

ABSTRACT
Small non-coding RNAs (sRNAs) are involved in the control of numerous cellular processes through various regulatory mechanisms, and in the past decade many studies have identified sRNAs in a multitude of bacterial species using RNA sequencing (RNA-seq). Here, we present the first genome-wide analysis of sRNA sequencing data in Rhodobacter capsulatus, a purple nonsulfur photosynthetic alphaproteobacterium. Using a recently developed bioinformatics approach, sRNA-Detect, we detected 422 putative sRNAs from R. capsulatus RNA-seq data. Based on their sequence similarity to sRNAs in a sRNA collection, consisting of published putative sRNAs from 23 additional bacterial species, and RNA databases, the sequences of 124 putative sRNAs were conserved in at least one other bacterial species; and, 19 putative sRNAs were assigned a predicted function. We bioinformatically characterized all putative sRNAs and applied machine learning approaches to calculate the probability of a nucleotide sequence to be a bona fide sRNA. The resulting quantitative model was able to correctly classify 95.2% of sequences in a validation set. We found that putative cis-targets for antisense and partially overlapping sRNAs were enriched with protein-coding genes involved in primary metabolic processes, photosynthesis, compound binding, and with genes forming part of macromolecular complexes. We performed differential expression analysis to compare the wild type strain to a mutant lacking the response regulator CtrA, an important regulator of gene expression in R. capsulatus, and identified 18 putative sRNAs with differing levels in the two strains. Finally, we validated the existence and expression patterns of four novel sRNAs by Northern blot analysis.
KEYWORDS: CtrA, logistic regression, Rhodobacter capsulatus, RNA-seq, small non-coding RNAs, sRNA-Detect
Introduction
Bacterial small non-coding RNAs (sRNAs) are regulatory RNAs that are heterogeneous in size (generally approximately 50 to 250 nucleotides) and structure. sRNAs are known to function in a number of regulatory processes such as inhibition and activation of translation, degradation and stabilization of mRNA, transcriptional interference, and control of protein activity. sRNAs are usually classified into five categories based on their regulatory mechanisms. Cis-encoded base-pairing RNAs are those that bind to their mRNA target with the highest degree of complementarity. An example of this type of sRNA is GadY, which is involved in the regulation of the acid response system of Escherichia coli.1,2 Riboswitches are cis-regulatory elements that directly bind a metabolite when abundance of this metabolite exceeds a threshold level. This binding induces a conformational change in the RNA to form a structure that affects transcription termination or translation initiation.3 Some riboswitches also function as sRNAs and are able to act in trans, such as the S-adenosylmethionine (SAM) riboswitches SreA and SreB of Listeria monocytogenes.4 These two riboswitches regulate the expression of the virulence regulator PrfA by pairing with the 5′ untranslated region (UTR) of its mRNA.4 Trans-encoded base-pairing small RNAs have limited complementarity to their target mRNA(s) and can, in some cases, regulate more than one target. A well-characterized example of a trans-encoded regulatory sRNA is RyhB, which is involved in the regulation of intracellular iron usage in bacteria such as E. coli.5 Protein modulator sRNAs are ones that counter the activities of mRNA-binding proteins. An example is CsrB, which is part of the carbon storage regulator (Csr) system in E. coli.6 The final category consists of the clustered regularly interspaced short palindromic repeat (CRISPR) RNAs (crRNAs), which are palindromes interspaced with short unique spacer sequences that act as a defense mechanism against homologous foreign DNA, such as that from viruses.7
Numerous cellular processes, such as metabolic reactions, quorum sensing, biofilm formation, stress responses, and pathogenesis, are controlled by sRNAs in various species of bacteria.8 In the last decade, high-throughput RNA sequencing (RNA-seq) methods have been employed to identify sRNAs on a genome-wide scale in numerous bacterial species (see Table 1 for some examples). In this work, we used sRNA-Detect9 to perform the first genome-wide detection of sRNAs from RNA-seq data in the purple nonsulfur alphaproteobacterium Rhodobacter capsulatus. This is an organism of interest for its metabolic versatility10 and production of a gene transfer agent.11 We performed comparative RNA-seq targeting sRNAs in the R. capsulatus wild type strain, SB1003, and a mutant strain, SBRM1, lacking the histidyl-aspartyl phosphorelay response regulator CtrA, and identified 422 putative sRNAs expressed in R. capsulatus in the early stationary growth phase when growing in photoheterotrophic conditions. Among these 422 putative sRNAs, we identified 18 sRNAs with differing levels in the two strains. Based on significant matches to sequences in the Rfam database,12 in the RNAcentral database,13 and in the bacterial small regulatory RNA database (BSRD),14 19 of the 422 putative sRNAs were assigned a predicted function. The transcript levels for selected sRNA candidates were validated by Northern blot analysis.
Table 1.
List of bacterial species and sRNAs used for comparative analysis.
| Species | Phylum or class | Genome assembly accession number | Number of sRNAs | Reference |
|---|---|---|---|---|
| Chlamydia trachomatis L2b/UCH-1/proctitis | Chlamydiae | NC_010280.2 | 46 | 52 |
| Clostridium difficile 630 | Firmicutes | NC_009089.1 | 253 | 53 |
| Streptococcus pneumoniae TIGR4 | Firmicutes | NC_003028.3 | 88 | 54 |
| Bacillus subtilis subsp. subtilis str. 168 | Firmicutes | NC_000964.3 | 84 | 55 |
| Corynebacterium glutamicum ATCC 13032 | Actinobacteria | NC_003450.3 | 805 | 56 |
| Mycobacterium tuberculosis H37Rv | Actinobacteria | NC_000962.3 | 258 | 57 |
| Propionibacterium acnes KPA171202 | Actinobacteria | AE017283.1 | 79 | 58 |
| Streptomyces venezuelae ATCC 10712 | Actinobacteria | NC_018750.1 | 175 | 59 |
| Streptomyces avermitilis MA-4680 | Actinobacteria | NC_003155.4 | 199 | 59 |
| Streptomyces coelicolor A3 | Actinobacteria | NC_003888.3 | 92 | 59 |
| Campylobacter jejuni RM1221, 81–176, 81116, and NCTC11168 | Epsilonproteobacteria | NC_003912.7, NC_008787.1, NC_009839.1, NC_002163.1 | 102 | 60 |
| Helicobacter pylori 26695 | Epsilonproteobacteria | NC_000915.1 | 276 | 61 |
| Neisseria gonorrhoeae FA 1090 | Betaproteobacteria | NC_002946.2 | 231 | 62 |
| Caulobacter crescentus sp K31 | Alphaproteobacteria | NC_010338.1 | 29 | 34 |
| Rhodobacter capsulatus SB1003 | Alphaproteobacteria | NC_014034.1 | 422 | This work |
| NC_014035.1 | ||||
| Agrobacterium tumefaciens | Alphaproteobacteria | NC_003062.2, NC_003063.2 | 187 | 63 |
| Rhodobacter sphaeroides 2.4.1 | Alphaproteobacteria | NC_007493.2, NC_007494.2 | 28 | 19,64 |
| Sinorhizobium meliloti 1021 | Alphaproteobacteria | NC_003047.1 | 150 | 65 |
| Vibrio cholerae O1 biovar El Tor str. N16961 | Gammaproteobacteria | NC_002505.1, NC_002506.1 | 480 | 66 |
| Pseudomonas aeruginosa UCBPP-PA14 | Gammaproteobacteria | NC_008463.1 | 165 | 67 |
| Escherichia coli str. K-12 substr. MG1655 | Gammaproteobacteria | NC_000913.2 | 309 | 68 |
| Erwinia amylovora ATCC 49946 | Gammaproteobacteria | NC_013971.1 | 40 | 69 |
| Yersinia pestis KIM10+ | Gammaproteobacteria | NC_004088.1 | 31 | 70 |
| Salmonella enterica subsp enterica serovar Typhimurium str. SL1344 | Gammaproteobacteria | NC_016810.1 | 113 | 71 |
We also collected genome sequences and published putative sRNAs from 23 additional bacterial species, which included representatives from the phyla Chlamydiae, Firmicutes, and Actinobacteria, and the Alpha-, Beta-, Gamma-, and Epsilonproteobacteria classes of the phylum Proteobacteria. This yielded a collection of 4,725 predicted sRNAs. Based on sequence comparisons, 124 of the 422 putative R. capsulatus sRNAs were conserved in at least one other bacterial species.
Finally, we characterized all putative sRNAs for four bioinformatics characteristics and then applied machine learning approaches to develop a quantitative model to calculate the probability of a given RNA sequence to be a bona fide sRNA. The model was able to correctly classify 95.2% of sequences in a validation set.
Results and discussion
Sequencing and detection of R. capsulatus sRNAs
We grew cultures under photoheterotrophic conditions to early in the stationary phase of growth so that the data collected would match with our most comprehensive collection of transcriptomic data from previous microarray studies.15,16 Sequencing of size-selected RNA, ≤ 200 nucleotides, from the genome-sequenced strain, SB1003, and its derived ctrA mutant strain, SBRM1,17 generated a total of 4.45 million reads. From these reads, 93.5% were uniquely mapped to the R. capsulatus genome. These sequence data have been submitted to the NCBI Gene Expression Omnibus (GEO) under accession number GSE82056.
Recently, we showed that sRNA-Detect, a new computational program for the detection of bacterial small transcripts from RNA-seq data, exhibits higher recall rates at comparable specificity levels than other standalone computational approaches.9 We used sRNA-Detect on our sequence data, and after removal of detected small transcripts located within annotated tRNAs (tRNAs) and rRNAs (rRNAs), we detected 422 potential sRNAs in R. capsulatus.
sRNAs with predicted functions or homologs
To annotate R. capsulatus putative sRNAs with predicted functions, we retrieved significant matches to R. capsulatus sRNAs from the Rfam, RNAcentral and BSRD databases. Based on these matches, we annotated 19 sRNAs with predicted functions (Tables 2 and S1). There were six riboswitches (including those binding thiamine pyrophosphate and cobalamin), three segments of transfer-mRNA (tmRNA), three segments of the catalytic RNA of ribonuclease P (RNase P RNA), the signal recognition particle (SRP) RNA (ffs), 6S RNA, an α-operon ribosome binding site, the cspA thermoregulator, the upstream sRNA of mraZ (UpsM)18 and an sRNA homologous to a validated Rhodobacter sphaeroides sRNA (RSs1386).19 Several sRNAs corresponding to fragments of the tmRNA and the RNase P RNA were predicted due to differences in read depth coverage across the full length of these transcripts.
Table 2.
List of functionally annotated sRNAs.
| Identifier(s) | Inferred Annotation |
|---|---|
| sRNA00627 | TPP riboswitch |
| sRNA00822 | Signal recognition particle (SRP) RNA (ffs) |
| sRNA00687, sRNA00526, sRNA00688, sRNA00508, sRNA01035 | Cobalamin riboswitch |
| sRNA00123 | α-operon ribosome binding site |
| sRNA00598 | Bacterial small signal recognition particle RNA |
| sRNA01208 | cspA thermoregulator |
| sRNA01158, sRNA01157, sRNA01156 | Transfer-messenger mRNA (fragment of) |
| sRNA01077 | Upstream sRNA of mraZ, UpsM |
| sRNA00648 | 6S RNA |
| sRNA00470 | Homologous sRNA to the Rhodobacter sphaeroides validated sRNA RSs1386 |
| sRNA01141, sRNA01140, sRNA01139 | Ribonuclease P catalytic RNA (fragment of) |
To investigate the extent of sequence conservation of putative R. capsulatus sRNAs in different bacterial species, we obtained sRNA sequences identified in recent studies of 23 other bacterial species (Table 1) and used BLAST (version 2.2.30+)20 to search for pairwise reciprocal best matches between the sRNAs of each of the other 23 bacterial species and the R. capsulatus sRNAs from this study. As differences in the characteristics of each study, including but not limited to differences in sequencing platforms, growth conditions, RNA extraction methods, and sRNA identification methods, lead to limitations in this analysis, we also searched for sequence conservation of R. capsulatus sRNAs in the genomes of these 23 other bacterial species. In total 124 (or 29%) of the 422 putative sRNAs had homologous sRNAs or were found to be conserved in the genome of at least one other bacterial species (Fig. 1). We organized these 124 sRNAs based on our level of confidence in their conservation. We referred to sRNAs with matches in at least one of the three RNA databases (Rfam, RNAcentral and BSRD) as hypothetical equivalogs, which represented 24 sRNAs that likely belong to a set of sRNAs conserved with respect to function. This category includes the 19 sRNAs for which we inferred a function. We classified sRNAs with homologs found in bacterial species belonging to other genera as inter-taxa homologs, which represented 40 sRNAs that are likely to be true functional sRNAs. The sRNAs whose sequences were only present in the genome of the related bacterial species R. sphaeroides were classified as intra-genus homologs, which represented 60 sRNAs. The remaining 298 putative R. capsulatus sRNAs appear to be species-specific. Not surprisingly, there are more intra-genus than inter-taxa homologs and, as already pointed out by Gomez-Lozano et al.,21 there is limited sRNA sequence conservation across different species.
Figure 1.
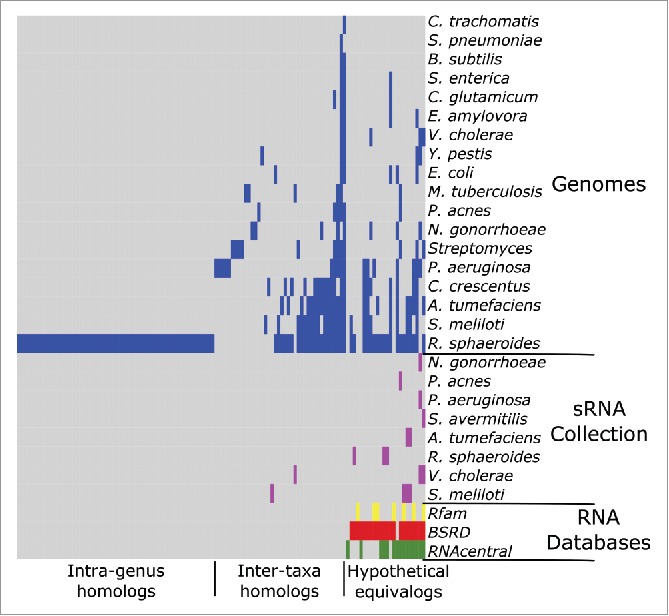
Map of 124 sRNAs in R. capsulatus with sequence conservation in other bacterial species. Sequence similarity searches were performed for all putative R. capsulatus sRNAs against three RNA databases and a panel of 23 bacterial species including representatives from the Chlamydiae, Firmicutes, and Actinobacteria phyla and the Alpha-, Beta-, Gamma-, and Epsilonproteobacteria classes of the phylum Proteobacteria. From right to left, three classes (hypothetical equivalogs, 24 sRNAs; inter-taxa homologs, 40 sRNAs; and intra-genus homologs, 60 sRNAs) proceed from nearly complete certainty about a putative sRNA's function to no functional information. Gray indicates no homologs (matches) were found for the sRNA in that organism or database.
Bioinformatic characterization of putative sRNAs in R. capsulatus
We characterized all 422 putative sRNAs in terms of their predicted secondary structures, their proximities to predicted promoter sites, their proximities to predicted Rho-independent terminators, and their genomic contexts. To be able to compare the features of the putative sRNAs with a null distribution, we randomly extracted sequences matching the length and strand of putative sRNAs from the R. capsulatus genome. There were at least 10 random sequences for each putative sRNA sequence. We used CentroidFold22 to predict the secondary structures of both the sRNA sequences and the random sequences, and to calculate the free energies of the folded structures. We found that the distribution of free energies of the sRNAs' secondary structures was shifted toward lower values than the distribution of free energies of the random sequences' secondary structures (Fig. 2A). The difference between the free energies of the sRNAs' secondary structures and the free energies of the random sequences' secondary structures was statistically significant (p = 5.9E-12, Mann-Whitney test). This indicates that our putative sRNAs tend to adopt more stable conformations than random genomic sequences.
Figure 2.
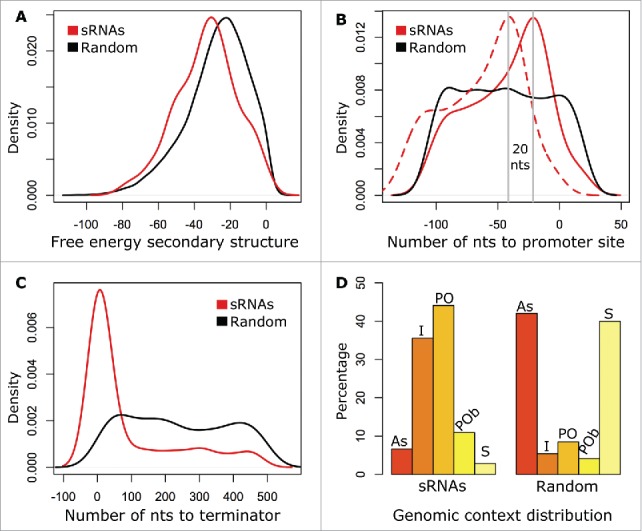
Characteristics of putative sRNAs in comparison with the null distribution. (A) Probability distribution of the free energy of the predicted secondary structures for the putative sRNAs (red line) and 4,400 random genomic sequences of matching length and orientation (black line). The average free energy of the sRNAs' predicted secondary structures is statistically significantly lower than the average free energy of the random sequences' secondary structures (p = 5.902E-12, Mann-Whitney test). (B) Density function of the number of nucleotides (nts) upstream from the predicted 5′ end of the putative sRNAs to −10 (solid red line) and −35 (dashed red line) predicted promoter sites in comparison with the number of nts from the 5′ end of random genomic sequences to −10 predicted promoter sites (solid black line). (C) As B, but number of nts downstream from the predicted 3′ end of the putative sRNAs (red line) and of random genomic sequences (black line) to predicted Rho-independent terminators. (D) Proportion of sRNAs (left) and random genomic sequences (right) in a specific class of genomic context (antisense (AS), 28 sRNAs; intergenic (I), 150 sRNAs; partial overlapping (PO), 186 sRNAs; partial overlapping on both ends (POb), 46 sRNAs; and sense (S), 12 sRNAs).
Using the BPROM program,23 we searched for putative promoters in the region spanning 150 nucleotides (nts) upstream to 20 nts downstream from the predicted 5′ ends of both the putative sRNAs and the random genomic sequences. Of the 422 putative sRNAs, 183 (43%) had predicted promoter sites, in contrast to 18.6% of the random sequences. Furthermore, there was a distinct peak at position −21.5 in the probability density function for the −10 promoter positions of putative sRNAs, whereas the random sequences had a uniform probability distribution for the −10 promoter positions (Fig. 2B). As sRNA-Detect tends to predict transcripts that lie within the boundaries of the actual sRNA (i.e., it misses some nucleotides at the 5′ and 3′ ends of the sRNAs),9 the average distance to the −10 and −35 promoter sites from the actual 5′ end of the sRNAs would be less than as estimated above. Our data indicates that many of the putative sRNAs have proximal promoter sites and supports the notion that they are independently transcribed.
Next, we used TransTermHP,24 a computational method to detect Rho-independent transcription terminators, to predict the locations of terminators in the R. capsulatus genome. We associated a terminator to a putative sRNA if the terminator was within 500 nts downstream from the predicted 3′ end of the sRNA as described by Kingsford et al.24 Of the 422 putative sRNAs, 130 (31%) had an associated predicted Rho-independent transcription terminator, whereas only 8.15% of the random genomic sequences did. Moreover, as depicted in Fig. 2C, there was a distinct peak in the probability that the 3′ ends of the sRNAs were located 7 nts from the closest downstream terminator, whereas the random sequences' density function had a uniform distribution.
Based on the putative sRNAs' genomic contexts, we classified the sRNAs as either “intergenic” if located in intergenic regions (IGRs), “antisense” if located within an annotated gene and transcribed on the strand opposite to this gene, “partially overlapping” if the 5′ or 3′ end of the sRNA overlaps the 5′ or 3′ end of an annotated gene, “partially overlapping on both ends” if the 5′ end of the sRNA overlaps an annotated gene and the 3′ end of the sRNA overlaps another annotated gene, or “sense” if located within an annotated gene and transcribed on the same strand as this gene (Fig. 3). 150 sRNAs were intergenic, 186 were partially overlapping and 46 were partially overlapping on both ends. These amounts were 6.6, 5.2 and 2.6 times more than expected, respectively, if the locations were randomly distributed over the genome (Fig. 2D). In contrast, 28 antisense sRNAs (asRNAs) and 12 sense sRNAs were detected, which is 6.3 and 14.1 times less than expected, respectively (Fig. 2D).
Figure 3.
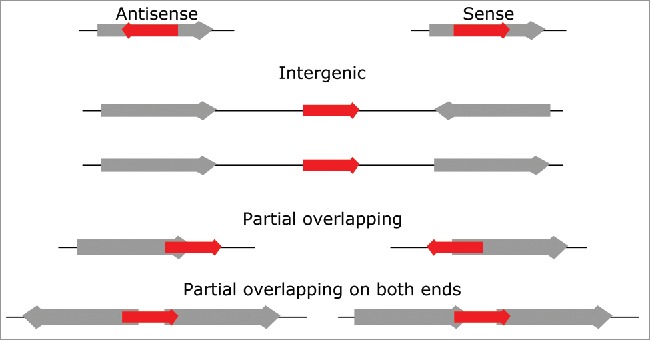
Schematic illustration of the different classes of genomic contexts of sRNAs. Genes are depicted as thick arrows with open reading frames (ORFs) shown in gray and sRNAs shown in red. Only a subset of all possible direction of transcription combinations are shown. Antisense RNAs (asRNAs) are within an ORF that is transcribed on the strand opposite to the asRNAs. Sense sRNAs are within an ORF that is transcribed on the same strand as the sRNA. Intergenic sRNAs are found in intergenic regions (IGRs) between ORFs. Partial overlapping sRNAs occur when the 5′ or 3′ end of the sRNA overlaps with the 5′ or 3′ end of an ORF. Partial overlapping on both ends sRNAs occur when the 5′ of the sRNA overlaps the 5′ or 3′ end of an ORF and the 3′ of the sRNA with the 5′ or 3′ end of another ORF.
As putative sRNAs had clearly distinct characteristics from random sequences, we decided to apply machine learning approaches (classifiers) to obtain a model to quantify the probability of a sequence being a bona fide sRNA. To derive the model, we selected as predictors (attributes) the free energy of the predicted secondary structure of the sRNA, the distance to a predicted promoter site, the distance to a Rho-independent terminator, and the sRNA genomic context. The genomic context included distance to the closest “left” neighboring ORF, distance to the closest “right” neighboring ORF, and whether the sRNA was on the same strand as the closest neighboring annotated ORFs. We refer to an annotated ORF located at the 5′ end of a sRNA on the forward strand or an annotated ORF located at the 3′ end of a sRNA on the reverse strand as “left,” and an annotated ORF located at the 3′ end of a sRNA on the forward strand or an annotated ORF located at the 5′ end of a sRNA on the reverse strand as “right” (illustrated in Fig. S1). To create the model, we considered those sRNAs with inter-taxa homologs in the sRNA collection or conserved in the genome of at least two other bacterial species, and sRNAs with hypothetical equivalogs (Fig. 1) as “bona fide sRNAs.” We randomly chose 33 of these 41 bona fide sRNAs as positive instances and 98 random sequences as negative instances to train the classifiers. We then evaluated the classifiers' performances on the remaining 8 bona fide sRNAs and 4322 random sequences. We applied three machine learning approaches, namely, logistic regression, linear discriminant analysis (LDA) and quadratic discriminant analysis (QDA).25 Among these three methods, logistic regression had the highest recall rates at the lowest false positive rate (Fig. S2A-C). Details about the logistic regression model obtained are given in the Materials and Methods section. At a probability cut-off of 0.6, the logistic regression model retrieved 66.25% of the positive test instances and only 4.7% of the random sequences. We then calculated the probability of being a bona fide sRNA using the logistic regression model for all 422 putative sRNAs. Of the 422 putative sRNAs, 109 (26%) scored a probability >0.6 (Fig. S2D). At the estimated false positive rate, only five of these 109 sRNAs would be expected to be false positives. We expect that assigning a confidence estimate for being a bona fide sRNA to a given putative sRNA will help prioritize sRNAs for experimental validation. A limitation of this analysis is that, as the majority of positive instances used to learn the logistic regression model were intergenic or partially overlapping sRNAs, the logistic regression model underestimates the probability of asRNAs being bona fide sRNAs. These analyses need to be replicated in other bacterial species with a larger number of confirmed sRNAs to corroborate these findings and obtain better performance estimates. Table S1 contains the full description of all putative sRNAs, including their estimated probabilities of being bona fide sRNAs.
Identification of a putative tRNA-derived sRNA locus
We observed a putative intergenic sRNA (sRNA00295) found to be conserved in the genomes of 16 other bacterial species without a homologous sRNA in the sRNA collection or the RNA databases. This sRNA lacked a homologous sRNA of known function and we decided to inspect it further. The sequence of sRNA00295 was identical to the 3′ region of the four tRNA-Met genes found in the R. capsulatus chromosome. The homology with the tRNAs makes interpreting the RNA-seq read data somewhat challenging, as reads originating from the tRNAs could be mapping onto this putative sRNA locus, and vice versa. However, a promoter site and Rho-independent terminator were predicted to flank this putative sRNA. We also checked this region in an additional unpublished data set based on differential RNA-seq (dRNA-seq), which identifies 5′ ends of RNAs that originate from transcription initiation as opposed to RNA processing,26 and a 5′ end was identified at this location (Grüll et al., unpublished). There have been recent discoveries of tRNA-derived sRNAs, which have been implicated in different regulatory processes.27,28 If genuine, this sRNA would instead represent an independent tRNA-derived fragment locus, and this warrants future investigation. Fig. 4 depicts sRNA00295's genomic context and predicted secondary structure.
Figure 4.
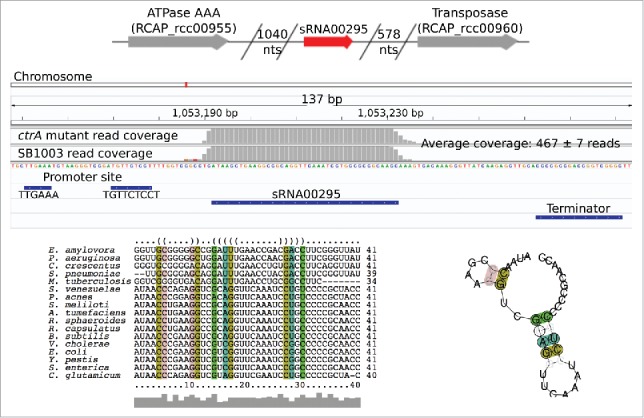
Genomic context and predicted secondary structure of a putative tRNA-derived sRNA locus (sRNA00295). The top panel shows sRNA00295's genomic context indicating distance to the closest protein-coding genes. The middle panel illustrates read depth coverage, location of predicted promoter site, and location of Rho-independent terminator (panel generated using Integrative Genomics Viewer version 2.3.72). The numbers of reads mapped for the SB1003 and the ctrA mutant strain were 472 and 462, respectively, as calculated by htseq-count. The bottom panel shows a multiple sequence alignment and predicted consensus secondary structure obtained using LocARNA. Colored nucleotides indicate correspondence between positions in the alignment and the RNA structure.
To gain insight into the likely functional role of this putative sRNA, we used the CopraRNA web server29 to predict sRNA00295's targets. Despite recent advances, most sRNA target prediction programs have a high false positive rate; CopraRNA, which requires at least three homologous sequences to predict targets, has twice the prediction accuracy of other sRNA target prediction programs.30 Table 3 shows the top 10 sRNA00295 targets predicted by CopraRNA (the complete CopraRNA results, which includes all 76 predicted targets are listed in Table S2). To quantify protein interactions among sRNA00295's 76 predicted targets, we used the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING, version 10.0) database of physical and functional interactions.31 Compared with similarly sized randomly selected protein sets, sRNA00295's 76 predicted targets have significantly more interactions among themselves (PPI enrichment p = 1.13E-08), with 28 interactions as compared with eight for random protein sets. We also tested for functional enrichment among sRNA00295's 76 predicted targets using STRING, but no functional enrichment was found.
Table 3.
Top 10 targets predicted by CopraRNA for sRNA00295 a putative tRNA-derived sRNA locus.
| Gene ID | Description |
|---|---|
| RCAP_rcc01474 | amino acid permease |
| RCAP_rcp00009 | LacI family transcriptional regulator |
| RCAP_rcc00101 | ABC transporter permease |
| RCAP_rcc02606 | mammalian cell entry domain-containing protein |
| RCAP_rcc00024 | glutaryl-CoA dehydrogenase |
| RCAP_rcc01400 | signal transduction histidine kinase |
| RCAP_rcc00616 | acriflavine resistance protein B |
| RCAP_rcc00505 | type II secretion system protein E |
| RCAP_rcc01291 | kinetochore Spc7 domain-containing protein |
| RCAP_rcc02771 | TetR family transcriptional regulator |
Functional and protein-interaction enrichment of potential cis-targets of putative antisense and partially overlapping sRNAs
To obtain insight into the biological processes potentially regulated by the antisense and partially overlapping putative sRNAs, we assumed that they were cis-acting and examined the 265 overlapping protein-coding mRNAs for functional and protein interaction enrichment using STRING. As antisense and partially overlapping sRNAs have been shown to also regulate gene expression in trans,32 this approach likely missed additional regulatory targets of these putative sRNAs. Nevertheless, the set of cis-targets showed a significant enrichment of genes involved in primary metabolic process (28 genes, FDR-corrected p = 1.97E-5), photosynthesis (16 genes, FDR-corrected p = 3.98E-5), compound binding (24 genes, FDR-corrected p = 0.004), and of genes encoding parts of macromolecular complexes (17 genes, FDR-corrected p = 3.2E-7). The complete functional enrichment results are provided in Table S3. We also investigated whether putative cis-targets were co-expressed based on previously determined R. capsulatus gene co-expression modules,16 and found that cis-targets showed a significant accumulation in two gene co-expression modules (13 genes in the midnightblue module, FDR-corrected p = 0.002; and 7 genes in the salmon4 module, FDR-corrected p = 0.003). Additionally, there were significantly higher interactions among the network of cis-targets (PPI enrichment p = 0), with 528 interactions as compared to 204 for random protein sets. This indicates that several of the likely cis-targets interact and are co-expressed, and supports the notion that sRNAs play a regulatory role in these processes.
Effects of loss of ctrA on sRNA expression
We investigated whether putative sRNAs were differentially expressed between two R. capsulatus strains: the genome-sequenced strain, SB1003, and its ctrA null mutant derivative, SBRM1. CtrA is a two-component/histidyl-aspartyl phosphorelay response regulator that affects many processes in R. capsulatus such as motility and gene transfer agent production.33 In Caulobacter crescentus, where it is an essential protein and controls many cell cycle-related process, CtrA was shown to regulate expression of sRNAs as part of its regulon.34 Fig. S3 illustrates the distribution of the normalized log2 fold change of the sRNAs' read counts between the two strains. Although more samples are required to have enough statistical power to identify statistically differentially expressed sRNAs, the vast majority of sRNAs do not appear to be differentially expressed. However, 18 sRNAs had an absolute log2 fold change >3, suggesting possible differential expression between the strains. Among these 18 sRNAs, there are 2 asRNAs, 7 intergenic, 8 partially overlapping, and 2 partially overlapping on both ends sRNAs. Nine of the 14 ORFs overlapped by the antisense and partially overlapping sRNAs were previously identified as affected by the loss of CtrA15 (p = 4.14E-10, Hypergeometric test), including genes encoding the flagellar protein MotB (rcc00006), the flagellar hook-associated protein FlgK (rcc00008), an Hpt domain-containing protein (rcc00180), and the DNA-protecting protein DprA (rcc03098). We also investigated whether these 14 ORFs overlapped by potential differentially expressed sRNAs were co-expressed based on previously determined R. capsulatus gene co-expression modules and, indeed, they were significantly over-represented in two modules: pink (6 genes, FDR-corrected p = 1.05E-5) and orange (3 genes, FDR-corrected p = 2.9E-4). The orange module was identified as associated with the production of RcGTA16 and the DprA protein is required for uptake of DNA from RcGTA particles by recipient cells,35 thereby adding sRNAs as another regulatory mechanism involved in controlling RcGTA-mediated gene exchange in R. capsulatus.11 As all of the potentially differentially expressed sRNAs are R. capsulatus-specific, we were unable to use CopraRNA to predict potential targets of the intergenic sRNAs.
Experimental validation of putative sRNAs using Northern blot analysis
We chose four putative sRNAs to evaluate by Northern blotting. These were sRNA00385, sRNA01029, sRNA00848, and sRNA01129, representing four R. capsulatus-specific intergenic sRNAs, three of which showed differential expression between the wild type and ctrA mutant strains, as evaluated by read counts in the RNA-seq data. We purposefully chose three of the targets due to their predicted differential expression to help with interpretation of the Northern blots as previous studies have detected multiple bands on Northern blots probed for sRNA detection.36 These differentially expressed sRNAs are also candidates for future investigation for potential roles in the regulation of CtrA-affected processes, such as the production of RcGTA. As expected due to the program's limitation with respect to correctly identifying the 5′ and 3′ boundaries of sRNAs,9 the bands detected for each of the sRNAs were larger than predicted by sRNA-Detect. Manual inspection of the sequence read data allowed us to estimate the boundaries and sizes of these sRNAs more accurately (Fig. 5) to match the sizes estimated on the Northern blots, and we identified putative promoter sequences for these sRNAs (Fig. 6) that agree with a previously identified R. capsulatus consensus promoter sequence.37
Figure 5.
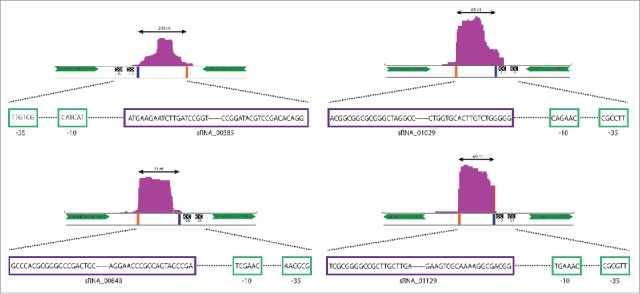
Read depth coverage plots and genomic locations for the experimentally confirmed sRNAs. Parts of the neighboring genes are shown with green arrows indicating their direction in the genome. Their relative distance to the coverage plots is not to scale. Predicted promoter −10 and −35 elements are depicted as black boxes with white arrows inside, and the sequences are given in Fig. 6 and below. The distance of the promoter relative to the 5′ end of the corresponding sRNA is also not to scale. The sRNA sequencing reads are presented as purple plots with an indication of the sRNA's predicted size on top. Blue bars mark the predicted 5′ ends and orange bars the predicted 3′ ends of the sRNAs. The sRNAs' 5′ and 3′ end sequences and the −10/−35 elements are shown underneath the plots in their respective 5′–3′ orientations.
Figure 6.
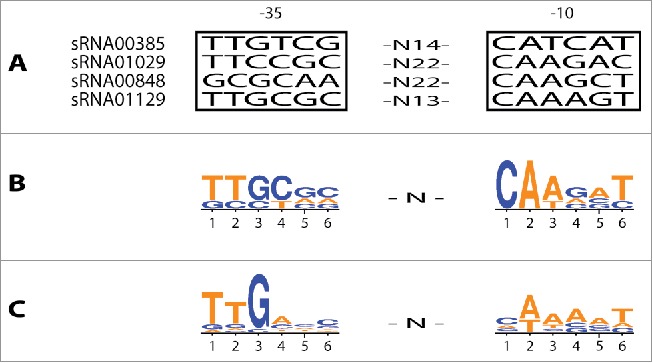
Identification of putative promoter −10 and −35 sequences for four experimentally confirmed sRNAs. (A) Predicted promoter sites upstream of each of the sRNAs. The nucleotide spacing between the motifs is indicated. (B) The frequency of bases found at each position is indicated by the size of the colored letters, created with Weblogo 3.0.51 (C) Consensus promoter sequence based on promoters identified in a previous study.37
sRNA00385 was predicted to have a size of 189 nts based on sRNA-Detect. Examination of the sequence reads for this region suggested an actual size of 249 nts (Fig. 5). A putative promoter site was found upstream of the predicted 5′ end (Figs. 5 and 6) although in this case the −10 site was centered 18 nts upstream of the predicted 5′ end, possibly indicating either poor read coverage at the 5′ end as frequently found in RNA-seq,38 or variable length spacing in the promoter elements.39,40 This putative sRNA showed similar, high levels of expression in the RNA-seq data from both strains. The Northern blot showed a major band at approximately 230 nts (Fig. 7). There were several additional bands detected on this blot, most of which were present in both strains. These presumably result from non-specific hybridization of the probe to additional RNAs, as has been observed in previous studies detecting sRNAs by this method.36
Figure 7.
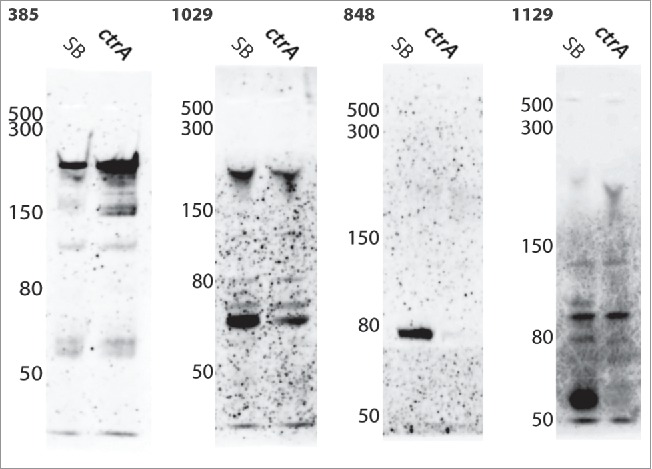
Northern blot images of the experimentally confirmed sRNAs. RNA from the genome-sequenced strain, SB1003, and the ctrA mutant strain were run in the left and right lane, respectively, of each gel. The sequences of the biotin-labeled probes are given in Table 4. The sizes for the corresponding ladder bands are indicated on the left of each blot image, and the number on top of each image identifies the corresponding sRNA probe.
sRNA01029 was predicted to have a size of 52 nts by sRNA-Detect. Inspection of the sequencing data for this sRNA suggested a size of 68 nts and a −10 element was identified centered 12 nts upstream of the predicted sRNA's 5′ end (Figs. 5 and 6). The sRNA was predicted to be more highly expressed in the wild type strain based on read count data (approximately 3:1, Table S1). The Northern blot showed several bands in both strains, with one band at approximately 65 nts that was present at higher levels in the wild type strain in comparison to the ctrA mutant (Fig. 7).
sRNA00848 was predicted to have a size of 71 nts by sRNA-Detect, with inspection of the sequencing data suggesting a size of 91 nts. A putative promoter sequence was identified upstream of the predicted 5′ end (Figs. 5 and 6) but, as with sRNA00385, the −10 sequence was centered more than 10 nts upstream (25 nts). This sRNA was only detected in the RNA-seq data from the wild type strain and the Northern blot showed a band at approximately 78 nts only in RNA from the wild type strain (Fig. 7).
sRNA01129 was predicted to have a size of 69 nts based on sRNA-Detect, and this matched the predicted size from manual inspection of the sequencing data. We found a putative promoter with a −10 element centered 7 nts upstream of the predicted 5′ end (Figs. 5 and 6). This sRNA was detected at a much higher level in the wild type strain RNA-seq data compared to the ctrA mutant (28:1, Table S1). The Northern blot showed a band at approximately 60 nts that was present in the wild type strain but not detected in the ctrA mutant (Fig. 7).
Conclusions
Using RNA-seq data we have identified 422 putative sRNAs in R. capsulatus: 24 sRNAs with hypothetical equivalogs, 40 sRNAs with putative inter-taxa homologs, 60 sRNAs with putative intra-genus homologs and 298 potential R. capsulatus-specific sRNAs. To help prioritize further investigations into these sRNAs, we have bioinformatically characterized these sRNAs and used logistic regression to quantify the probability of a putative sRNA being a bona fide sRNA. Using the logistic regression model, 109 (or 26%) of the 422 putative sRNAs were assigned a probability greater than 0.6 of being a bona fide sRNA; at the estimated false positive rate of 4.8%, only five out of these 109 sRNAs are expected to be false positives. Analysis of a strain lacking the important response regulator CtrA identified 18 putative sRNAs that were differentially expressed relative to the wild type strain. This indicates that effects on the levels of sRNAs is another means by which the CtrA phosphorelay regulates processes in R. capsulatus. We experimentally confirmed the existence of four of the putative sRNAs by Northern blot analysis, and validated the differential expression that was predicted from the RNA-seq data analysis for three of these. The abundance of sRNAs detected in R. capsulatus indicates that a potential extra layer of regulatory complexity exists in this species. Revealing the functional roles of these sRNAs will improve our understanding of the mechanisms R. capsulatus employs to regulate its physiology.
Materials and methods
R. capsulatus growth and RNA isolation
R. capsulatus cultures were grown under anaerobic phototrophic conditions at 35°C in complex YPS medium41 until four hours after reaching stationary phase. The culture was mixed 5:1 with 95% ethanol and 5% saturated phenol,42 the cells were pelleted by centrifugation, the supernatant was removed, and the cell pellets were frozen in dry ice/ethanol and stored at −80°C until RNA isolations were performed. sRNA purification was performed with the NucleoSpin® miRNA kit (MACHEREY-NAGEL) following the manufacturer's protocol for purification of the small RNA fraction (<200 nts).
Library preparation and sequencing
The isolated small RNA fraction was used for RNA library preparation for sequencing using an Ion Torrent Personal Genome Machine (PGM; Thermo-Fisher). The RNA quality was checked prior to library preparation using an Agilent Bioanalyzer (Agilent Technologies). Library preparation followed the manufacturer's recommendations for small RNA libraries with the RNA-seq Kit v2 (Thermo-Fisher). The library was amplified using an Ion Torrent One Touch 2 system. The samples were loaded individually on 316 v2 chips and sequenced with the number of flows set to 550.
Processing of RNA-seq data
The RNA-seq data quality was verified using the FastQC tool (version 0.10.0) and reads were filtered and trimmed using the fastq_quality_trimmer available in the FASTX Toolkit (version 0.0.13.2) with a quality threshold of 22 and minimum read length of 28 nucleotides. Filtered and trimmed reads were mapped to the R. capsulatus genome using the Torrent mapper tmap (version 3.0.1), executed with the parameters: -B 18 -a 2 -v stage1 map1 map2 map3. Mapping statistics were obtained using samtools.43
Detection of sRNAs from RNA-seq data
sRNAs were predicted from mapped RNA-seq data using sRNA-Detect.9 sRNA-Detect constructs a coverage vector using the function GenomicArray available in HTSeq44 (version 0.5.4p5) and then goes through the genomic intervals in the coverage vector and finds continuous segments between 20 and 250 nucleotides long with similar numbers of reads, with a maximum percentage change of 3% allowed in the average number of reads. A minimum of 10 reads across all samples was required to consider a transcript as expressed. sRNA-Detect is available at www.cs.mun.ca/∼lourdes. Predicted transcripts overlapping to tRNAs and rRNAs were removed from the putative sRNA set using the tool intersectBed available in BEDtools45 (version 2.25).
Collection and analysis of sRNAs from other bacterial species
Published studies performing genome-wide identification of sRNAs using RNA-seq data were identified (Table 1), genomic coordinates of the putative sRNAs were collected, and the corresponding sRNA sequences were obtained using the tool fastaFromBed available in BEDtools.
Bioinformatic analysis of sRNAs
Sequence conservation of putative sRNAs was determined by identifying reciprocal best BLAST matches between pairs of species (Table S4). The program blastn (version 2.2.30+) was executed with an E-value cut-off of 1E-4, a best_hit_overhang of 0.1 and task mode of “blastn.” Rfam matches were obtained using the batch search functionality in the Rfam database (version 12.1). If an sRNA had multiple Rfam matches only the most significant match was considered. All 30581 sRNA sequences in BSRD were downloaded (May 2015) and a BLAST database was created with these sequences. BSRD matches per sRNA were obtained using blastn with the same settings as for the homology search. If an sRNA had multiple BSRD matches only the match with the lowest E-value was considered. The RNAcentral database (release 5) was downloaded (May 2016) and nhmmer46 (version 3.1b2) with an E-value cutoff of 1E-3 was used to identify RNAcentral matches for each putative sRNA. If an sRNA had multiple RNAcentral matches only the most significant match was considered. CentroidFold with parameters -e “CONTRAfold” and -g 4 was used to predict the secondary structure of putative sRNAs and random genomic sequences. Sequences of the sRNAs including 150 nts upstream of the predicted 5′ end were obtained using slopBed and fastaFromBed and promoter sites were predicted using BPROM with default values. Rho-independent terminators in R. capsulatus genome were predicted using TransTermHP with default values and providing an annotation file with the coordinates of the protein-coding genes. The numbers of reads mapped to the putative sRNAs per strain were calculated using htseq-count available in HTSEQ. Normalized log2 fold changes between the two R. capsulatus strains were obtained using edgeR47 (version 3.12.1). All results were compiled, processed and visualized using R (version 3.2.4).
To apply machine learning approaches, we represented a putative sRNA or a random genomic sequence as a numerical vector X consisting of seven numerical predictors (input variables); namely, free energy of the secondary structure, distance ranging from [−150, 20] nts to the −10 predicted promoter site (if no promoter site was predicted in that range a value of −1000 was used), distance to terminator ranging from [0, 500] nts (if no terminator was predicted within this distance range a value of 1000 was used), distance (−∞, 0] nts to closest left ORF, a binary number indicating whether the RNA is transcribed on the same strand as its left ORF (1 if transcribed on same strand), distance [0, +∞) to closest right ORF, and a binary number indicating whether the RNA is transcribed on the same strand as its right ORF. For training the classifiers, 33 of the 41 putative sRNAs deemed as bona fide sRNAs were randomly selected as positive instances, and 98 of the 4420 random genomic sequences were randomly selected as negative instances. The remaining sequences were used for testing. Logistic regression was applied using the R function glm (with family = binomial), and cross-validation was performed using the function cv.glm from the R package boot (version 1.3-18). LDA and QDA were applied using the lda and qda functions from the R package MASS (version 7.3-45). Performance measurements were calculated using the R package ROCR48 (version 1.0-7). For the classifiers' performance comparison, we used recall and false positive rates. Recall indicates the proportion of testing positive instances that are predicted to be bona fide sRNAs by a given approach at a certain probability threshold (i.e., true positives (TP) divided by the total number of positive instances (P)). The false positive rate is the proportion of negative instances that are predicted to be bona fide sRNAs by a given approach at a certain probability threshold (i.e., false positives (FP) divided by the total number of negative instances (N)). The logistic regression estimates the parameter θ to model p(X) = e ˆ(θ0+ θ1X1+ … + θpXp) / (1 + e ˆ(θ0 + θ1X1+ … + θpXp)) where X is the vector of attributes representing an instance, e is the base of the natural logarithm, p is the number of attributes in X, and Xi is the value of attribute i. The parameter θ was chosen to maximize the likelihood function. The value of the estimated parameters was θ = [−2.02, −0.037, −5.8e-4, −2.1e-3, −0.011, 0.25, 5e-3, 0.38]. Intuitively, the model learnt makes sense; for instance, decreasing the free energy increases the probability of being a bona fide sRNA, and decreasing the distance to a terminator increases the probability of being a bona fide sRNA. We used these parameters' values to calculate the probability of being a bona fide sRNA for all putative sRNAs. A probability cut-off of 0.6 was chosen as the optimal cut-off to have high recall while maintaining a low false positive rate.
Detection of sRNAs by Northern blotting
Purified sRNA was eluted in 30 µl of nuclease-free water. The water was subsequently evaporated using a vacuum centrifuge (Thermo Scientific, Savant DNA120 SpeedVac Concentrator) for 30 minutes at high vacuum setting. The RNA was then dissolved in 20 µl of nuclease-free water to increase the initial concentration.
A denaturing 15% polyacrylamide gel containing 7 M urea was used to separate the sRNAs. The gel was pre-run for 30 minutes at 18 mA (100 V) in 1X TBE buffer (89 mM Tris, 89 mM boric acid, 20 mM EDTA; pH 8.0). A total of 10 µg of RNA was prepared in a 10 µl sample for electrophoresis and mixed with 5 µl of 3X loading buffer (95% (v/v) formamide, 20 mM EDTA, Bromphenol blue and Cyanol xylene) such that paired wild type and mutant samples contained the same amount of RNA. The low-range single stranded RNA ladder (NEB; N0364S) was included for size reference. The gels were run for 80 minutes at 18 mA (100 V) in 1X TBE buffer. After electrophoresis, the lanes containing one set of samples with a corresponding ladder were cut from the gel and stained in ethidium bromide for 15 minutes before taking an image. The remaining gel was cut into pieces containing paired wild type and mutant sRNA samples and each pair transferred to a Hybond-N+ nylon membrane (Amersham) by electro-blotting in 1X SSC buffer (3 M NaCl, 30 mM Sodium Citrate) for 2 hours at 250 mA (4 V). The RNA was cross-linked to the membranes by exposing them to 120000 μJ cm−2 (UVC500 UV Crosslinker; Hoefer) and the membranes were then dried at 50 °C for 30 minutes.
The membranes were rolled with hybridization mesh and pre-hybridized for 3 hours in 10 ml pre-hybridization solution containing 10 μg ml−1 of salmon sperm DNA at 40 °C in a hybridization oven (Model 5420; VWR). After the pre-hybridization step, 50 μg ml−1 of biotin-labeled probe49 was added directly to the pre-hybridization solution and the membranes were hybridized with the probe for 16 hours at 40 °C. Probe sequences are given in Table 4. After hybridization, the membranes were washed twice in 2X SSC/0.1% SDS for 15 minutes at 40 °C, with a final wash in 0.1X SSC for 15 minutes at room temperature.50 The Chemiluminescent Nucleic Acid Detection Module (catalog # 89880; Thermo-Fisher) was used for probe detection following the manufacturer's recommendations. Images were captured using an ImageQuant LAS4000 (General Electric Canada). The resulting images were adjusted for brightness and contrast using Adobe Photoshop CC 2017. The images of the ethidium bromide-stained portions of the corresponding gels were used to construct standard curves to allow estimation of the sizes of bands detected on the blots.
Table 4.
Biotin-labeled probes for detection of selected sRNAs on Northern blots.
| sRNA | Oligo sequence (5′ to 3′) |
|---|---|
| sRNA00385 | BIO-GCGCAGTTGACGCGCCGTCT |
| sRNA01029 | BIO-GGAAACCGGGCGCGGGAACC |
| sRNA00848 | BIO-TCAAGCCTCTGAGGAAGGTC |
| sRNA01129 | BIO-GGGGCTGTTGACCGCCCGCC |
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
We thank J. Thomas Beatty for assistance with the Northern blotting.
Funding
MG was supported in part by a fellowship from the Memorial University of Newfoundland School of Graduate Studies. The research was supported by grants to LPC and ASL from the Natural Sciences and Engineering Research Council (NSERC) and from the Memorial University of Newfoundland Faculty of Science to MEM.
References
- 1.Castanie-Cornet MP, Penfound TA, Smith D, Elliott JF, Foster JW. Control of acid resistance in Escherichia coli. J Bacteriol 1999; 181:3525-35; PMID:10348866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Biase D, Tramonti A, Bossa F, Visca P. The response to stationary-phase stress conditions in Escherichia coli: Role and regulation of the glutamic acid decarboxylase system. Mol Microbiol 1999; 32:1198-211; PMID:10383761; https://doi.org/ 10.1046/j.1365-2958.1999.01430.x [DOI] [PubMed] [Google Scholar]
- 3.Serganov A, Nudler E. A decade of riboswitches. Cell 2013; 152:17-24; PMID:23332744; https://doi.org/ 10.1016/j.cell.2012.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loh E, Dussurget O, Gripenland J, Vaitkevicius K, Tiensuu T, Mandin P, Repoila F, Buchrieser C, Cossart P, Johansson J. A trans-acting riboswitch controls expression of the virulence regulator PrfA in Listeria monocytogenes. Cell 2009; 139:770-9; PMID:19914169; https://doi.org/ 10.1016/j.cell.2009.08.046 [DOI] [PubMed] [Google Scholar]
- 5.Hantke K. Iron and metal regulation in bacteria. Curr Opin Microbiol 2001; 4:172-7; PMID:11282473 [DOI] [PubMed] [Google Scholar]
- 6.Romeo T. Global regulation by the small RNA-binding protein CsrA and the non-coding RNA molecule CsrB. Mol Microbiol 1998; 29:1321-30; PMID:9781871; https://doi.org/ 10.1046/j.1365-2958.1998.01021.x [DOI] [PubMed] [Google Scholar]
- 7.Storz G, Vogel J, Wassarman KM. Regulation by small RNAs in bacteria: Expanding frontiers. Mol Cell 2011; 43:880-91; PMID:21925377; https://doi.org/ 10.1016/j.molcel.2011.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michaux C, Verneuil N, Hartke A, Giard JC. Physiological roles of small RNA molecules. Microbiology 2014; 160:1007-19; PMID:24694375; https://doi.org/ 10.1099/mic.0.076208-0 [DOI] [PubMed] [Google Scholar]
- 9.Pena-Castillo L, Gruell M, Mulligan ME, Lang AS. Detection of bacterial small transcripts from RNA-seq data: A comparative assessment. Pac Symp Biocomput 2016; 21:456-67; PMID:26776209 [PubMed] [Google Scholar]
- 10.Imhoff JF, Genus I. Rhodobacter In: Brenner DJ, Krieg NR, Staley JT, Garrity GM, eds. Bergey's Manual of Systematic Bacteriology. 2nd ed. New York: Springer, 2005:161; https://doi.org/ 10.1002/9781118960608.gbm00862 [DOI] [Google Scholar]
- 11.Lang AS, Zhaxybayeva O, Beatty JT. Gene transfer agents: Phage-like elements of genetic exchange. Nat Rev Microbiol 2012; 10:472-82; PMID:22683880; https://doi.org/ 10.1038/nrmicro2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, Eddy SR, Gardner PP, Bateman A. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res 2013; 41:D226-32; PMID:23125362; https://doi.org/ 10.1093/nar/gks1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.RNAcentral Consortium RNAcentral: An international database of ncRNA sequences. Nucleic Acids Res 2015; 43:D123-9; PMID:25352543; https://doi.org/ 10.1093/nar/gku991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li L, Huang D, Cheung MK, Nong W, Huang Q, Kwan HS. BSRD: A repository for bacterial small regulatory RNA. Nucleic Acids Res 2013; 41:D233-8; PMID:23203879; https://doi.org/ 10.1093/nar/gks1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mercer RG, Callister SJ, Lipton MS, Pasa-Tolic L, Strnad H, Paces V, Beatty JT, Lang AS. Loss of the response regulator CtrA causes pleiotropic effects on gene expression but does not affect growth phase regulation in Rhodobacter capsulatus. J Bacteriol 2010; 192:2701-10; PMID:20363938; https://doi.org/ 10.1128/JB.00160-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pena-Castillo L, Mercer RG, Gurinovich A, Callister SJ, Wright AT, Westbye AB, Beatty JT, Lang AS. Gene co-expression network analysis in Rhodobacter capsulatus and application to comparative expression analysis of Rhodobacter sphaeroides. BMC Genomics 2014; 15:730, 2164-15-730; PMID:25164283; https://doi.org/ 10.1186/1471-2164-15-730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lang AS, Beatty JT. A bacterial signal transduction system controls genetic exchange and motility. J Bacteriol 2002; 184:913-8; PMID:11807050; https://doi.org/ 10.1128/jb.184.4.913-918.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weber L, Thoelken C, Volk M, Remes B, Lechner M, Klug G. The conserved Dcw gene cluster of R. sphaeroides is preceded by an uncommonly extended 5′ leader featuring the sRNA UpsM. PLoS One 2016; 11:e0165694; PMID:27802301; https://doi.org/ 10.1371/journal.pone.0165694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berghoff BA, Glaeser J, Sharma CM, Vogel J, Klug G. Photooxidative stress-induced and abundant small RNAs in Rhodobacter sphaeroides. Mol Microbiol 2009; 74:1497-512; PMID:19906181; https://doi.org/ 10.1111/j.1365-2958.2009.06949.x; 10.1111/j.1365-2958.2009.06949.x [DOI] [PubMed] [Google Scholar]
- 20.Shiryev SA, Papadopoulos JS, Schaffer AA, Agarwala R. Improved BLAST searches using longer words for protein seeding. Bioinformatics 2007; 23:2949-51; PMID:17921491; https://doi.org/ 10.1093/bioinformatics/btm479 [DOI] [PubMed] [Google Scholar]
- 21.Gomez-Lozano M, Marvig RL, Molina-Santiago C, Tribelli PM, Ramos JL, Molin S. Diversity of small RNAs expressed in Pseudomonas species. Environ Microbiol Rep 2015; 7:227-36; PMID:25394275; https://doi.org/ 10.1111/1758-2229.12233 [DOI] [PubMed] [Google Scholar]
- 22.Hamada M, Kiryu H, Sato K, Mituyama T, Asai K. Prediction of RNA secondary structure using generalized centroid estimators. Bioinformatics 2009; 25:465-73; PMID:19095700; https://doi.org/ 10.1093/bioinformatics/btn601 [DOI] [PubMed] [Google Scholar]
- 23.Solovjev V, Salamov A. Automatic annotation of microbial genomes and metagenomic sequences In: Li RW, ed. Metagenomics and its Applications in Agriculture, Biomedicine and Environmental Studies. Nova Science Publishers, 2011:61. [Google Scholar]
- 24.Kingsford CL, Ayanbule K, Salzberg SL. Rapid, accurate, computational discovery of rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol 2007; 8:R22; PMID:17313685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.James G, Witten D, Hastie T, Tibshirani R. An Introduction to Statistical Learning: With Applications in R. New York: Springer; 2014; https://doi.org/ 10.1007/978-1-4614-7138-7 [DOI] [Google Scholar]
- 26.Sharma CM, Vogel J. Differential RNA-seq: The approach behind and the biological insight gained. Curr Opin Microbiol 2014; 19:97-105; PMID:25024085; https://doi.org/ 10.1016/j.mib.2014.06.010 [DOI] [PubMed] [Google Scholar]
- 27.Lee YS, Shibata Y, Malhotra A, Dutta A. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev 2009; 23:2639-49; PMID:19933153; https://doi.org/ 10.1101/gad.1837609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raina M, Ibba M. tRNAs as regulators of biological processes. Front Genet 2014; 5:171; PMID:24966867; https://doi.org/ 10.3389/fgene.2014.00171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wright PR, Richter AS, Papenfort K, Mann M, Vogel J, Hess WR, Backofen R, Georg J. Comparative genomics boosts target prediction for bacterial small RNAs. Proc Natl Acad Sci U S A 2013; 110:E3487-96; PMID:23980183; https://doi.org/ 10.1073/pnas.1303248110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pain A, Ott A, Amine H, Rochat T, Bouloc P, Gautheret D. An assessment of bacterial small RNA target prediction programs. RNA Biol 2015; 12:509-13; PMID:25760244; https://doi.org/ 10.1080/15476286.2015.1020269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al.. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015; 43:D447-52; PMID:25352553; https://doi.org/ 10.1093/nar/gku1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caldelari I, Chao Y, Romby P, Vogel J. RNA-mediated regulation in pathogenic bacteria. Cold Spring Harb Perspect Med 2013; 3:a010298; PMID:24003243; https://doi.org/ 10.1101/cshperspect.a010298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mercer RG, Quinlan M, Rose AR, Noll S, Beatty JT, Lang AS. Regulatory systems controlling motility and gene transfer agent production and release in Rhodobacter capsulatus. FEMS Microbiol Lett 2012; 331:53-62; PMID:22443140; https://doi.org/ 10.1111/j.1574-6968.2012.02553.x [DOI] [PubMed] [Google Scholar]
- 34.Landt SG, Abeliuk E, McGrath PT, Lesley JA, McAdams HH, Shapiro L. Small non-coding RNAs in Caulobacter crescentus. Mol Microbiol 2008; 68:600-14; PMID:18373523; https://doi.org/ 10.1111/j.1365-2958.2008.06172.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brimacombe CA, Ding H, Beatty JT. Rhodobacter capsulatus DprA is essential for RecA-mediated gene transfer agent (RcGTA) recipient capability regulated by quorum-sensing and the CtrA response regulator. Mol Microbiol 2014; 92:1260-78; PMID:24784901; https://doi.org/ 10.1111/mmi.12628 [DOI] [PubMed] [Google Scholar]
- 36.Thomason MK, Bischler T, Eisenbart SK, Forstner KU, Zhang A, Herbig A, Nieselt K, Sharma CM, Storz G. Global transcriptional start site mapping using differential RNA sequencing reveals novel antisense RNAs in Escherichia coli. J Bacteriol 2015; 197:18-28; PMID:25266388; https://doi.org/ 10.1128/JB.02096-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leung MM, Brimacombe CA, Beatty JT. Transcriptional regulation of the Rhodobacter capsulatus response regulator CtrA. Microbiology 2013; 159:96-106; PMID:23154973; https://doi.org/ 10.1099/mic.0.062349-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang W, Wei Z, Lam TW, Wang J. Next generation sequencing has lower sequence coverage and poorer SNP-detection capability in the regulatory regions. Sci Rep 2011; 1:55; PMID:22355574; https://doi.org/ 10.1038/srep00055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guzina J, Djordjevic M. Promoter recognition by extracytoplasmic function sigma factors: Analyzing DNA and protein interaction motifs. J Bacteriol 2016; 198:1927-38; PMID:27137497; https://doi.org/ 10.1128/JB.00244-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hook-Barnard IG, Hinton DM. Transcription initiation by mix and match elements: Flexibility for polymerase binding to bacterial promoters. Gene Regul Syst Bio 2007; 1:275-93; PMID:19119427 [PMC free article] [PubMed] [Google Scholar]
- 41.Weaver PF, Wall JD, Gest H. Characterization of Rhodopseudomonas capsulata. Arch Microbiol 1975; 105:207-16; PMID:1103769; https://doi.org/ 10.1007/BF00447139 [DOI] [PubMed] [Google Scholar]
- 42.Jahn CE, Charkowski AO, Willis DK. Evaluation of isolation methods and RNA integrity for bacterial RNA quantitation. J Microbiol Methods 2008; 75:318-24; PMID:18674572; https://doi.org/ 10.1016/j.mimet.2008.07.004 [DOI] [PubMed] [Google Scholar]
- 43.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup . The sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25:2078-9; PMID:19505943; https://doi.org/ 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anders S, Pyl PT, Huber W. HTSeq–a python framework to work with high-throughput sequencing data. Bioinformatics 2015; 31:166-9; PMID:25260700; https://doi.org/ 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quinlan AR, Hall IM. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010; 26:841-2; PMID:20110278; https://doi.org/ 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wheeler TJ, Eddy SR. Nhmmer: DNA homology search with profile HMMs. Bioinformatics 2013; 29:2487-9; PMID:23842809; https://doi.org/ 10.1093/bioinformatics/btt403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robinson MD, McCarthy DJ, Smyth GK. edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010; 26:139-40; PMID:19910308; https://doi.org/ 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sing T, Sander O, Beerenwinkel N, Lengauer T. ROCR: Visualizing classifier performance in R. Bioinformatics 2005; 21:3940-1; PMID:16096348; https://doi.org/ 10.1093/bioinformatics/bti623 [DOI] [PubMed] [Google Scholar]
- 49.Huang Q, Mao Z, Li S, Hu J, Zhu Y. A non-radioactive method for small RNA detection by northern blotting. Rice (N Y) 2014; 7:26, 014-0026-1. Epub 2014 Oct 1; PMID:26224555; https://doi.org/ 10.1186/s12284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rio DC. Northern blots for small RNAs and microRNAs. Cold Spring Harb Protoc 2014; 2014:793-7; PMID:24987143; https://doi.org/ 10.1101/pdb.prot080838 [DOI] [PubMed] [Google Scholar]
- 51.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: A sequence logo generator. Genome Res 2004; 14:1188-90; PMID:15173120; https://doi.org/ 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Albrecht M, Sharma CM, Reinhardt R, Vogel J, Rudel T. Deep sequencing-based discovery of the Chlamydia trachomatis transcriptome. Nucleic Acids Res 2010; 38:868-77; PMID:19923228; https://doi.org/ 10.1093/nar/gkp1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Soutourina OA, Monot M, Boudry P, Saujet L, Pichon C, Sismeiro O, Semenova E, Severinov K, Le Bouguenec C, Coppee JY, et al.. Genome-wide identification of regulatory RNAs in the human pathogen Clostridium difficile. PLoS Genet 2013; 9:e1003493; PMID:23675309; https://doi.org/ 10.1371/journal.pgen.1003493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Acebo P, Martin-Galiano AJ, Navarro S, Zaballos A, Amblar M. Identification of 88 regulatory small RNAs in the TIGR4 strain of the human pathogen Streptococcus pneumoniae. RNA 2012; 18:530-46; PMID:22274957; https://doi.org/ 10.1261/rna.027359.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rasmussen S, Nielsen HB, Jarmer H. The transcriptionally active regions in the genome of Bacillus subtilis. Mol Microbiol 2009; 73:1043-57; PMID:19682248; https://doi.org/ 10.1111/j.1365-2958.2009.06830.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mentz A, Neshat A, Pfeifer-Sancar K, Puhler A, Ruckert C, Kalinowski J. Comprehensive discovery and characterization of small RNAs in Corynebacterium glutamicum ATCC 13032. BMC Genomics 2013; 14:714; 2164-14-714; PMID:24138339; https://doi.org/ 10.1186/1471-2164-14-714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miotto P, Forti F, Ambrosi A, Pellin D, Veiga DF, Balazsi G, Gennaro ML, Di Serio C, Ghisotti D, Cirillo DM. Genome-wide discovery of small RNAs in Mycobacterium tuberculosis. PLoS One 2012; 7:e51950; PMID:23284830; https://doi.org/ 10.1371/journal.pone.0051950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin YF, A DR, Guan S, Mamanova L, McDowall KJ. A combination of improved differential and global RNA-seq reveals pervasive transcription initiation and events in all stages of the life-cycle of functional RNAs in Propionibacterium acnes, a major contributor to wide-spread human disease. BMC Genomics 2013; 14:620; 2164-14-620; PMID:24034785; https://doi.org/ 10.1186/1471-2164-14-620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moody MJ, Young RA, Jones SE, Elliot MA. Comparative analysis of non-coding RNAs in the antibiotic-producing Streptomyces bacteria. BMC Genomics 2013; 14:558; 2164-14-558; PMID:23947565; https://doi.org/ 10.1186/1471-2164-14-558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dugar G, Herbig A, Forstner KU, Heidrich N, Reinhardt R, Nieselt K, Sharma CM. High-resolution transcriptome maps reveal strain-specific regulatory features of multiple Campylobacter jejuni isolates. PLoS Genet 2013; 9:e1003495; PMID:23696746; https://doi.org/ 10.1371/journal.pgen.1003495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, Sittka A, Chabas S, Reiche K, Hackermuller J, Reinhardt R, et al.. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 2010; 464:250-5; PMID:20164839; https://doi.org/ 10.1038/nature08756 [DOI] [PubMed] [Google Scholar]
- 62.McClure R, Tjaden B, Genco C. Identification of sRNAs expressed by the human pathogen Neisseria gonorrhoeae under disparate growth conditions. Front Microbiol 2014; 5:456; PMID:25221548; https://doi.org/ 10.3389/fmicb.2014.00456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wilms I, Overloper A, Nowrousian M, Sharma CM, Narberhaus F. Deep sequencing uncovers numerous small RNAs on all four replicons of the plant pathogen Agrobacterium tumefaciens. RNA Biol 2012; 9:446-57; PMID:22336765; https://doi.org/ 10.4161/rna.17212; 10.4161/rna.17212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berghoff BA, Glaeser J, Sharma CM, Zobawa M, Lottspeich F, Vogel J, Klug G. Contribution of Hfq to photooxidative stress resistance and global regulation in Rhodobacter sphaeroides. Mol Microbiol 2011; 80:1479-95; PMID:21535243; https://doi.org/ 10.1111/j.1365-2958.2011.07658.x [DOI] [PubMed] [Google Scholar]
- 65.Schluter JP, Reinkensmeier J, Daschkey S, Evguenieva-Hackenberg E, Janssen S, Janicke S, Becker JD, Giegerich R, Becker A. A genome-wide survey of sRNAs in the symbiotic nitrogen-fixing alpha-proteobacterium Sinorhizobium meliloti. BMC Genomics 2010; 11:245, 2164-11-245; PMID:20398411; https://doi.org/ 10.1186/1471-2164-11-245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bradley ES, Bodi K, Ismail AM, Camilli A. A genome-wide approach to discovery of small RNAs involved in regulation of virulence in Vibrio cholerae. PLoS Pathog 2011; 7:e1002126; PMID:21779167; https://doi.org/ 10.1371/journal.ppat.1002126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wurtzel O, Yoder-Himes DR, Han K, Dandekar AA, Edelheit S, Greenberg EP, Sorek R, Lory S. The single-nucleotide resolution transcriptome of Pseudomonas aeruginosa grown in body temperature. PLoS Pathog 2012; 8:e1002945; PMID:23028334; https://doi.org/ 10.1371/journal.ppat.1002945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shinhara A, Matsui M, Hiraoka K, Nomura W, Hirano R, Nakahigashi K, Tomita M, Mori H, Kanai A. Deep sequencing reveals as-yet-undiscovered small RNAs in Escherichia coli. BMC Genomics 2011; 12:428, 2164-12-428; PMID:21864382; https://doi.org/ 10.1186/1471-2164-12-428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zeng Q, Sundin GW. Genome-wide identification of hfq-regulated small RNAs in the fire blight pathogen Erwinia amylovora discovered small RNAs with virulence regulatory function. BMC Genomics 2014; 15:414, 2164-15-414; PMID:24885615; https://doi.org/ 10.1186/1471-2164-15-414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beauregard A, Smith EA, Petrone BL, Singh N, Karch C, McDonough KA, Wade JT. Identification and characterization of small RNAs in Yersinia pestis. RNA Biol 2013; 10:397-405; PMID:23324607; https://doi.org/ 10.4161/rna.23590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kroger C, Dillon SC, Cameron AD, Papenfort K, Sivasankaran SK, Hokamp K, Chao Y, Sittka A, Hebrard M, Handler K, et al.. The transcriptional landscape and small RNAs of Salmonella enterica serovar typhimurium. Proc Natl Acad Sci U S A 2012; 109:E1277-86; PMID:22538806; https://doi.org/ 10.1073/pnas.1201061109 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.