

ABSTRACT
Ischemic events, common in many diseases, result from decreased blood flow and impaired delivery of oxygen and glucose to tissues of the body. While much is known about the cellular transcriptional response to ischemia, much less is known about the posttranscriptional response to oxygen and glucose deprivation. The goal of this project was to investigate one such posttranscriptional response, the regulation of mRNA stability. To that end, we have identified several novel ischemia-related mRNAs that are synergistically stabilized by oxygen and glucose deprivation including VEGF, MYC, MDM2, and CYR61. This increase in mRNA half-life requires the synergistic effects of both low oxygen (1%) as well as low glucose (≤ 1 g/L) conditions. Oxygen or glucose deprivation alone fails to initiate the response, as exposure to either high glucose (4 g/L) or normoxic conditions inhibits the response. Furthermore, in response to hypoxia/hypoglycemia, the identified mRNAs are released from the RNA binding protein KHSRP which likely contributes to their stabilization.
KEYWORDS: Hypoxia, hypoglycemia, mRNA stability, KHSRP
Introduction
Ischemia is a frequent complicating factor in many cardiovascular diseases (CVD) such as heart attack and stroke. While ischemia results in the deprivation of many nutrients, lack of oxygen and glucose arguably have the greatest impact on the metabolism of the cell. Indeed, glucose deprivation can result in a myriad of cellular effects including cell cycle arrest, apoptosis, and autophagy.1 In response to glucose deprivation, pathways such as mTOR/AKT and AMPK along with transcription factors such as MYC are activated to mitigate the detrimental effects.2-4 The cellular response to hypoxia has also been well studied and much is known about the transcriptional response that is initiated upon exposure to low oxygen conditions. The main mediators of this response are the Hypoxia Inducible Factor (HIF) family of transcription factors, with the oxygen-dependent HIF-specific prolyl hydroxylases (PHDs) and the mitochondria playing a critical role in the regulation of the HIF-α subunits.5-11
Interestingly, glucose levels have been shown to impact the hypoxic stabilization and activation of the HIF pathway in multiple mammalian cell types.12-15 Indeed, while depletion of glucose can prevent hypoxic HIF-1α accumulation in some cell types,13 increased levels of glucose can also lead to attenuation of the hypoxic HIF transcriptional response through effects on both HIF-α stabilization and activation.12,14,15 Furthermore, these cellular effects can have widespread implications for the organism. For example, hyperglycemia is a common result of poorly controlled diabetes and can result in decreased HIF-α stabilization, contributing to many diabetic complications such as decreased vascular density and impaired wound healing.16-19 In addition, increased blood glucose has also been shown to impair the ability to recover from an acute hypoxic challenge such as a myocardial infarction,20,21 and is a recognized risk factor for CVD.22,23 Thus it is important to understand the interplay between glucose levels and the hypoxic response as it relates to all aspects of gene expression.24
Although the transcriptional response to hypoxia is well established, the posttranscriptional response to oxygen and glucose deprivation is less understood.25 Increases in the stability of individual mRNAs, such as VEGF and GLUT1, have been associated with hypoxic exposure.26 Indeed, the hypoxic posttranscriptional regulation of VEGF has been studied in multiple cell types.27-32 VEGF posttranscriptional regulation is reported to involve AU Rich Elements (AREs), and multiple RNA binding proteins (RBPs) have been implicated in its regulation including HuR, PTB, hnRNPL, and others.27,31,33-36 Specific sequences have also been identified in the VEGF mRNA 3′ UTR and ORF that contribute to this hypoxic regulation,27,31-34 but this analysis has not been extended to other mRNAs. Interestingly, glucose deprivation alone, or in conjunction with severe hypoxia/anoxia (≤ 0.1% O2), has been shown to lead to stabilization of the VEGF mRNA,37-39 but the effects of hypoglycemia with moderate hypoxia have not been investigated.
As hypoxia/hypoglycemia can have many different effects on gene expression, it is important to understand all aspects of the cellular response to develop the most effective therapeutics. Therefore, we set out to investigate the understudied posttranscriptional response to oxygen and glucose deprivation. Our overall finding is that oxygen and glucose deprivation synergistically increase the mRNA stability of genes that are important for the ischemic response.
Results
The discovery that glucose levels modulated the hypoxic response came from attempts to replicate previous studies which reported that VEGF mRNA half-life significantly increased in response to exposure to 1% oxygen.29,31,33,40 However, intermittent success during preliminary investigations revealed that VEGF mRNA was only stabilized when cells were plated at higher densities. Reasoning that another nutrient must be impacting the hypoxic response, several culture media components were analyzed for their impact on VEGF mRNA stabilization. While changes in pyruvate, glutamine, and serum levels had no effect on the stabilization of VEGF mRNA (data not shown), depriving rat C6 glioma cells of glucose and oxygen simultaneously resulted in robust stabilization of VEGF as well as MYC and MDM2 (Fig. 1A). It should be noted that throughout this paper, half-life measurements are limited to the p165A isoform of VEGFA (VEGF165) which is one of the most abundant pro-angiogenic isoforms.35
Figure 1.

Oxygen and glucose deprivation synergistically stabilize mRNA. C6 (A) or Hep3B (B) cells exposed to 24 hours of Normoxia or Hypoxia (1% O2) in the presence of 4, 1, or 0 g/L glucose before being treated with Actinomycin D for 0, 1 or 2 hours. mRNA levels of VEFG165, MYC and MDM2 were determined via qPCR, normalized to GAPDH and 0 hour time point and half-lives determined. (C, D) Relative mRNA expression (fold change) in response to hypoxic conditions (Hypox) expressed as fold change from normoxic values (Norm). Data represents average of N≥ 4 ± SD for each condition. *P ≤ 0.05 from respective Normoxic condition.
For all of the experiments, cells were treated as described in the material and methods. Briefly, cells maintained in high glucose (4.0 g/L) media were plated at 0.5 × 106 cell per well of a 6 well plate and allowed to recover for 18–24 hours. The next day, media was removed and replaced with media of defined glucose concentration and cells were placed in the hypoxia hood set for 24 hours. Actinomycin D (ActD) treatment was then performed over a 2 hour time course. ActD time course was limited to 2 hours because in preliminary experiments, mRNAs with half-lives of approximately one hour or less only displayed exponential decay in response to ActD for approximately 2 to 4 hours (Fig. S8A) before beginning to level off. Because of this, mRNA half-lives calculated from a full 8 hour time course were grossly over-estimated when compared with half-lives calculated from only the first 2 hours of the time course which better predicted the actual time taken to decay 50% of the mRNA (Fig. S8B). Furthermore, all calculated half-lives greater than 6 hours or those calculated to have a negative half-life (infinitely stable) were converted to 6 hours which was the maximum half-life reliably calculated by this method (data not shown).
Concomitant oxygen and glucose deprivation triggers increased mRNA stability
Initial experiments revealed that hypoxic mRNA stabilization exhibited an inverse dose dependence on glucose in a variety of cell types. As shown in Fig. 1A, under normoxic conditions, altering the glucose levels from 0 to 4 g/L had no effect on the steady-state half-lives of VEGF, MYC, or MDM2 mRNA in rat C6 glioma cells. Similarly, when C6 cells were incubated under hypoxic conditions (1% O2) in the presence of high glucose (4 g/L) media for 24 hours there was no significant change in the mRNA half-life of VEGF, MYC or MDM2. However, when the glucose concentration was decreased to either 1 g/L (low glucose) or 0 g/L (no glucose), there was a significant increase in VEGF, MYC, and MDM2 mRNA half-lives in the cells cultured under hypoxia for 24 hours (Fig. 1A; corresponding decay curves can be found in Fig. S1A). These increases in mRNA stability were mirrored by changes in overall mRNA abundance (Fig. 1C) with hypoxic induction of mRNA inversely correlated with glucose concentration (R2≈ −0.8) for all 3 mRNAs.
This glucose dependence of hypoxic mRNA stabilization was not limited to rat C6 glioma cells as it was observed in several other cell types from both human and mouse. However, the sensitivity of the response did vary somewhat between the cell types. For example, the human hepatocellular carcinoma cell line, Hep3B, required greater glucose deprivation to see the mRNA stabilization. At both 4 and 1 g/L glucose there was no significant stabilization of VEGF, MYC, or MDM2 mRNA under hypoxia, however when there was a complete lack of glucose (0 g/L), significant hypoxic stabilization of all 3 messages was seen (Fig. 1B; corresponding decay curves can be found in Fig. S1B). Interestingly, similar to the C6 cells, the hypoxic increase of VEGF mRNA abundance was also inversely correlated with the glucose levels (R2 = −0.98), while MYC (R2 = −0.75) and MDM2 (R2 = −0.04) mRNA levels were less correlated and actually decreased in abundance under hypoxia (Fig. 1D).
The human embryonic kidney cell line, HEK293T, showed a similar glucose response to the C6 rat glioma cells. Indeed, while exposure of HEK293T cells to 24 hours of hypoxia in the presence of 4 g/L glucose had no significant effect on VEGF or MYC mRNA half-life (Fig. 2A), depletion of glucose to 1 g/L or less resulted in significant stabilization of both messages. Interestingly, MDM2 had a much longer basal half-life (4–6 hrs) in HEK293T cells, and as such, in the experimental conditions tested, no increase in half-life in response to hypoxic exposure was able to be measured (Fig. 2A; corresponding decay curves in Fig. S2A). Contrary to what was seen with the C6 cells and the half-life results, there was a positive correlation between overall mRNA abundance and glucose levels for VEGF (R2 = 0.96) and MYC (R2 = 0.84) and the extent of the hypoxic increase was much lower (Fig. 2C).
Figure 2.

Oxygen and glucose deprivation synergistically stabilize mRNA in HEK293T cells. HEK293T cells exposed to 24 hours of Normoxia and either 1% Oxygen (A) or 3% Oxygen (B) in the presence of 4, 1, or 0 g/L glucose before being treated with Actinomycin D for 0, 1 or 2 hours. mRNA levels of VEFG165, MYC, MDM2, and CYR61 were determined via qPCR, normalized to GAPDH and 0 hour time point and half-lives determined. (C, D) Relative mRNA expression (fold change) in response to hypoxic conditions (Hypox) expressed as fold change from normoxic values (Norm). Data represents average of N = 4 ± SD for each condition. *P ≤ 0.05 from respective Normoxic condition.
mRNA stability is dependent on Oxygen level
Having determined that the response was dependent on the extent of glucose deprivation, the dependency on the severity of the oxygen deprivation was also tested. As with 1% oxygen, high levels of glucose (4 g/L) coupled with exposure to 3% oxygen for 24 hours, did not result in any significant change in the half-life of either VEGF, MYC or CYR61, another mRNA target revealed by preliminary global analysis (described in Fig. 5). However, 3% oxygen with 1 g/L glucose induced a somewhat variable increase in MYC and VEGF mRNA half-life while not affecting CYR61, while complete glucose deprivation (0 g/L) resulted in a more consistent and significant increase in the half-life of VEGF and MYC mRNA (Fig. 2B; corresponding decay curves in Fig. S2B) while CYR61 showed a trend of increased mRNA half-life. Furthermore, in all cases the extent of the hypoxic stabilization was less at 3% oxygen than it was at 1% oxygen. For example, at 1 g/L glucose MYC mRNA was stabilized only 2.4-fold under 3% oxygen while incubation at 1% oxygen resulted in a 7.6-fold increase in mRNA half-life. Similar results were seen with VEGF and CYR61 suggesting that the extent of ischemic mRNA stabilization is dependent on the severity of both the oxygen and glucose deprivation. Again, mRNA abundance of VEGF, MYC, and CYR61 did not correlate with glucose levels or mRNA half-life, although CYR61 did show greater hypoxic induction in 0 and 1 g/L glucose when compared with 4 g/L (Fig. 2D).
Figure 5.
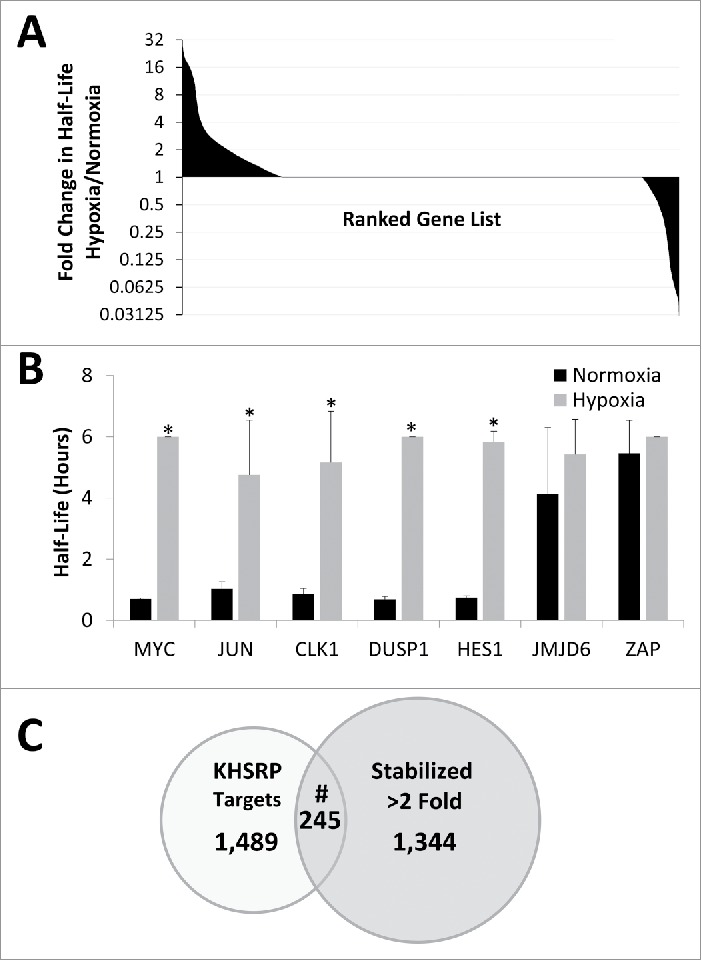
Global changes in mRNA stabilization in response to hypoxia/hypoglycemia. (A) Ranked gene list of half-lives of all mRNAs expressed in all conditions from deep sequencing of HEK293T cells exposed to 24 hours of normoxia or hypoxia (1% O2) in the presence of 1 g/L glucose for 24 hours before being treated with Actinomycin D for 0, 1 or 2 hours. Note log 2 scale of y axis. Data represent an N = 1. (B) Real time verification of mRNA half-lives determined from deep sequencing experiment. Data represents average of N = 4 ± SD for each condition. *P ≤ 0.05 from respective Normoxic condition. (C) Overlap of all mRNAs identified as stabilized greater than 2-fold in our study and KHSRP targets identified in Winzen et al. #Representation factor: 1.5, p < 1.064e−12 via normal approximation of exact hypergeometric probability.
mRNA stability affected by glucose level but not by osmolarity
Because there was such a dramatic difference in the mRNA stability response between 1 and 4 g/L in the previous experiments, glucose doses ranging from 1 to 4 g/L glucose were also tested. As shown in Fig. 3A, VEGF, MYC, and CYR61 mRNAs all showed a dose dependent increase in hypoxic mRNA half-life starting around 2 g/L, with significant increases over normoxic half-life evident at 1 g/L glucose for all 3 mRNAs (corresponding decay curves can be found in Fig. S3A). Although there was a significant increase in VEGF mRNA level with hypoxic exposure, that increase did not correlate with mRNA half-life or glucose level (Fig. 3C). CYR61 however, showed a significant increase in mRNA levels under hypoxic conditions only in the 1 g/L glucose condition and there was a strong correlation between mRNA abundance and half-life (R2 = 0.98) and a negative correlation with glucose dose (R2 = −0.81).
Figure 3.

mRNA stabilization is dependent on glucose concentration but not osmolarity. (A) HEK293T cells exposed to 24 hours of Normoxia or Hypoxia (1% O2) in the presence of 4, 3, 2, or 1 g/L glucose before being treated with Actinomycin D for 0, 1 or 2 hours. (B) HEK293T cells exposed to 24 hours of Normoxia or Hypoxia (1% O2) in the presence of glucose free DMEM alone (0) or supplemented with 4.5 g/L glucose or mannitol before being treated with Actinomycin D for 0, 1 or 2 hours. (A, B) mRNA levels of VEFG165, MYC, and CYR61 were determined via qPCR, normalized to GAPDH and 0 hour time point and half-lives determined. (C, D) Relative mRNA expression (fold change) in response to hypoxic conditions (Hypox) expressed as fold change from normoxic values (Norm). Data represents average of N = 5 (A, C) or 4 (B, D) ± SD for each condition. *P ≤ 0.05 from respective Normoxic condition.
To verify that the observed effects were due to a metabolic response and not simply due to a change in osmolarity between the various glucose doses, cells were also exposed to identical doses of mannitol. As shown in Fig. 3B, HEK293T cultures in glucose free media (0 g/L) exhibit robust mRNA stabilization of all 3 targets tested in response to hypoxia. Addition of 4.5 g/L glucose to the media during the hypoxic exposure completely blocked the hypoxic mRNA stabilization response. However, addition of 4.5 g/L mannitol had minimal effect on the stabilization with all 3 targets showing an increase in hypoxic mRNA half-life similar to glucose free media suggesting the inhibition of the mRNA stabilization response is not due to the increase in osmolarity (corresponding decay curves can be found in Fig. S3B). Interestingly, CYR61 was once again the only mRNA to show mRNA abundance changes that positively correlated (R2 = 0.86) with the mRNA stability results (Fig. 3D).
Involvement of RNA binding proteins
Previous studies have identified HuR as well as hnRNPL as regulators of VEGF's mRNA stability under hypoxic conditions.27,31,33,36,41,42 To test the involvement of these RNA binding proteins in the ischemic mRNA stabilization, siRNA-mediated knockdown was used. Preliminary experiments suggested that that greatest mRNA and protein knockdown occurred after 72 hours of siRNA treatment (data not shown). Therefore, HEK293T cells were transfected with siRNAs targeting either HuR or hnRNPL or with a negative control siRNA for 48 hours before exposing the cells to 24 hours of hypoxia/hypoglycemia (1% O2/1 g/L glucose) followed by ActD decay analysis. Despite robust knockdown of both HuR and hnRNPL protein levels (Fig. 4C), VEGF and MYC mRNA continued to be significantly stabilized under ischemic conditions (Fig. 4A, B; corresponding decay curves and mRNA expression in Fig. S4). Similar results were seen with siRNA-mediated knockdown of hNRNPA1/B2, and PTBP and with CRISPR-mediated knockout of HuR (data not shown). Furthermore, ribonucleoprotein immunoprecipitations followed by real time PCR detection of the targets (RIP-RT) failed to detect any changes in the ability of either HuR or hnRNPL to bind VEGF, MYC, or other known target mRNAs (data not shown), suggesting that these RNA binding proteins are not directly involved in the changes to mRNA stability seen in response to ischemia.
Figure 4.

Silencing of either HuR or hnRNP L has no effect on ischemic stabilization of mRNA. HEK293T cells were transfected with siRNA targeting the RNA binding proteins HuR (A), hnRNP L (B), or a negative control siRNA (Neg) for 48 hours before being exposed to 24 hours of Normoxia or Hypoxia (1% O2) in the presence of 1 g/L glucose. Cells were treated with Actinomycin D for 0, 1 or 2 hours, mRNA levels of VEFG165 and MYC were determined via qPCR, normalized to GAPDH and 0 hour time point and half-lives determined. Data represents average of N = 4 (HuR) or N = 3 (hnRNP L) ± SD for each condition. *P ≤ 0.05 from respective Normoxic condition. (C) Representative western blots showing extent of knockdown (% protein remaining) for either HuR or hnRNP L normalized to tubulin loading control.
Multiple mRNAs are stabilized in response to hypoxia/hypoglycemia
To identify additional mRNAs that are stabilized in response to hypoxia, a deep sequencing experiment with HEK293T cells treated with ActD for 0, 1, or 2 hours was conducted. RNA from 4 biologic replicates was combined into one cDNA library and one technical replicate was run. Raw and processed deep sequencing data for all genes can be found in Supplemental File 2. Despite its limited statistical power, this experiment did provide valuable insight into this phenomena. For example, out of the 17,275 genes identified as expressed in all 6 conditions (Normoxic, Hypoxic, 0, 1, 2 hrs ActD), 1,589 mRNAs (9.2%) showed a 2-fold or greater increase in mRNA half-life in response to 24 hours of hypoxia/hypoglycemia (1% O2/1 g/L glucose; Fig. 5A). In addition to CYR61 (see Fig. 3), qPCR was used to verify several other novel mRNAs identified by this analysis that were also stabilized in response to oxygen and glucose deprivation. As shown in Fig. 5B, 4 out of 6 mRNAs tested showed significant stabilization in response to hypoxia/hypoglycemia similar to MYC, while 2 that had fairly long half-lives under the normoxic condition did not show any significant increase under hypoxia/hypoglycemia (corresponding decay curves and mRNA expression can be found in Fig. S5B). Furthermore, while the majority of the mRNAs did not show a detectable change in stability, a subset of mRNAs (716, 4.1%) were also identified as less stable in response to oxygen and glucose deprivation (Fig. 5A).
The list of mRNAs found to be stabilized by hypoxia/hypoglycemia was also used to identify potential regulators of this response. First, all mRNAs that increased their mRNA half-life 2-fold or greater in response to oxygen and glucose deprivation were identified and compiled into a list. Then lists of published RNA binding protein targets identified through global methods were compiled and the overlap between the RBP targets and the list of stabilized mRNAs was analyzed. As shown in Fig. 5C, this approach identified the RNA binding protein KHSRP as one potential regulator of ischemic mRNA stabilization. Indeed, of the 1,734 KHSRP targets identified in Winzen et al,43 245, or 14%, overlapped with the 1,589 ischemia-stabilized mRNAs, an overlap that was highly significant via normal approximation of exact hypergeometric probability (Representation factor: 1.5, p < 1.064e−12).
Role of KHSRP in the ischemic mRNA stabilization
To investigate whether KHSRP's binding to target mRNAs was altered in response to hypoxia/hypoglycemia, RIP-RT was performed using beads labeled with normal rabbit serum as a negative control. Interestingly, unlike HuR and hnRNPL, KHSRP binding to these mRNA targets was affected by hypoxic/hypoglycemic exposure. As shown in Fig. 6A, when compared with a normal rabbit serum control IP, KHSRP-specific antibody pull-downs from normoxic HEK293T cells cultured in the presence of low glucose (1 g/L) were enriched for known mRNA targets such as β-catenin (BCAT), as well as our mRNAs of interest, VEGF, MYC and CYR61 while non-targets such as Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) showed no such enrichment. However, when the cells were exposed to 24 hours of hypoxia/hypoglycemia (1% O2 and 1 g/L glucose) there was a dramatic decrease in KHSRP binding to mRNA targets that showed stabilization under the same conditions while BCAT mRNA remained associated with KHSRP. This was not due to differing amounts of protein being pulled down as western blots for KHSRP indicated similar amounts of KHSRP protein in both the inputs as well as the KHSRP IP's (Fig. 6B) suggesting that the difference may be due to an alteration in the ability of KHSRP to recognize these specific mRNAs.
Figure 6.

KHSRP binding to specific mRNAs is inhibited by oxygen and glucose deprivation. HEK293T cells were exposed to 24 hours of normoxia or hypoxia (1% O2) in the presence of 1 g/L glucose for 24 hours and then subjected to KHSRP immunoprecipitation. (A) Fold enrichments calculated from immunoprecipitated mRNA levels normalizing to GAPDH as a negative control and the normal rabbit serum negative control IP. (B) Representative Western blot showing equivalent amounts of KHSRP protein being immunoprecipitated (IP) under Normoxic (N) or Hypoxic (H) conditions, antibody heavy chain (Ab HC) was also detected. (C) Representative western blot showing extent of KHSRP protein knockdown in normoxic conditions after 72 hours siRNA treatment as compared with negative control siRNA (Neg). GAPDH protein levels were used as a loading control. (D) HEK293T cells were transfected with siRNA targeting KHSRP or negative control siRNA (Neg) for 48 hours before being exposed to 24 hours of Normoxia or Hypoxia (1% O2) in the presence of 1 g/L glucose. Cells were treated with Actinomycin D for 0, 1 or 2 hours, mRNA levels of VEFG165 and MYC were determined via qPCR, normalized to GAPDH and 0 hour time point and half-lives determined. Data represents average of N = 3 ± SD for each condition. *P ≤ 0.05 from respective Normoxic condition.
To test whether KHSRP was necessary for the ischemic stabilization of these mRNAs, siRNA-mediated knockdown was performed. Despite a robust reduction in KHSRP protein and mRNA levels (Fig. 6C and Fig. S6C) there was no significant inhibition of the ischemic stabilization of VEGF, MYC, or CYR61 mRNAs (Fig. 6D; corresponding decay curves and mRNA abundance can be found in Fig. S6). In addition, knockout of KHSRP by CRISPR had no significant effect on the hypoxic stabilization of these messages when compared with cells treated with a negative control CRISPR construct (Fig. S7) suggesting that other RNA binding proteins or posttranscriptional regulators are likely to be involved.
Summary
Overall, these studies have revealed that in response to oxygen and glucose deprivation, short-lived mRNAs such as VEGF, MYC, CYR61, and others are stabilized, while high levels of glucose appear to inhibit the response. This response is dose dependent on both oxygen and glucose but not osmolarity. Finally, while the RNA binding proteins HuR and hnRNPL do not appear to be involved in the response, KHSRP does exhibit differential binding to VEGF, MYC, and CYR61 mRNAs in response to hypoxia/hypoglycemia although knockdown/knockout of KHSRP is not sufficient to inhibit the response.
Discussion
The response to hypoxia involves many different changes in gene expression.25 The transcriptional changes that occur through the actions of the Hypoxia Inducible Factor family have been well studied, however, there are also many posttranscriptional events that likely play a key role in adapting to the nutrient deprivation as well.41,42 It has been well established that in response to severe oxygen deprivation there is an inhibition of global cap-dependent translation with a subset of important mRNAs continuing to be translated through several proposed mechanisms.25,44-48 Recently, global changes in alternative splicing have also been shown to be involved in the hypoxic response, although the therapeutic potential of these findings is yet to be explored.49,50 Finally, it has been shown that hypoxia alone leads to an increase in mRNA half-life of messages such as VEGF and GLUT1, although we have had difficulty replicating those findings.26,27,31,51 We have now expanded upon these studies to show that oxygen and glucose deprivation synergize during cellular ischemia to initiate a response that results in the stabilization of several mRNAs that are known to be important for the adaptation to the ischemic conditions including VEGF, MYC, and CYR61.
Our results clearly show that the amount of glucose present can impact the hypoxic response at the posttranscriptional level. We have been able to demonstrate that simultaneous deprivation of oxygen and glucose results in the stabilization of multiple mRNAs while high glucose or oxygen levels prevent the response. While this is the first time that oxygen and glucose have been shown to synergistically regulate gene expression at the posttranscriptional level, glucose has been shown to affect the HIF transcriptional response to hypoxia. Indeed, multiple studies have shown that decreased glucose levels result in increased transcription of known HIF transcriptional targets including VEGF.20,24,38,52,53 It is worth noting however, that many of those studies relied on steady-state mRNA levels to arrive at their conclusions and it is possible that an increase in mRNA stability was also contributing to the increased mRNA content. While we did also see some correlations between mRNA steady-state levels and glucose concentration or half-life, the effect varied between cell types and among mRNAs suggesting that there are multiple layers of regulation occurring. This may be feedback mechanisms designed to limit overall mRNA abundance or perhaps crosstalk between the transcriptional and posttranscriptional pathways. These relationships will need to be explored in future studies to help resolve this apparent disconnect.
Despite our best efforts we were unable to determine a role for either HuR or hNRNPL in this hypoxic/hypoglycemic response. This is curious as multiple studies have shown a role for these RBPs in regulating VEGF mRNA stability in response to a variety of conditions, including hypoxia.27,31,33-36,42 It is possible that differences in cell types underlie this phenomena, although it may also be the experimental conditions. In our experiments, we used a hypoxic workstation which allows us to conduct the entire experiment, including ActD treatment and harvesting in Trizol, in a hypoxic environment, preventing any reoxygenation from occurring. Although it is unclear for all of the studies, at least some were conducted using hypoxic incubators which would necessitate opening (and hence reoxygenation) during ActD treatment and harvest. Thus it is possible that while HuR appears to not affect the hypoxic stabilization, perhaps it is involved in a hypoxic cycling or reoxygenation response.
This inability to identify the posttranscriptional player(s) responsible for the effect prompted us to run a limited global analysis with the idea that if related subsets of mRNAs are being regulated similarly maybe their identity could help direct our efforts. Our results suggest that related subsets of mRNAs may be regulated in concert, with many pathways related to angiogenesis, cardiovascular disease, and cancer being over-represented in the stabilized mRNAs. This supports the idea that there may be hypoxic posttranscriptional RNA operons (PTROs) that could be identified and exploited for novel therapeutic treatments of these diseases.54-57 Thus, these results may have broad reaching implications for the role of mRNA stabilization in both physiologic as well as pathological conditions.
We were also able to identify KHSRP as a potential RNA binding protein involved in this response. KHSRP has been shown to bind AU-Rich Elements (ARE) in mRNAs and target them for degradation by the exosome.58-61 This role would explain the mRNA stabilization response seen in our system. Under normoxic conditions, KHSRP would bind and direct the degradation of the mRNAs. Upon ischemic exposure, KHSRP is no longer able to bind these mRNA and they become stabilized. What controls this switch in binding upon hypoxic exposure is still unknown but likely involves cellular signaling pathways.
Many pathways such as mTOR/AKT and AMPK along with transcription factors such as MYC are activated upon glucose deprivation to mitigate the detrimental effects of the nutrient deficit.2-4 AMPK in particular has been shown to be necessary and sufficient for an increase in VEGF and GLUT1 mRNA stability in response to glucose deprivation.37 In the brain, glucose deprivation has been shown to synergize with hypoxia to increase the stabilization of GLUT151,62 Furthermore, the p38 and JNK pathways has been implicated in the hypoxic stabilization of VEGF message via HuR in rat glioma cells, as well as other transcriptional, and posttranscriptional hypoxic responses.63-68
Interestingly, it has been shown that phosphorylation of KHSRP can differentially regulate its ability to bind to specific subsets of mRNAs.43,69,70 For example, during C2C12 myogenic differentiation, phosphorylation of Threonine 692 by the P38 MAP kinase alters KHSRP's RNA binding specificity such that it no longer binds to a subset of mRNAs important for myogenic differentiation, thus leading to their stabilization.69 Suprisingly, not all KHSRP targets are released suggesting that the P38 phosphorylation impacts only a specific subset of mRNAs. This is mirrored in our data with VEGF and MYC being released while B-catenin remains bound by KHSRP under hypoxia/hypoglycemia. Studies are currently underway to determine if P38 or other kinases may be involved in the ischemic stabilization of the identified mRNAs.
While our RNP-IP data clearly show that hypoxia/hypoglycemia can alter KHSRP's ability to bind VEGF, MYC, and CYR61 mRNA, knockout of KHSRP appeared to have no effect on their hypoxic mRNA stabilization. This could be due to a couple of reasons. First, the extent of KHSRP knockdown may have been insufficient to alter the response. While we calculated an approximately 80% knockdown of KHSRP protein levels by siRNA and almost 100% with CRISPR, we saw no change in the steady mRNA half-life, mRNA level, or protein content of VEGF, MYC, β-catenin, or other known KHSRP targets suggesting that either we need even greater levels of knockdown or that other regulators may be compensating for the loss of KHSRP in the HEK293 cells. It is also possible that multiple RBPs, and/or micro RNAs may be playing a role in this response and that only by altering all of them at once may we be able to affect the stabilization response.
Unfortunately, the functional aspects of this response have eluded us. Despite great effort we have been unable to observe any changes in protein level that can be attributed to the ischemic mRNA stabilization (data not shown), suggesting that it may play another role. It is well known that when exposed to hypoxic conditions severe enough to deplete cellular ATP levels, generalized cap-dependent translation is inhibited while certain mRNAs (including VEGF) continue to be translated.25,44-46,48,71 It may be that the mRNA stabilization response we are seeing is somehow coupled to this decrease in translation. It is also possible that the depletion of oxygen and glucose may induce stress granule formation which might also be sequestering the VEGF and MYC mRNAs and protecting them from degradation.45 However, if this were true, we might expect that restoration of oxygen and glucose levels to the cell might then lead to an increase in VEGF and MYC translation using the stabilized mRNA. However, our preliminary experiments suggest that this is not the case. Future studies will investigate whether our culture conditions are indeed affecting cellular ATP levels to an extent to induce stress granules and if so what effect that might have on the mRNA degradation machinery.
Overall we have shown that synergistic oxygen and glucose deprivation can lead to the stabilization of several mRNAs, including VEGF, MYC, and CYR61. Future studies will delve into dissecting the signaling pathway further as well as identifying the posttranscriptional regulators and mRNA sequences responsible for this effect. It is our hope that an increased understanding of all aspects of the gene expression response to ischemia will aid in the development of new therapeutics.
Material and methods
Cell lines
HEK293T (HEK293T/17; CRL-11268), HEP3B (Hep 3B2.1–7; HB-8064), and C6(C6; CCL-107) cell lines were obtained directly from ATCC and maintained in high glucose (4 g/L) DMEM (Corning/Mediatech, 15–017-CV) supplemented with 10% FBS (Atlanta Biologicals, 511150), 2mM Glutamine (Corning/Mediatech, 25–050CI), and 1X Pen/Strep (Corning/Mediatech, 30–002-CI) and passaged when approximately 85–90% confluent. Cells were tested for mycoplasma upon receipt. For experiments, cells were plated at 0.5 × 106 cell per well of a 6 well plate (CytoOne, USA Scientific, CC7682–7506) in high glucose (4.0 g/L) media and allowed to attach/recover for 18–24 hours. The next day, the media was removed and replaced with media of defined glucose concentration. Hypoxic treatments were performed in a Ruskin InVivo 400 Hypoxia Hood (The Baker Company) maintained at 37°C, 5% CO2, 70% humidity and 1% oxygen. All other chemical reagents were obtained from Sigma-Aldrich unless otherwise specified.
RNA extraction
Trizol (Life Technologies, 15596018) was used for all RNA extractions according to the manufacturer's protocol. For RNA extraction from ribonucleoprotein immunoprecipitations (RNP-IP), GlycoBlue (Life Technologies, AM9516) was added as a carrier during the precipitation step. RNA quality and quantity was determined via NanoDrop 1000 (ThermoFisher Scientific).
PCR
Reverse transcription was performed on 1 µg of total RNA in a 20 µl reaction with the iScript cDNA synthesis kit (Bio-Rad Laboratories, 170–8891). After diluting cDNA 5-fold, quantitative real-time PCR was performed using a Roche Lightcycler 96 with Fast Start Essential DNA Green (Roche Diagnostics Corporation, 06–924–204–001) and primers from Integrated DNA Technologies, Inc. Primers used are listed in Table S1. Primer efficiency was verified to be over 95% for all primer sets used. Quantification of mRNA was performed via ΔΔCT analysis using GAPDH mRNA and the respective control condition for normalization. All real-time PCR primer sets were designed so the products would span at least one intron (> 1kb when possible), and amplification of a single product was confirmed by agarose gel visualization and/or melting curve analysis.
mRNA Decay rates
mRNA levels were determined by real-time quantitative PCR at 0, 1, or 2, hours after the addition of 5 µg/ml Actinomycin D (Sigma Aldrich, A9415). GAPDH mRNA and time 0, untreated controls were used for ΔΔCT normalization. Half-lives were determined by first calculating the slope of the natural log (Ln) of the concentration versus time data and then using that value (λ) to calculate half-life via the equation HL = (Ln(2)/λ). All half-lives >6 hours or those calculated to have a negative half-life (infinitely stable) were converted to 6 hours which was the maximum half-life reliably calculated by this method.
Western blots
Whole cell lysates were prepared in whole cell extract buffer (WCEB: 50 mM Tris pH 7.4, 150 mM NaCl, 5 mM EDTA, 0.1% SDS, and protease inhibitor cocktail (ThermoFisher Scientific, 88660)). Equal amounts of protein (30–50 μg) were electrophoresed on a mini-PROTEAN any KD acrylamide gel (Bio-Rad Laboratories, 456–9034) and transferred to Hybond ECL nitrocellulose (GE Healthcare, RPN303D). Transfer was verified via Ponceau S staining then blot was blocked with 5% nonfat dry milk (LabScientific, 732–291–1940) in Tris buffered saline with 0.1% Tween 20 (TBST) for one hour at room temperature, followed by primary antibody in blocking buffer overnight at 4°C. After washing extensively with TBST, blots were incubated for 1–2 hours at room temperature with appropriate HRP-linked secondary antibody (GE Healthcare), washed again with TBST, developed using Pierce ECL Western Blotting Substrate (ThermoFisher Scientific, 32106), and exposed to film for detection. Primary antibodies used, their catalog numbers and their concentrations can be found in Table S2.
siRNA transfections
siRNAs (Silencer or Silencer Select; Life Technologies) were transfected using Lipofectamine 2000 (Life Technologies, 11668–019) as per manufacturer's protocol using 100 pM siRNAs/well of a 6 well plate. For each target, 2 independent siRNAs were used to confirm results. Part numbers and siRNA IDs can be found in Table S3.
Ribonucleoprotein immunoprecipitation
Ribonucleoprotein Immunoprecipitation (RNP-IP) reactions were performed as described previously.72,73 Briefly, cells were lysed in polysomal lysis buffer (PLB: 100 mM KCl, 5 mM MgCl2, 10 mM HEPES (pH 7.0), 0.5% NP40, 1 mM DTT, 100 units/ml RNase Out (Life Technologies, 10777–019) and protease inhibitor cocktail), pre-cleared, and protein concentration determined via Pierce BCA Assay (ThermoFisher Scientific, 23225). 4 mg of cell lysate were added to protein G sepharose beads (Sigma-Aldrich, P3296) in NT2 (50 mM Tris (pH 7.4), 150 mM NaCl, 1 mM MgCl2, 0.05% Nondiet P-40) pre-bound to either KHSRP Antibody (Bethyl Laboratories, A302–022A) or normal rabbit serum (Sigma-Aldrich, R9133) as a negative control, and the reaction mixtures were tumbled for 4 hours at 4°C. Beads were thoroughly washed with NT2 and then extracted in either WCEB for Western blotting or 1 ml of Trizol for RNA isolation.
Deep sequencing and data analysis
For deep sequencing analysis, 2.5 µg of total RNA from 4 biologic replicates were combined into one sample for sequencing due to financial constraints. The RNA was further purified via Ambion Purelink column (ThermoFisher Scientific, 12183020) and submitted to Beckman Coulter Genomics for library generation, sequencing and gene and mRNA isoform expression analysis. Six libraries (Normoxia and Hypoxia at 0, 1, 2 hrs ActD) were constructed and paired-end sequenced via Illumina technology. 186,651,585 read pairs (1.1 lane equivalent) were produced, resulting in >30 million read pairs per sample. Data was returned as Fragments Per Kilobase of exon per Million fragments (FPKM) as calculated by Cuffdif based on Cufflinks gene identification from TopHat-aligned sequences. For analysis details see the Study Report from Beckman Coulter Genomics in Supplemental File 1. Data were then filtered to only retain genes that had a non-zero value in all 6 of the samples and half-lives were determined by calculating the slope of the natural log (Ln) of the concentration vs. time data and then using that value (λ) to calculate half-life via the equation HL = (Ln(2)/λ). Finally all half-life values less than zero (infinitely stable) or greater than 6 hours (the upper limit of our experimental design) were converted to 6 hours to allow for downstream analysis. Raw and processed deep sequencing data for all genes can be found in Supplemental File 2.
Statistical analysis
All experiments were performed on at least 3 separate occasions to generate biologic replicates. qPCR was performed at least twice on each cDNA for technical verification of data. Half-lives were calculated for each biologic replicate and then averaged together to determine final value and standard deviation of experiment. Statistical significance was calculated by a 2-tailed, paired Student's t-test comparing experimental to control conditions. A P-value below 0.05 was defined as statistically significant. For mRNA abundance, statistical significance was calculated by a 2-tailed, paired Student's t-test comparing normoxic and hypoxic dCT values. A P-value below 0.05 was defined as statistically significant. Pearson correlation coefficients were used to determine the relationship of mRNA abundance with mRNA stability and glucose concentrations and absolute R2 values ≥ 0.80 were considered reportable.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgements
We would like to thank Jack D. Keene (R01 CA157268) and Laura Simone Bisogno (F31 CA185992) in the Keene laboratory at Duke University Medical Center for supporting preliminary experiments related to this study. We would also like to thank Matt Friedersdorf and Jeff Blackinton in the Keene laboratory, as well as Brett Keiper at ECU for helpful discussions during the course of this project. EMJ is currently affiliated with the Department of Biology, University of North Carolina Wilmington, Wilmington, NC. TCK is currently affiliated with University of Tennessee, Pediatric Residency Program, Memphis, TN.
Funding
This work was supported by East Carolina University Start-Up Funds provided to KDM. Additional support provided by the Brody School of Medicine Summer Biomedical Research Program [EMJ] and the Brody School of Medicine Summer Scholars Program [TCK].
References
- 1.Bursch W, Karwan A, Mayer M, Dornetshuber J, Frohwein U, Schulte-Hermann R, Fazi B, Di Sano F, Piredda L, Piacentini M, et al.. Cell death and autophagy: Cytokines, drugs, and nutritional factors. Toxicology 2008; 254:147-57; PMID: 18694801; https://doi.org/ 10.1016/j.tox.2008.07.048 [DOI] [PubMed] [Google Scholar]
- 2.Spasic MR, Callaerts P, Norga KK. AMP-activated protein kinase (AMPK) molecular crossroad for metabolic control and survival of neurons. Neuroscientist 2009; 15:309-16; PMID: 19359670; https://doi.org/ 10.1177/1073858408327805 [DOI] [PubMed] [Google Scholar]
- 3.Hardie DG. Sensing of energy and nutrients by AMP-activated protein kinase. Am J Clin Nutr 2011; 93:891S-6; PMID: 21325438; https://doi.org/ 10.3945/ajcn.110.001925 [DOI] [PubMed] [Google Scholar]
- 4.Ferretti AC, Larocca MC, Favre C. Nutritional stress in eukaryotic cells: oxidative species and regulation of survival in time of scarceness. Mol Genet Metab 2012; 105:186-92; PMID: 22192525; https://doi.org/ 10.1016/j.ymgme.2011.11.007 [DOI] [PubMed] [Google Scholar]
- 5.Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell 2010; 40:294-309; PMID: 20965423; https://doi.org/ 10.1016/j.molcel.2010.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simon MC, Liu L, Barnhart BC, Young RM. Hypoxia-induced signaling in the cardiovascular system. Annu Rev Physiol 2008; 70:51-71; PMID: 17850210; https://doi.org/ 10.1146/annurev.physiol.70.113006.100526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simon MC, Ramirez-Bergeron D, Mack F, Hu CJ, Pan Y, Mansfield K. Hypoxia, HIFs, and cardiovascular development. Cold Spring Harb Symp Quant Biol 2002; 67:127-32; PMID: 12858533; https://doi.org/ 10.1101/sqb.2002.67.127 [DOI] [PubMed] [Google Scholar]
- 8.Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005; 1:401-8; PMID: 16054089; https://doi.org/ 10.1016/j.cmet.2005.05.001 [DOI] [PubMed] [Google Scholar]
- 9.Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT, Simon MC. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab. 2005; 1:393-9; PMID: 16054088; https://doi.org/ 10.1016/j.cmet.2005.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem 2000; 275:25130-8; PMID: 10833514; https://doi.org/ 10.1074/jbc.M001914200 [DOI] [PubMed] [Google Scholar]
- 11.Chandel NS, Schumacker PT. Cellular oxygen sensing by mitochondria: old questions, new insight. J Appl Physiol 2000; 88:1880-9; PMID: 10797153; https://doi.org/ 10.1063/1.1303764 [DOI] [PubMed] [Google Scholar]
- 12.Staab A, Loffler J, Said HM, Katzer A, Beyer M, Polat B, Einsele H, Flentje M, Vordermark D. Modulation of glucose metabolism inhibits hypoxic accumulation of hypoxia-inducible factor-1alpha (HIF-1alpha). Strahlenther Onkol 2007; 183:366-73; PMID: 17609869; https://doi.org/ 10.1007/s00066-007-1649-6 [DOI] [PubMed] [Google Scholar]
- 13.Vordermark D, Kraft P, Katzer A, Bolling T, Willner J, Flentje M. Glucose requirement for hypoxic accumulation of hypoxia-inducible factor-1alpha (HIF-1alpha). Cancer Lett 2005; 230:122-33; PMID: 16253768; https://doi.org/ 10.1016/j.canlet.2004.12.040 [DOI] [PubMed] [Google Scholar]
- 14.Gao W, Ferguson G, Connell P, Walshe T, O'Brien C, Redmond EM, Cahill PA. Glucose attenuates hypoxia-induced changes in endothelial cell growth by inhibiting HIF-1alpha expression. Diab Vasc Dis Res 2014; 11:270-80; PMID:24853909; https://doi.org/12234775 10.1177/1479164114533356 [DOI] [PubMed] [Google Scholar]
- 15.Malhotra R, Tyson DG, Sone H, Aoki K, Kumagai AK, Brosius FC 3rd. Glucose uptake and adenoviral mediated GLUT1 infection decrease hypoxia-induced HIF-1alpha levels in cardiac myocytes. J Mol Cell Cardiol 2002; 34:1063-73; PMID: 12234775; https://doi.org/ 10.1006/jmcc.2002.2047 [DOI] [PubMed] [Google Scholar]
- 16.Rivard A, Silver M, Chen D, Kearney M, Magner M, Annex B, Peters K, Isner JM. Rescue of diabetes-related impairment of angiogenesis by intramuscular gene therapy with adeno-VEGF. Am J Pathol 1999; 154:355-63; PMID: 10027394; https://doi.org/ 10.1016/S0002-9440(10)65282-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sarkar K, Fox-Talbot K, Steenbergen C, Bosch-Marce M, Semenza GL. Adenoviral transfer of HIF-1alpha enhances vascular responses to critical limb ischemia in diabetic mice. Proc Natl Acad Sci U S A 2009; 106:18769-74; PMID: 19841279; https://doi.org/ 10.1073/pnas.0910561106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Botusan IR, Sunkari VG, Savu O, Catrina AI, Grunler J, Lindberg S, Pereira T, Ylä-Herttuala S, Poellinger L, Brismar K, et al.. Stabilization of HIF-1alpha is critical to improve wound healing in diabetic mice. Proc Natl Acad Sci U S A 2008; 105:19426-31; PMID: 19057015; https://doi.org/ 10.1073/pnas.0805230105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bento CF, Pereira P. Regulation of hypoxia-inducible factor 1 and the loss of the cellular response to hypoxia in diabetes. Diabetologia 2011; 54:1946-56; PMID: 21614571; https://doi.org/ 10.1007/s00125-011-2191-8 [DOI] [PubMed] [Google Scholar]
- 20.Catrina SB, Okamoto K, Pereira T, Brismar K, Poellinger L. Hyperglycemia regulates hypoxia-inducible factor-1alpha protein stability and function. Diabetes 2004; 53:3226-32; PMID: 15561954; https://doi.org/ 10.2337/diabetes.53.12.3226 [DOI] [PubMed] [Google Scholar]
- 21.Malmberg K, Norhammar A, Wedel H, Ryden L. Glycometabolic state at admission: important risk marker of mortality in conventionally treated patients with diabetes mellitus and acute myocardial infarction: long-term results from the Diabetes and Insulin-Glucose Infusion in Acute Myocardial Infarction (DIGAMI) study. Circulation 1999; 99:2626-32; PMID: 10338454; https://doi.org/ 10.1161/01.CIR.99.20.2626 [DOI] [PubMed] [Google Scholar]
- 22.Broder HJ, Nesto RW. Glucose, insulin, and potassium for metabolic support in acute myocardial infarction: is the jury still out?. Rev Cardiovasc Med 2006; 7 Suppl 2:S44-50; PMID: 17224877. [PubMed] [Google Scholar]
- 23.Leiter LA. From hyperglycemia to the risk of cardiovascular disease. Rev Cardiovasc Med 2006; 7 Suppl 2:S3-9; PMID: 17224875. [PubMed] [Google Scholar]
- 24.Xiao H, Gu Z, Wang G, Zhao T. The possible mechanisms underlying the impairment of HIF-1alpha pathway signaling in hyperglycemia and the beneficial effects of certain therapies. Int J Med Sci 2013; 10:1412-21; PMID: 23983604; https://doi.org/ 10.7150/ijms.5630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu L, Simon MC. Regulation of transcription and translation by hypoxia. Cancer Biol Ther 2004; 3:492-7; PMID:15254394; https://doi.org/10849653 10.4161/cbt.3.6.1010 [DOI] [PubMed] [Google Scholar]
- 26.Paulding WR, Czyzyk-Krzeska MF. Hypoxia-induced regulation of mRNA stability. Adv Exp Med Biol 2000; 475:111-21; PMID: 10849653; https://doi.org/ 10.1007/0-306-46825-5_11 [DOI] [PubMed] [Google Scholar]
- 27.Levy AP. Hypoxic regulation of VEGF mRNA stability by RNA-binding proteins. Trends Cardiovasc Med 1998; 8:246-50; PMID: 14987559; https://doi.org/ 10.1016/S1050-1738(98)00020-6 [DOI] [PubMed] [Google Scholar]
- 28.Levy AP, Levy NS, Goldberg MA. Hypoxia-inducible protein binding to vascular endothelial growth factor mRNA and its modulation by the von Hippel-Lindau protein. J Biol Chem 1996; 271:25492-7; PMID: 8810320; https://doi.org/ 10.1074/jbc.271.41.25492 [DOI] [PubMed] [Google Scholar]
- 29.Levy AP, Levy NS, Goldberg MA. Post-transcriptional regulation of vascular endothelial growth factor by hypoxia. J Biol Chem 1996; 271:2746-53; PMID: 8576250; https://doi.org/ 10.1074/jbc.271.5.2746 [DOI] [PubMed] [Google Scholar]
- 30.Levy AP, Levy NS, Loscalzo J, Calderone A, Takahashi N, Yeo KT, Koren G, Colucci WS, Goldberg MA. Regulation of vascular endothelial growth factor in cardiac myocytes. Circ Res 1995; 76:758-66; PMID: 7728992; https://doi.org/ 10.1161/01.RES.76.5.758 [DOI] [PubMed] [Google Scholar]
- 31.Levy NS, Chung S, Furneaux H, Levy AP. Hypoxic stabilization of vascular endothelial growth factor mRNA by the RNA-binding protein HuR. J Biol Chem 1998; 273:6417-23; PMID: 9497373; https://doi.org/ 10.1074/jbc.273.11.6417 [DOI] [PubMed] [Google Scholar]
- 32.Levy NS, Goldberg MA, Levy AP. Sequencing of the human vascular endothelial growth factor (VEGF) 3′ untranslated region (UTR): conservation of five hypoxia-inducible RNA-protein binding sites. Biochim Biophys Acta 1997; 1352:167-73; PMID: 9199248; https://doi.org/ 10.1016/S0167-4781(97)00052-3 [DOI] [PubMed] [Google Scholar]
- 33.Goldberg-Cohen I, Furneauxb H, Levy AP. A 40-bp RNA element that mediates stabilization of vascular endothelial growth factor mRNA by HuR. J Biol Chem 2002; 277:13635-40; PMID: 11834731; https://doi.org/ 10.1074/jbc.M108703200 [DOI] [PubMed] [Google Scholar]
- 34.Dibbens JA, Miller DL, Damert A, Risau W, Vadas MA, Goodall GJ. Hypoxic regulation of vascular endothelial growth factor mRNA stability requires the cooperation of multiple RNA elements. Mol Biol Cell 1999; 10:907-19; PMID: 10198046; https://doi.org/ 10.1091/mbc.10.4.907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arcondeguy T, Lacazette E, Millevoi S, Prats H, Touriol C. VEGF-A mRNA processing, stability and translation: a paradigm for intricate regulation of gene expression at the post-transcriptional level. Nucleic Acids Res 2013; 41:7997-8010; PMID: 23851566; https://doi.org/ 10.1093/nar/gkt539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yao P, Potdar AA, Ray PS, Eswarappa SM, Flagg AC, Willard B, Fox PL. The HILDA complex coordinates a conditional switch in the 3′-untranslated region of the VEGFA mRNA. PLoS Biol 2013; 11:e1001635; PMID: 23976881; https://doi.org/ 10.1371/journal.pbio.1001635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yun H, Lee M, Kim SS, Ha J. Glucose deprivation increases mRNA stability of vascular endothelial growth factor through activation of AMP-activated protein kinase in DU145 prostate carcinoma. J Biol Chem 2005; 280:9963-72; PMID: 15640157; https://doi.org/ 10.1074/jbc.M412994200 [DOI] [PubMed] [Google Scholar]
- 38.Stein I, Neeman M, Shweiki D, Itin A, Keshet E. Stabilization of vascular endothelial growth factor mRNA by hypoxia and hypoglycemia and coregulation with other ischemia-induced genes. Mol Cell Biol 1995; 15:5363-8; PMID: 7565686; https://doi.org/ 10.1128/MCB.15.10.5363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang K, Breen EC, Wagner PD. Hu protein R-mediated posttranscriptional regulation of VEGF expression in rat gastrocnemius muscle. Am J Physiol Heart Circ Physiol 2002; 283:H1497-504; PMID: 12234802; https://doi.org/ 10.1152/ajpheart.00813.2001 [DOI] [PubMed] [Google Scholar]
- 40.Pages G, Berra E, Milanini J, Levy AP, Pouyssegur J. Stress-activated protein kinases (JNK and p38/HOG) are essential for vascular endothelial growth factor mRNA stability. J Biol Chem 2000; 275:26484-91; PMID: 10849421; https://doi.org/ 10.1074/jbc.M002104200 [DOI] [PubMed] [Google Scholar]
- 41.Gorospe M, Tominaga K, Wu X, Fahling M, Ivan M. Post-Transcriptional control of the Hypoxic response by RNA-Binding proteins and MicroRNAs. Front Mol Neurosci 2011; 4:7; PMID: 21747757; https://doi.org/ 10.3389/fnmol.2011.00007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Masuda K, Abdelmohsen K, Gorospe M. RNA-binding proteins implicated in the hypoxic response. J Cell Mol Med 2009; 13:2759-69; PMID: 19583805; https://doi.org/ 10.1111/j.1582-4934.2009.00842.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Winzen R, Thakur BK, Dittrich-Breiholz O, Shah M, Redich N, Dhamija S, Kracht M, Holtmann H. Functional analysis of KSRP interaction with the AU-rich element of interleukin-8 and identification of inflammatory mRNA targets. Mol Cell Biol 2007; 27:8388-400; PMID: 17908789; https://doi.org/ 10.1128/MCB.01493-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fahling M. Surviving hypoxia by modulation of mRNA translation rate. J Cell Mol Med 2009; 13:2770-9; PMID: 19674191; https://doi.org/ 10.1111/j.1582-4934.2009.00875.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fahling M. Cellular oxygen sensing, signalling and how to survive translational arrest in hypoxia. Acta Physiol (Oxf) 2009; 195:205-30; PMID: 18764866; https://doi.org/ 10.1111/j.1748-1716.2008.01894.x [DOI] [PubMed] [Google Scholar]
- 46.Uniacke J, Holterman CE, Lachance G, Franovic A, Jacob MD, Fabian MR, Payette J, Holcik M, Pause A, Lee S. An oxygen-regulated switch in the protein synthesis machinery. Nature 2012; 486:126-9; PMID: 22678294; https://doi.org/ 10.1038/nature11055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van den Beucken T, Koritzinsky M, Wouters BG. Translational control of gene expression during hypoxia. Cancer Biol Ther 2006; 5:749-55; PMID:16861930; https://doi.org/18430730 10.4161/cbt.5.7.2972 [DOI] [PubMed] [Google Scholar]
- 48.Young RM, Wang SJ, Gordan JD, Ji X, Liebhaber SA, Simon MC. Hypoxia-mediated selective mRNA translation by an internal ribosome entry site-independent mechanism. J Biol Chem 2008; 283:16309-19; PMID: 18430730; https://doi.org/ 10.1074/jbc.M710079200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu X, Wu R, Shehadeh LA, Zhou Q, Jiang C, Huang X, Zhang L, Gao F, Liu X, Yu H, et al.. Severe hypoxia exerts parallel and cell-specific regulation of gene expression and alternative splicing in human mesenchymal stem cells. BMC genomics 2014; 15:303; PMID: 24758227; https://doi.org/ 10.1186/1471-2164-15-303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sena JA, Wang L, Heasley LE, Hu CJ. Hypoxia regulates alternative splicing of HIF and non-HIF target genes. Mol Cancer Res 2014; 12(9):1233-43; PMID:24850901; https://doi.org/10206432 10.1158/1541-7786.MCR-14-0149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bruckner BA, Ammini CV, Otal MP, Raizada MK, Stacpoole PW. Regulation of brain glucose transporters by glucose and oxygen deprivation. Metabolism 1999; 48:422-31; PMID: 10206432; https://doi.org/ 10.1016/S0026-0495(99)90098-7 [DOI] [PubMed] [Google Scholar]
- 52.Elanchezhian R, Palsamy P, Madson CJ, Mulhern ML, Lynch DW, Troia AM, Usukura J, Shinohara T. Low glucose under hypoxic conditions induces unfolded protein response and produces reactive oxygen species in lens epithelial cells. Cell Death Dis 2012; 3:e301; PMID:22513875; https://doi.org/18268023 10.1038/cddis.2012.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thomas JD, Dias LM, Johannes GJ. Translational repression during chronic hypoxia is dependent on glucose levels. RNA 2008; 14:771-81; PMID: 18268023; https://doi.org/ 10.1261/rna.857308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Simone LE, Keene JD. Mechanisms coordinating ELAV/Hu mRNA regulons. Curr Opin Genet Dev 2013; 23:35-43; PMID: 23312841; https://doi.org/ 10.1016/j.gde.2012.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mansfield KD, Keene JD. The ribonome: a dominant force in co-ordinating gene expression. Biol Cell 2009; 101:169-81; PMID:19152504; https://doi.org/17572691 10.1042/BC20080055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Keene JD. RNA regulons: coordination of post-transcriptional events. Nat Rev Genet 2007; 8:533-43; PMID: 17572691; https://doi.org/ 10.1038/nrg2111 [DOI] [PubMed] [Google Scholar]
- 57.Keene JD, Lager PJ. Post-transcriptional operons and regulons co-ordinating gene expression. Chromosome Res 2005; 13:327-37; PMID: 15868425; https://doi.org/ 10.1007/s10577-005-0848-1 [DOI] [PubMed] [Google Scholar]
- 58.Briata P, Chen CY, Giovarelli M, Pasero M, Trabucchi M, Ramos A, Gherzi R. KSRP, many functions for a single protein. Front Biosci (Landmark Ed) 2011; 16:1787-96; PMID:23178464; https://doi.org/23178464 10.2741/3821 [DOI] [PubMed] [Google Scholar]
- 59.Briata P, Chen CY, Ramos A, Gherzi R. Functional and molecular insights into KSRP function in mRNA decay. Biochim Biophys Acta 2013; 1829:689-94; PMID: 23178464; https://doi.org/ 10.1016/j.bbagrm.2012.11.003 [DOI] [PubMed] [Google Scholar]
- 60.Gherzi R, Chen CY, Ramos A, Briata P. KSRP controls Pleiotropic cellular functions. Semin Cell Dev Biol 2014; 14(5):571-83; PMID: 24845017; https://doi.org/ 10.1016/j.molcel.2004.05.002 [DOI] [PubMed] [Google Scholar]
- 61.Gherzi R, Lee KY, Briata P, Wegmuller D, Moroni C, Karin M, Chen CY. A KH domain RNA binding protein, KSRP, promotes ARE-directed mRNA turnover by recruiting the degradation machinery. Mol Cell 2004; 14:571-83; PMID: 15175153; https://doi.org/ 10.1016/j.molcel.2004.05.002 [DOI] [PubMed] [Google Scholar]
- 62.Boado RJ, Pardridge WM. Glucose deprivation causes posttranscriptional enhancement of brain capillary endothelial glucose transporter gene expression via GLUT1 mRNA stabilization. J Neurochem 1993; 60:2290-6; PMID: 8098356; https://doi.org/ 10.1111/j.1471-4159.1993.tb03516.x [DOI] [PubMed] [Google Scholar]
- 63.Venigalla RK, Turner M. RNA-binding proteins as a point of convergence of the PI3K and p38 MAPK pathways. Front Immunol 2012; 3:398; PMID: 23272005; https://doi.org/ 10.3389/fimmu.2012.00398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Emerling BM, Platanias LC, Black E, Nebreda AR, Davis RJ, Chandel NS. Mitochondrial reactive oxygen species activation of p38 mitogen-activated protein kinase is required for hypoxia signaling. Mol Cell Biol 2005; 25:4853-62; PMID: 15923604; https://doi.org/ 10.1128/MCB.25.12.4853-4862.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fan B, Wang YX, Yao T, Zhu YC. p38 Mitogen-activated protein kinase mediates hypoxia-induced vascular endothelial growth factor release in human endothelial cells. Sheng Li Xue Bao 2005; 57:13-20; PMID: 15719130. [PubMed] [Google Scholar]
- 66.Jackson RM, Gupta C. Hypoxia and kinase activity regulate lung epithelial cell glutathione. Exp Lung Res 2010; 36:45-56; PMID: 20128681; https://doi.org/ 10.3109/01902140903061795 [DOI] [PubMed] [Google Scholar]
- 67.Kulisz A, Chen N, Chandel NS, Shao Z, Schumacker PT. Mitochondrial ROS initiate phosphorylation of p38 MAP kinase during hypoxia in cardiomyocytes. Am J Physiol Lung Cell Mol Physiol 2002; 282:L1324-9; PMID: 12003789; https://doi.org/ 10.1152/ajplung.00326.2001 [DOI] [PubMed] [Google Scholar]
- 68.Zhang CL, Song F, Zhang J, Song QH. Hypoxia-induced Bcl-2 expression in endothelial cells via p38 MAPK pathway. Biochem Biophys Res Commun 2010; 394:976-80; PMID: 20307495; https://doi.org/ 10.1016/j.bbrc.2010.03.102 [DOI] [PubMed] [Google Scholar]
- 69.Briata P, Forcales SV, Ponassi M, Corte G, Chen CY, Karin M, Puri PL, Gherzi R. p38-dependent Phosphorylation of the mRNA decay-promoting factor KSRP controls the stability of select myogenic transcripts. Mol Cell 2005; 20:891-903; PMID: 16364914; https://doi.org/ 10.1016/j.molcel.2005.10.021 [DOI] [PubMed] [Google Scholar]
- 70.Frevel MA, Bakheet T, Silva AM, Hissong JG, Khabar KS, Williams BR. p38 Mitogen-activated protein kinase-dependent and -independent signaling of mRNA stability of AU-rich element-containing transcripts. Mol Cell Biol 2003; 23:425-36; PMID: 12509443; https://doi.org/ 10.1128/MCB.23.2.425-436.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thomas JD, Johannes GJ. Identification of mRNAs that continue to associate with polysomes during hypoxia. RNA 2007; 13:1116-31; PMID: 17488873; https://doi.org/ 10.1261/rna.534807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Keene JD, Komisarow JM, Friedersdorf MB. RIP-Chip: the isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat Protoc 2006; 1:302-7; PMID: 17406249; https://doi.org/ 10.1038/nprot.2006.47 [DOI] [PubMed] [Google Scholar]
- 73.Tenenbaum SA, Lager PJ, Carson CC, Keene JD. Ribonomics: identifying mRNA subsets in mRNP complexes using antibodies to RNA-binding proteins and genomic arrays. Methods 2002; 26:191-8; PMID: 12054896; https://doi.org/ 10.1016/S1046-2023(02)00022-1 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.