solution to maintain blood flow supply to match tissue demand, however the long-term ramifications of substituting ROS for NO, a well-established anti-atherogenic mediator, as the dilator molecule could be at the forefront of endothelial dysfunction and pro-atherosclerotic mechanisms.
Perspectives
Our findings highlight an important role for endothelial NADPH oxidase in mediating flow-mediated dilation in the adipose microcirculation during coronary artery disease. In addition, this study provides evidence of cross-talk between NADPH oxidase and mitochondria, such that RIRR of hydrogen peroxide from mitochondria is responsible for FID in the microcirculation of patients with CAD. Future studies are needed to further our understanding of signaling pathways involving NADPH oxidase and mitochondria in human vasculature.
Acknowledgments
National Institutes of Health grant funding HL085614, HL095701 and HL130513 to SAP; American Heart Association Fellowship 16POST27000011 to ISF
References
- 1.Koller A, Sun D, Huang A, Kaley G. Corelease of nitric oxide and prostaglandins mediates flow- dependent dilation of rat gracilis muscle arterioles. Am J Physiol. 1994;267:H326–32. doi: 10.1152/ajpheart.1994.267.1.H326. [DOI] [PubMed] [Google Scholar]
- 2.Sun D, Huang A, Smith CJ, Stackpole CJ, Connetta JA, Shesely EG, et al. Enhanced release of prostaglandins contributes to flow-induced arteriolar dilation in eNOS knockout mice. Circ Res. 1999;85(3):288–93. doi: 10.1161/01.res.85.3.288. [DOI] [PubMed] [Google Scholar]
- 3.Huang A, Sun D, Wu Z, Yan C, Carroll MA, Jiang H, et al. Estrogen elicits cytochrome P450--mediated flow-induced dilation of arterioles in NO deficiency: role of PI3K-Akt phosphorylation in genomic regulation. Circ Res. 2004;94(2):245–52. doi: 10.1161/01.RES.0000111525.96232.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shimokawa H, Matoba T. Hydrogen peroxide as an endothelium-derived hyperpolarizing factor. Pharmacol Res. 2004;49(6):543–9. doi: 10.1016/j.phrs.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 5.Miura H, Bosnjak JJ, Ning G, Saito T, Miura M, Gutterman DD. Role for hydrogen peroxide in flow-induced dilation of human coronary arterioles. Circ Res. 2003;92(2):e31–e40. doi: 10.1161/01.res.0000054200.44505.ab. [DOI] [PubMed] [Google Scholar]
- 6.Phillips SA, Hatoum OA, Gutterman DD. The mechanism of flow-induced dilation in human adipose arterioles involves hydrogen peroxide during CAD. Am J Physiol Heart Circ Physiol. 2007;292(1):H93–100. doi: 10.1152/ajpheart.00819.2006. [DOI] [PubMed] [Google Scholar]
- 7.Fantuzzi G, Mazzone T. Adipose tissue and atherosclerosis: exploring the connection. Arterioscler Thromb Vasc Biol. 2007;27(5):996–1003. doi: 10.1161/ATVBAHA.106.131755. [DOI] [PubMed] [Google Scholar]
- 8.Gao YJ, Takemori K, Su LY, An WS, Lu C, Sharma AM, et al. Perivascular adipose tissue promotes vasoconstriction: the role of superoxide anion. Cardiovasc Res. 2006;71(2):363–73. doi: 10.1016/j.cardiores.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 9.Matoba T, Shimokawa H, Nakashima M, Hirakawa Y, Mukai Y, Hirano K, et al. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J Clin Invest. 2000;106(12):1521–30. doi: 10.1172/JCI10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y, Bubolz AH, Mendoza S, Zhang DX, Gutterman DD. H2O2 is the transferrable factor mediating flow-induced dilation in human coronary arterioles. Circ Res. 2011;108(5):566–73. doi: 10.1161/CIRCRESAHA.110.237636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Phillips SA, Hatoum OA, Gutterman DD. The mechanism of flow-induced dilation in human adipose arterioles involves hydrogen peroxide during CAD. Am J Physiol Heart Circ Physiol. 2007;292(1):H93–100. doi: 10.1152/ajpheart.00819.2006. [DOI] [PubMed] [Google Scholar]
- 12.Djordjevic T, Pogrebniak A, BelAiba RS, Bonello S, Wotzlaw C, Acker H, et al. The expression of the NADPH oxidase subunit p22phox is regulated by a redox-sensitive pathway in endothelial cells. Free Radic Biol Med. 2005;38(5):616–30. doi: 10.1016/j.freeradbiomed.2004.09.036. [DOI] [PubMed] [Google Scholar]
- 13.Lee SB, Bae IH, Bae YS, Um HD. Link between mitochondria and NADPH oxidase 1 isozyme for the sustained production of reactive oxygen species and cell death. J Biol Chem. 2006;281(47):36228–35. doi: 10.1074/jbc.M606702200. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC, Gutterman DD. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ Res. 2003;93(6):573–80. doi: 10.1161/01.RES.0000091261.19387.AE. [DOI] [PubMed] [Google Scholar]
- 15.Laurindo FR, de Pedro MA, Barbeiro HV, Pileggi F, Carvalho MH, Augusto O, et al. Vascular free radical release. Ex vivo and in vivo evidence for a flow-dependent endothelial mechanism. Circ Res. 1994;74(4):700–9. doi: 10.1161/01.res.74.4.700. [DOI] [PubMed] [Google Scholar]
- 16.Sorescu GP, Song H, Tressel SL, Hwang J, Dikalov S, Smith DA, et al. Bone morphogenic protein 4 produced in endothelial cells by oscillatory shear stress induces monocyte adhesion by stimulating reactive oxygen species production from a nox1-based NADPH oxidase. Circ Res. 2004;95(8):773–9. doi: 10.1161/01.RES.0000145728.22878.45. [DOI] [PubMed] [Google Scholar]
- 17.Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O2− and systolic blood pressure in mice. Circ Res. 2001;89(5):408–14. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 18.Celermajer DS, Sorensen KE, Bull C, Robinson J, Deanfield JE. Endothelium-dependent dilation in the sytemic arteries of asymptomatic subjects relates to coronary risk factors and their interaction. J Am Coll Cardiol. 1994;24:1468–74. doi: 10.1016/0735-1097(94)90141-4. [DOI] [PubMed] [Google Scholar]
- 19.Brookes P, Darley-Usmar VM. Hypothesis: the mitochondrial NO(*) signaling pathway, and the transduction of nitrosative to oxidative cell signals: an alternative function for cytochrome C oxidase. Free Radic Biol Med. 2002;32(4):370–4. doi: 10.1016/s0891-5849(01)00805-x. [DOI] [PubMed] [Google Scholar]
- 20.Beyer AM, Durand MJ, Hockenberry J, Gamblin TC, Phillips SA, Gutterman DD. An Acute Rise in Intraluminal Pressure Shifts the Mediator of Flow-Mediated Dilation from Nitric Oxide to Hydrogen Peroxide in Human Arterioles. American journal of physiology Heart and circulatory physiology. 2014 doi: 10.1152/ajpheart.00557.2014. ajpheart 00557 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doughan AK, Dikalov SI. Mitochondrial redox cycling of mitoquinone leads to superoxide production and cellular apoptosis. Antioxid Redox Signal. 2007;9(11):1825–36. doi: 10.1089/ars.2007.1693. [DOI] [PubMed] [Google Scholar]
- 22.Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC, Gutterman DD. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ Res. 2003;93(6):573–80. doi: 10.1161/01.RES.0000091261.19387.AE. [DOI] [PubMed] [Google Scholar]
- 23.Takabe W, Jen N, Ai L, Hamilton R, Wang S, Holmes K, et al. Oscillatory shear stress induces mitochondrial superoxide production: implication of NADPH oxidase and c-Jun NH2-terminal kinase signaling. Antioxid Redox Signal. 2011;15(5):1379–88. doi: 10.1089/ars.2010.3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hwang J, Ing MH, Salazar A, Lassegue B, Griendling K, Navab M, et al. Pulsatile versus oscillatory shear stress regulates NADPH oxidase subunit expression: implication for native LDL oxidation. Circ Res. 2003;93(12):1225–32. doi: 10.1161/01.RES.0000104087.29395.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li JM, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol. 2004;287(5):R1014–R30. doi: 10.1152/ajpregu.00124.2004. [DOI] [PubMed] [Google Scholar]
- 26.Cai H. NAD(P)H oxidase-dependent self-propagation of hydrogen peroxide and vascular disease. Circ Res. 2005;96(8):818–22. doi: 10.1161/01.RES.0000163631.07205.fb. [DOI] [PubMed] [Google Scholar]
- 27.Wyss CA, Koepfli P, Namdar M, Siegrist PT, Luscher TF, Camici PG, et al. Tetrahydrobiopterin restores impaired coronary microvascular dysfunction in hypercholesterolaemia. Eur J Nucl Med Mol Imaging. 2005;32(1):84–91. doi: 10.1007/s00259-004-1621-y. [DOI] [PubMed] [Google Scholar]
- 28.Fukuda Y, Teragawa H, Matsuda K, Yamagata T, Matsuura H, Chayama K. Tetrahydrobiopterin restores endothelial function of coronary arteries in patients with hypercholesterolaemia. Heart. 2002;87(3):264–9. doi: 10.1136/heart.87.3.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zielonka J, Kalyanaraman B. Hydroethidine- and MitoSOX-derived red fluorescence is not a reliable indicator of intracellular superoxide formation: another inconvenient truth. Free Radic Biol Med. 2010;48(8):983–1001. doi: 10.1016/j.freeradbiomed.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robinson KM, Janes MS, Beckman JS. The selective detection of mitochondrial superoxide by live cell imaging. Nat Protoc. 2008;3(6):941–7. doi: 10.1038/nprot.2008.56. [DOI] [PubMed] [Google Scholar]
- 31.Selemidis S, Dusting GJ, Peshavariya H, Kemp-Harper BK, Drummond GR. Nitric oxide suppresses NADPH oxidase-dependent superoxide production by S-nitrosylation in human endothelial cells. Cardiovascular research. 2007;75(2):349–58. doi: 10.1016/j.cardiores.2007.03.030. [DOI] [PubMed] [Google Scholar]