


Abstract
We have developed a novel inducible Cre mutant with enhanced recombinase activity to mediate genetic switching events. The protein, designated Cre*PR, is composed of a new Cre mutant at the N-terminus followed by the ligand-binding domain (LBD) of the progesterone receptor (PR). The response to low doses of inducer is significantly enhanced by elongating the C-terminus of the PR LBD from amino acid 891 to 914. The mutant Cre lacks the first 18 amino acids and contains a Val→Ala substitution at position 336, thereby destroying a cryptic splice donor at the 3′-end of Cre. The latter mutation reduces unwanted background recombinase activity in the absence of the synthetic ligand RU486 by a factor of at least 10 to an almost undetectable level. Thus, the recombinase activity turns out to be inducible by a factor of >200. We expect Cre*PR to serve as a valuable tool for conditional expression of genes both in vitro and in vivo.
INTRODUCTION
Gene targeting is a powerful method to explore gene function in vivo. However, this technique has several inherent limitations, such as embryonic lethality and compensatory effects by redundant genes. In order to overcome these problems inducible recombination based on site-specific recombinases has been employed (1). Temporal control allows the analysis of gene function before and after modification of the respective gene within the same mouse. In order to achieve temporal regulation chimeric proteins of recombinases have been used fused to mutants of ligand-binding domains (LBD) of steroid receptors (2–5). Cre–LBD fusion proteins can be activated by synthetic derivatives of the corresponding steroid. Inducible site-specific recombination has been used to target genes in B cells (6), brain (7), keratinocytes (8) and smooth muscle (9) by employing Cre fusions to LBDs originating from either the progesterone or estrogen receptor.
Some of these studies demonstrated, however, that the Cre–LBD system has two major limitations. First, induced recombination in the respective cell population is not complete even after applying high doses of inducer (6,7). Secondly, Cre–LBD fusion proteins exhibit unwanted residual recombinase activity in the absence of inducer in vivo (7) and, particularly, in vitro (4,10,11). Leakiness of the system is a major drawback, since every recombination event is irreversible and therefore even low background recombinase activity may result in high accumulation of recombined target genes. Unambiguous switching of genes using the Cre–LBD system is hindered by ligand-independent recombinase activity (4,10). It has been suggested that basal activity may result from proteolysis of Cre–LBD fusion proteins (4) or from cryptic splicing of the corresponding mRNA. Both events could result in constitutively active Cre molecules when the inactivating LBD moeity is absent.
For many applications both in vitro and in vivo an inducible Cre recombination system displaying negligible activity in the absence of inducer would be highly desirable. In this study we enhanced the range of inducibility of the Cre–PR system by minimizing the risk of cryptic splicing and by increasing the sensitivity to the inducer as well as decreasing the accessibility to proteases. By deleting the linker region between Cre and the PR LBD stepwise and by mutating a distinct cryptic splice donor site within Cre we generated a novel Cre–PR mutant which shows a significantly reduced, almost undetectable background Cre activity associated with dramatically enhanced inducibility. This novel Cre*PR system might serve as an improved and valuable tool for any experimental design in which conditional gene expression is involved.
MATERIALS AND METHODS
Plasmid constructions
The coding regions of PR650–891 and PR650–914 were amplified by PCR from the template pPAPGLVP (12) using the 5′-primer 5PR650 (5′-TTTAAGGATCCACCATGGGCGCCCTGGATGCTGTTGCTC-3′) with 3′-primers 3PR891 (5′-AAATTGATATCTATCCGTACGCATGCATGCAGTACAGATGAAGTTGTTT-3′) and 3PR914 (5′-AAATTGATATCTATCCGTACGCATGCATAGCAATAACTTCAGACATCATTT-3′) and were cloned into the BamHI and EcoRV sites of the pNN265E-bpA vector (Ralf Kühn, Artemis Pharmaceuticals, Cologne). For amplification of PR676–891 and PR676–914 the primer 5PR676 (5′-TTTAAGGATCCACCATGGGCGCCTCACCAGGTCAAGACATACA-3′) was used in combination with 3PR891 and 3PR914. In these new PR LBD-containing vectors the phage Cre gene starting from amino acid 19 was cloned into the SfoI site after amplification using pPGK-Cre-bpA (Kurt Fellenberg, University of Cologne) as template and 5cre19 (5′-TTTAAGGCGCCACGAGTGATGAGGTTCGCA-3′) and 3cre343 (5′-AAATTGGCGCCATCGCCATCTTCCAGCAGG-3′) as primers. Cre19V336A was obtained using 5cre19 and 3creV336A (5′-AAATTGGCGCCATCGCCATCTTCCAGCAGGCGCGCCATTGCCCC-3′). Constructs containing the humanized versions of Cre, hCre2 and hCre19, were amplified from pBluehCre (kindly provided by P.Seeburg and F.Stewart) using the primers 5hCre2 (5′-GGGGGATCCACCATGGGTGCCTCCAACCTGCTGACTGTG-3′) and 5hCre19 (5′-TTTAAGGATCCACCATGGGTGCCACCTCTGATGAAGTC-3′) together with 3hCre343 (5′-CCCTTGGCGCCGTCCCCATCCTCGAGCAG-3′) and then cloned into the BamHI and SfoI sites of pNNPR676–914. The mutant humanized CreV336A was amplified using the primers 5hCre19 and 3hCreV336A (5′-AAATTGGCGCCGTCCCCATCCTCGAGCAGCCTCGCCATGGCCCC-3′) and then cloned as described above into pNNPR676–914 and pNNPR650–914. To obtain pNNCrePR1, pCrePR1 (3) (kindly provided by Günther Schütz, German Cancer Research Center, Heidelberg) was XhoI digested, blunted using T4 DNA polymerase and BglII digested. The resulting 1.8 kb fragment, containing the coding region of CrePR1, was cloned into the BamHI and EcoRV sites of the pNN265E-bpA vector. All PCR products and junctions were confirmed by sequencing.
Cre recombinase activity assay
CV1-5B cells were cultured in Dulbecco’s modified Eagles’s medium containing 10% fetal calf serum and 100 U/ml penicillin and 0.1 mg/ml streptomycin. Aliquots of 8 × 104 cells were plated on a 6-well plate, grown for 24 h and then transfected with 1 µg of the test DNAs plus 1 µg pHD2-AP (encoding alkaline phosphatase) (3) to determine transfection efficiency. Transfection was performed using Fugene6 (Roche) according to the manufacturer’s protocol. Twelve hours after transfection cells were trypsinized and split into two wells of a 12-well plate containing medium with or without 100 nM RU486 (Sigma). Seventy-two hours after transfection cells were fixed in phosphate-buffered saline containing 4% formaldehyde and stained overnight with X-Gal staining solution as described (3). After heat inactivation of endogenous alkaline phosphatase (AP) activity (30 min at 65°C) cells were stained for AP activity using Fast Red tablets (Roche). In each well blue and red cells of three different areas (2 mm2) were counted. The ratio of blue to red cells was determined and the value of authentic Cre (i.e. without fusion partner) was set to 100%. Each experiment was performed at least in triplicate.
Dose–response assay
Samples of 8 × 105 CV1-5B cells were plated on 9 cm dishes, grown for 24 h and then transfected with 5 µg of the test plasmids. Twelve hours after transfection cells were trypsinized and split into seven wells of two 6-well plates containing different concentrations of RU486. Seventy-two hours after transfection cells were fixed, stained for β-galactosidase and counted as described above. Relative β-galactosidase activity was expressed as the percentage of the maximum activity after substraction of the activity obtained in the absence of ligand. Each experiment was performed in duplicate and mean values are given in Figures 2B and 6B.
Figure 2.
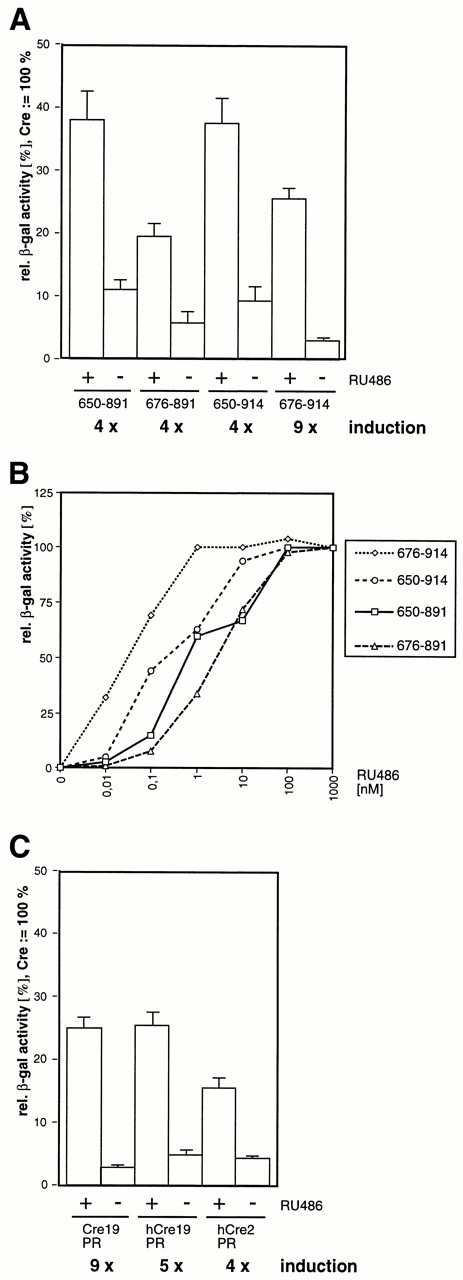
Quantitative analysis of recombinase activities of the different Cre–PR fusion proteins investigated. (A) Reporter cells were transfected with different Cre19–PR expression vectors and cultured in the presence (+) or absence (–) of 100 nM RU486. Three days later the relative β-galactosidase activity was determined as described in Materials and Methods. The columns show the mean values of at least four different transfections with standard deviations indicated by vertical lines. (B) Dose–response analysis of different Cre19–PR fusions. Reporter cells were transfected with the indicated constructs and cultured in the presence of various concentrations of RU486. No β-galactosidase activity was detected in the presence of 1 µM progesterone. (C) Comparison of activities of different Cre fused to PR676–914. The mean values from six different transfections are depicted with standard deviations indicated as vertical lines. Cre19, truncated Cre lacking the first 18 amino acids; hCre, humanized Cre; Cre2, full-length Cre.
Figure 6.
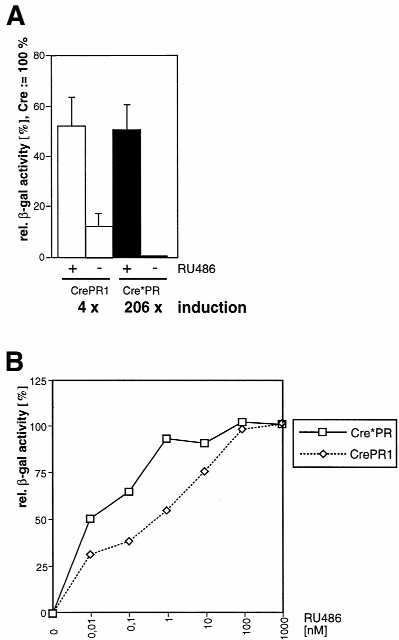
Comparative analysis of inducible Cre recombinase activities of the improved Cre*PR system and the previously published CrePR1 system (3,7). (A) Reporter cells were transfected with CrePR1 or Cre*PR expression vectors and cultured in the presence (+) or absence (–) of 100 nM RU486. Three days later the relative β-galactosidase activity was determined as described in Materials and Methods. The columns show the mean values of three different transfections with standard deviations indicated by vertical lines. (B) Dose–response analysis of CrePR1 and Cre*PR. Reporter cells were transfected with the indicated constructs and cultured in the presence of various concentrations of RU486.
RT–PCR
Total RNA was isolated from pNN-CrePR650–914-transfected cells using the RNeasy mini kit (Qiagen). Approximately 4 µg DNase I-digested RNA was used for cDNA synthesis according to Pharmacia protocols (first strand cDNA synthesis kit). Aliquots of 10 µl of this reaction were used for the first strand synthesis with either creRT (5′-GGAGATCATGCAAGCTGGT-3′) or hcreRT (5′-CTGAAATCATGCAGGCTGG-3′) in combination with PRRTL (5′-ATTAGATCAGGTGCAAAATACA-3′). The cycle program consisted of 30 cycles of 96°C for 45 s, 52°C for 60 s and 72°C for 60 s. After the first round of amplification only the non-spliced bands were visible by ethidium bromide staining. Agarose gel pieces were cut out at the height of expected sizes of the aberrantly spliced products. DNA was isolated and used for a semi-nested PCR (conditions as above) with either creRT or hcreRT in combination with PRRTS (5′-TCAGTGGTGGAATCAACTGT-3′). After amplification DNA fragments were separated in an agarose gel, isolated from the gel and cloned into pGEM-T-easy (Promega), followed by sequencing.
Splice site analysis
Cryptic splice sites were identified using the program GeneFinder (http://dot.imgen.bcm.tmc.edu:9331/gene-finder/gf.html). The scores of the corresponding sites were calculated by the program Splice Site Score Calculation (http://www2.imcb.osaka-u.ac.jp/splice/score.html). The score expresses how well the cryptic splice site fits the consensus sequence. For example, a 100% match to the mammalian 3′-splice site corresponds to a score of 14.2. A perfect 5′-splice site would have a score of 12.6. The mean scores of the 5′- and 3′-splice sites in constitutive exons were 8.1 and 7.9, respectively.
RESULTS
Comparison of the activity of different inducible Cre recombinases
Cre recombinase activity of Cre–PR fusion proteins is dependent on the presence of the synthetic steroid RU486 (3). A fusion of NLS–Cre (Cre containing a nuclear localization signal, NLS) to the PR LBD ranging from amino acid 641 to 891 has been used to achieve inducible recombination in brain (7). However, residual Cre recombinase activity in the absence of inducer and incomplete recombination in the presence of inducer were observed. The C-terminal deletion mutant PR891 is insensitive to the natural ligand progesterone but still has high affinity for the synthetic ligand RU486 (13). In order to further improve the inducibility of the system we generated a set of new constructs. Besides PR891 mutants we also cloned C-terminally extended versions ending with amino acid 914, because a previous report suggested that PR914 mutants display higher sensitivity to RU486 (12). At the N-terminus of the PR LBD we introduced two new truncations, resulting in constructs starting with amino acids 650 and 676, respectively. By this modification we expected tighter regulation and decreased accessibility to proteases due to the shorter linker between Cre and PR. Altogether we cloned four new PR LBDs exhibiting two different N- and C-termini, i.e. PR650–891, PR676–891, PR650–914 and PR676–914 (Fig. 1). As Cre-encoding cassettes we used both wild-type phage Cre and a cDNA which had been optimized for expression in mammalian cells by changing the codon usage (kindly provided by P.Seeburg, R.Sprengel and F.Stewart). In addition to full-length Cre we also constructed N-terminally truncated Cre lacking the first 18 amino acids, which do not exhibit a defined structure (14) and are therefore considered not to be essential for recombinase activity. Cre fusion genes have been cloned into the vector pNN265E-bpA (kindly provided by Ralf Kühn) which contains a CMV promoter, a 5′-splice substrate (15) and a bovine polyadenylation signal.
Figure 1.
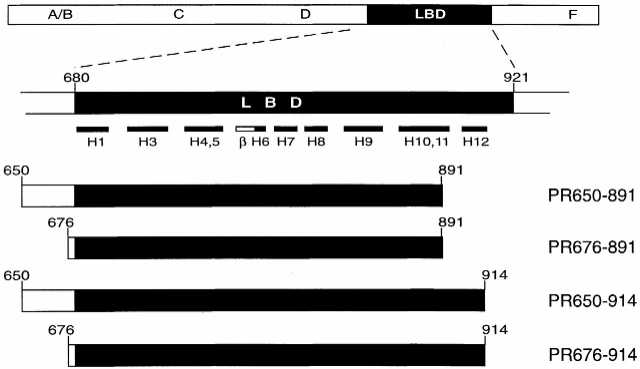
Schematic representation of the ligand-binding domain (LBD) of the progesterone receptor and mutants thereof used in this study. LBDs are represented as black boxes and linker regions as white boxes. Numbers represent amino acids. Structural motifs are depicted as H1–H12 (α-helices) and β (β-sheets). Structural data are derived from Williams and Sigler (19).
For quantitation of Cre recombinase activity we used the Cre reporter cell line CV1-5B containing a stably integrated single copy reporter gene that responds to Cre recombinase activity by LacZ activation (3). After transient transfection of the new constructs into the CV1-5B cell line we first confirmed correct expression of Cre fusions by western blotting (data not shown). In order to compare the Cre activities of the different fusion constructs we transfected CV1-5B cells and cultured them in the absence or presence of 100 nM RU486. Cre activities were quantitated by counting cells positive for the β-galactosidase activity of the reporter gene as described in Materials and Methods.
Both CrePR650 mutants show similar Cre recombinase activity in the presence of the synthetic ligand RU486. However, this high recombinase activity of ∼40% (relative to authentic Cre) is also accompanied by substantial activity in the absence of inducer (∼10%) (Fig. 2A). Constructs containing N-terminally truncated PR, Cre–PR676–891 and Cre–PR676–914, exhibit significantly less recombinase activity in the absence of inducer, although the maximum activity is also significantly lower as compared to the corresponding Cre–PR650 constructs. Dose–response analysis was performed in order to estimate the different affinities of the Cre fusions for RU486. We therefore transfected CV1-5B cells and measured Cre activity in the presence of various concentrations of inducer. This analysis shows that Cre–PR676–914 is half-maximally activated by ∼0.04 nM RU486, whereas Cre–PR676–891 needs at least a 100-fold higher concentration to reach the same level of activation (Fig. 2B). Cre–PR650–914 also responds at lower concentrations to the inducer than Cre–PR650–891, indicating that sensitivity to RU486 is increased by elongating the C-terminus of PR from 891 to 914. None of the constructs was activated in the presence of 1 µM progesterone (data not shown). Based on these results, we concentrated on the construct displaying the lowest c50 value (concentration of half-maximal activity) and the highest inducibility, namely Cre–PR676–914.
All constructs used so far contained a Cre recombinase from which we deleted the first 18 amino acids. In order to investigate the influence of both N-terminal truncation and optimization of the codon usage for Cre we fused PR676–914 to a codon optimized cDNA (kindly provided by P.Seeburg, R.Sprengel and F.Stewart), in the following designated hCre, encoding either full-length Cre (Cre2) or a truncated Cre lacking the first 18 amino acids (Cre19). A comparison of the recombinase activities of these constructs to the corresponding truncated Cre fusion originating from phage cDNA shows that both truncated Cre19–PR variants containing either hCre or phage Cre display definitely higher recombinase activity (26%) than the full-length hCre (16%) in the presence of inducer (Fig. 2C). The activity in the absence of inducer is slightly higher in the case of the hCre constructs (∼5%) as compared to the construct originating from phage Cre (3%).
Analyzing aberrant splice events
Since the Cre–PR system is intended to be used as a genetic switch in mice, one has to take into account that aberrant splicing of the Cre–PR-encoding mRNA may occur depending on the sequence context of the integrated construct. For example, mRNAs encoding for the tetracycline repressor fused to VP16 were found to be aberrantly spliced via several cryptic sites (16). It has been suggested that aberrant splicing might occur within Cre due to a potential splice acceptor site (17). In order to further enhance expression and inducibility of our constructs we analyzed the cryptic splice pattern of Cre–PR fusion genes in more detail. For this purpose we performed a computer-aided analysis of different Cre–PR mRNAs using the programs GeneFinder and Splice Site Score Calculator. According to this analysis phage Cre contains four distinct sites displaying high similarity to the consensus splice donor sequences. Two cryptic donor sites occur at the 3′-end of the gene (Fig. 3A). The fusion partner PR LBD contains at least three sequences within the region encoding for amino acids 650–914 showing high similarity to the splice acceptor consensus sequence. In order to investigate the actual potency of the cryptic splice sites we prepared RNA from Cre–PR650–914-transfected cells and performed RT–PCR analysis (data not shown). After cloning and sequencing the products we identified one aberrant splice product in which two distinct cryptic splice sites had been recognized by the splicing machinery, resulting in a synthetic intron ranging from the 5.8 splice donor site at the 3′-end of Cre to the 11.2 splice acceptor site at the 5′-end of PR (see Fig. 3A). The respective translation product of the alternatively spliced mRNA is C-terminally truncated due to a premature stop.
Figure 3.
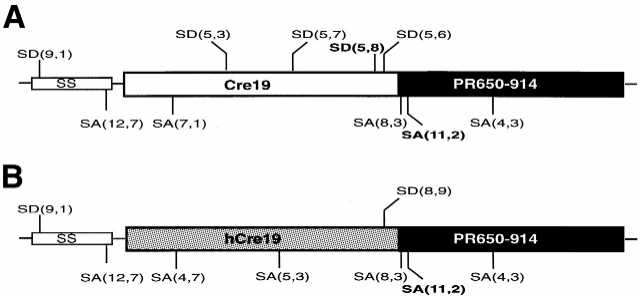
Map of cryptic splice sites within mRNAs of (A) Cre19–PR650–914 and (B) hCre19–PR650–914. Potential splice donors (SD) and acceptors (SA) are shown together with calculated scores in parentheses, which express how well the splice sites fit the consensus sequence (see Materials and Methods for more details). Sites that proved to function as splice sites in a physiological context are shown in bold. SS, splice substrate.
Given the experimental observation that Cre–PR fusion genes are indeed aberrantly spliced in a physiological context, we tried to prevent at least the sort of unwanted splicing that could result in truncated, constitutively active forms of Cre–PR due to (partial) loss of PR. Three of the potential splice donor sites had been destroyed by introducing mammalian codon usage. However, one cryptic donor site was still present in the hCre gene (Fig. 3B). Since a silent mutation is insufficient to unambiguously suppress the risk of splicing at this position we introduced an amino acid substitution. We mutated the codon encoding for amino acid 336 from GTG (Val) to GCG (Ala). This conservative amino acid exchange is expected to completely hinder splicing at this position, since a GT is indispensable for the splicing event (Fig. 4).
Figure 4.
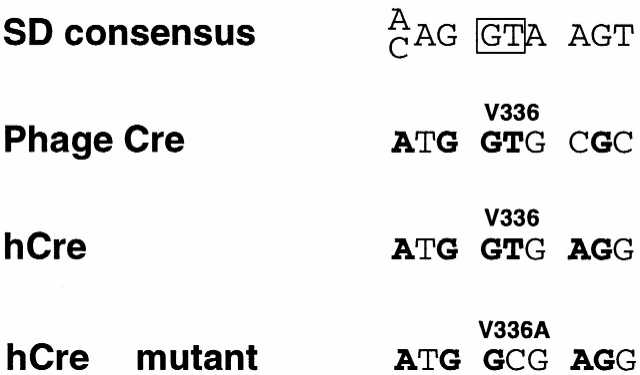
Sequence alignment of codons 335–337 of phage Cre, humanized Cre and the V336A mutant thereof compared to the consensus sequence of splice donor sites. The boxed GT indicates the nucleotides that are critical for splicing. Bases in bold indicate identity to the consensus sequence.
Activity of CreV336A–PR constructs
We introduced the V336A mutation into hCre fused to either PR676–914 or PR650–914 in order to compare the inducibility of the resulting recombinase activities to those of the respective unmutated constructs by transient transfection. Unmutated Cre–PR676–914 shows a recombinase activity of 2.5% in the absence of RU486, whereas the mutant hCreV336A–PR676–914 displays an at least 10-fold decreased background recombinase activity (Fig. 5). Given the low number of positive events in this case the quantified value of 0.2% is close to the lower detection limit resulting in an exceptional increase in inducibility from a level of 10- to 128-fold. No significant difference is observed with respect to the maximum activity of both PR676–914 constructs (∼26%), irrespective of the mutation. As demonstrated above, the PR650–914 construct showed an activity significantly higher than that of the N-terminally truncated PR676–914 version, but a higher background activity also became obvious. However, by introducing the V336A mutation into the hCre–PR650–914 construct (designated Cre*PR) the background activity was reduced 25-fold to a negligible level, but a maximum activity of >40% was still observed, altogether resulting in an induction factor of >200. In order to evaluate our improvements with respect to the previously described system, CrePR1 (3,7), we also cloned the CrePR1 coding cassette into the pNN265E-bpA vector and performed a side-by-side comparison with our optimized Cre*PR system. Cre*PR exhibits a dramatically reduced background recombinase activity as compared to CrePR1, whereas both constructs showed a similar maximum activity of ∼50% in the presence of the inducer (Fig. 6A). However, our dose–response analysis demonstrated that the c50 value of Cre*PR is at least 10 times lower than that of CrePR1 (Fig. 6B). This result again reflects the positive effect of elongation of the PR LBD C-terminus with respect to sensitivity of the inducible recombination system.
Figure 5.
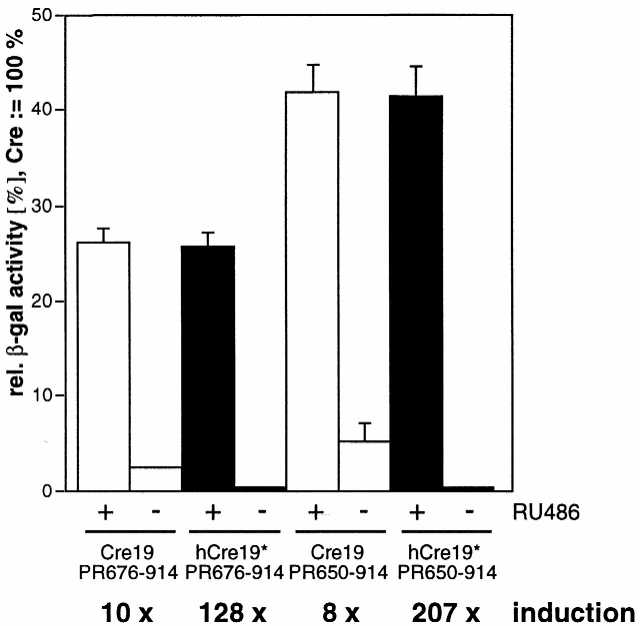
Recombinase activities of different Cre–PR fusion proteins containing a V336A mutation within Cre as compared to corresponding unmutated constructs. Black columns represent mutated Cre and white columns correspond to unmutated phage Cre. hCre19*, humanized Cre19 containing the V336A mutation.
DISCUSSION
Our investigations were aimed at developing an inducible Cre recombinase system which would show high recombinase activity after induction but minimal activity in the absence of the inducer. In order to achieve tight regulation we extensively tested modifications of the fusion protein construct. (i) Partial deletion of the linker region between Cre and the PR LBD resulted in a fusion protein exhibiting an ∼2-fold lower recombinase activity in the absence of inducer. There are two possible explanations for this observation: the resulting fusion protein may be less accessible to proteases due to the truncated linker between Cre and PR and/or the regulatory potential may be expected to increase with proximity of the LBD to the Cre fusion partner. (ii) By extension of the C-terminus of the PR LBD we expected to achieve an enhanced response of Cre–PR fusions to the synthetic ligand, since it has been reported that elongation from amino acid 891 to 914 increases the activation potential of PR (12). Indeed, it turned out that Cre19–PR676–914 reached half-maximum activation at a 100-fold lower concentration of RU486 than Cre19–PR676–891, whereas no activation was observed in the presence of the natural ligand progesterone. Up to now up to eight daily injections of 2.5 mg RU486 have had to be applied to obtain recombination in mice (7). These doses can result in unwanted antagonistic effects, particularly in pregnant mice. Our improved system might overcome this limitation, since the dose–response characteristics of the C-terminally extended Cre–PR are significantly enhanced in vitro and we expect substantially lower doses of inducer to be required for efficient recombination in vivo. A correlation between in vitro and in vivo data regarding dose–response characteristics has been shown for an improved version of a fusion between Cre and the LBD of the estrogen receptor (18). (iii) In order to enhance expression of the fusion protein we deleted the first 18 amino acids of Cre, which we considered not to be essential for recombinase activity. It turned out that a truncated Cre fusion protein definitely shows higher activity as compared to a fusion protein containing full-length Cre.
The improved fusion proteins, however, still exhibit substantial Cre activity, varying from 3 to 5% in the absence of inducer, disturbing unambiguous gene switching. The residual activity may result from proteolytic cleavage or/and aberrant splicing. Both of these events could lead to C-terminally truncated CrePR fusions exhibiting uncontrolled Cre activity. We showed that two cryptic splice sites within the Cre–PR mRNA are functional in eukaryotic cells after transient transfection. Even if the supposed low probability of this event may result in <1% aberrantly spliced mRNAs this could serve as an explanation for a background activity of a few percent. As far as the use in transgenic mice is concerned, the risk of aberrant splicing is expected to be even higher as compared to transient expression in cell culture, depending on the particular sequence context of the locus where the transgene is integrated. In order to overcome this limitation we destroyed one distinct cryptic splice donor site at the 3′-end of the Cre gene, the only one that is also present in the optimized hCre gene. The mutation also introduces a conservative amino acid exchange at this position, namely Val336Ala. This mutated form of hCre when fused to PR650–914 (designated Cre*PR) exhibits negligible background activity, whereas maximum activity is not influenced by the mutation and still reaches >40%. The resulting range of inducibility is >200-fold, whereas previously published comparably active Cre–PR constructs display inducibility factors of not more than 40 (3,7). We performed a side-by-side comparison of our Cre*PR construct with the previously published construct, namely CrePR1 (3,7), by using the same vector backbone and identical assay conditions. This analysis clearly demonstrated that Cre*PR exhibits an ∼50-fold lower background activity than CrePR1, resulting in a dramatically expanded range of inducibility. Moreover, the dose–response characteristics of Cre*PR are significantly enhanced by at least a 10 times lower c50 value as compared to CrePR1. To our knowledge this is the first time that highly efficient inducible Cre recombination is achieved in vitro with almost undetectable recombination activity in the absence of inducer. Other Cre–LBD systems, such as the mER–Cre–mER system, employing a fusion of Cre with two LBDs of the estrogen receptor (4), also display very low backgrounds, but are also significantly less active in the presence of inducer than Cre–PR fusion proteins (7). Our improved system will be useful for experiments involving conditional gene expression both in vitro and in vivo where leakiness should not be tolerated. For example, inducible and irreversible control of gene expression using an older version of a Cre–LBD fusion was recently reported. The system was limited by spontaneous expression of the reporter gene, apparently due to ligand-independent recombinase activity of the Cre–LBD (10). Employment of our improved system could overcome this limitation. Also given the response to lower doses of inducer, we consider Cre*PR a promising tool for conditional gene targeting in mice, particularly for applications where background activity is not desired. The generation of Cre*PR transgenic mice and analysis in vivo are in progress.
Acknowledgments
ACKNOWLEDGEMENTS
We are indebted to P.Seeburg, R.Sprengel and F.Stewart for providing pBluehCre and G.Schütz for the reporter cell line CV1-5B and pCrePR1. We thank R.Kühn and F.Schwenk for helpful discussions and P.Dröge and B.Jungnickel for critical reading of the manuscript. This work was supported by funds from the Volkswagen Foundation (I/76353), the Körber Foundation (Körber Award for European Science) and the European Union (QLG1-1999-00202).
References
- 1.Kühn R., Schwenk,F., Aguet,M. and Rajewsky,K. (1995) Inducible gene targeting in mice. Science, 269, 1427–1429. [DOI] [PubMed] [Google Scholar]
- 2.Logie C. and Stewart,A.F.(1995) Ligand-regulated site-specific recombination. Proc. Natl Acad. Sci. USA, 92, 5940–5944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kellendonk C., Tronche,F., Monaghan,A.P., Angrand,P.O., Stewart,A.F. and Schütz,G. (1996) Regulation of Cre recombinase activity by the synthetic steroid RU486. Nucleic Acids Res., 24, 1404–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y., Riesterer,C., Ayrall,A.-M., Sablitzky,F., Littlewood,T.D. and Reth,M. (1996) Inducible site-directed recombination in mouse embryonic stem cells. Nucleic Acids Res., 24, 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feil R., Brocard,J., Mascrez,B., LeMeur,M., Metzger,D. and Chambon,P. (1996) Ligand-activated site-specific recombination in mice. Proc. Natl Acad. Sci. USA, 93, 10887–10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwenk F., Kühn,R., Angrand,P., Rajewsky,K. and Stewart A.F. (1998) Temporally and spatially regulated somatic mutagenesis in mice. Nucleic Acids Res., 26, 1427–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kellendonk C., Tronche,F., Casanova,E., Anlag,K., Opherk,C. and Schütz,G. (1999) Inducible site-specific recombination in the brain. J. Mol. Biol., 285, 175–182. [DOI] [PubMed] [Google Scholar]
- 8.Li M., Indra,A.K., Warot,X., Brocard,J., Messaddeq,N., Kato,S., Metzger,D. and Chambon,P. (2000) Skin abnormalities generated by temporally controlled RXRα mutations in mouse epidermis. Nature, 407, 633–636. [DOI] [PubMed] [Google Scholar]
- 9.Kuhbandner S., Brummer,S., Metzger,D., Chambon,P., Hofmann,F. and Feil,R. (2000) Temporally controlled somatic mutagenesis in smooth muscle. Genesis, 28, 15–22. [DOI] [PubMed] [Google Scholar]
- 10.Fuhrmann-Benzakein E., Garcia-Gabay,I., Pepper,M.S., Vassalli,J.D. and Herrera,P.L. (2000) Inducible and irreversible control of gene expression using a single transgene. Nucleic Acids Res., 28, e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng T.H., Chang,C.R., Joy,P., Yablok,S. and Gartenberg,M.R. (2000) Controlling gene expression in yeast by inducible site-specific recombination. Nucleic Acids Res., 28, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y., Xu,J., Pierson,T., O’Malley,B.W. and Tsai,S.Y. (1997) Positive and negative regulation of gene expression in eukaryotic cells with an inducible transcriptional regulator. Gene Ther., 4, 432–441. [DOI] [PubMed] [Google Scholar]
- 13.Vegeto E., Allan,G.F., Schrader,W.T., Tsai,M.J., McDonnell,D.P. and O’Malley,B.W. (1992) The mechanism of RU486 antagonism is dependent on the conformation of the carboxy-terminal tail of the human progesterone receptor. Cell, 69, 703–713. [DOI] [PubMed] [Google Scholar]
- 14.Guo F., Gopaul,D.N. and Van Duyne,G.D. (1997) Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature, 389, 40–46. [DOI] [PubMed] [Google Scholar]
- 15.Choi T., Huang,M., Gorman,C. and Jaenisch,R. (1991) A generic intron increases gene expression in transgenic mice. Mol. Cell. Biol., 11, 3070–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Urlinger S., Baron,U., Thellmann,M., Hasan,M.T., Bujard,H. and Hillen,W. (2000) Exploring the sequence space for tetracycline-dependent transcriptional activators: novel mutations yield expanded range and sensitivity. Proc. Natl Acad. Sci. USA, 97, 7963–7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gorski J.A. and Jones,K.R. (1999) Efficient bicistronic expression of cre in mammalian cells. Nucleic Acids Res., 27, 2059–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Indra A.K., Warot,X., Brocard,J., Bornert,J.M., Xiao,J.H., Chambon,P. and Metzger,D. (1999) Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic Acids Res., 27, 4324–4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams S.P. and Sigler,P.B. (1998) Atomic structure of progesterone complexed with its receptor. Nature, 393, 392–396. [DOI] [PubMed] [Google Scholar]