

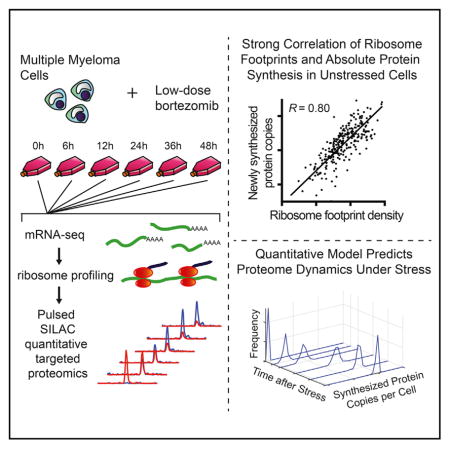
SUMMARY
Ribosome profiling is a widespread tool for studying translational dynamics in human cells. Its central assumption is that ribosome footprint density on a transcript quantitatively reflects protein synthesis. Here, we test this assumption using pulsed-SILAC (pSILAC) high-accuracy targeted proteomics. We focus on multiple myeloma cells exposed to bortezomib, a first-line chemotherapy and proteasome inhibitor. In the absence of bortezomib, we found that direct measurement of protein synthesis by pSILAC correlated well with indirect measurement of synthesis from ribosome footprint density. This correlation, however, broke down at high bortezomib doses. By developing a statistical model integrating longitudinal proteomic and mRNA-sequencing measurements, we found that proteomics could directly detect global alterations in translational rate caused by bortezomib; these changes are not detectable by ribosomal profiling alone. Further, by incorporating pSILAC data into a gene expression model, we predict cell-stress specific proteome remodeling events. These results demonstrate that pSILAC provides an important complement to ribosome profiling in measuring proteome dynamics.
In Brief
Direct measurement of protein synthesis by pulsed-SILAC mass spectrometry after cancer therapy reveals proteomic effects of global translational repression and predicts proteome remodeling under cellular stress.
INTRODUCTION
Dynamic changes in the cancer proteome control tumor growth, proliferation, metastasis, and response to therapy. Targeting aberrant mRNA translation in cancer has recently garnered significant interest as a therapeutic strategy (Boussemart et al., 2014; Hsieh et al., 2012; Wolfe et al., 2014). Furthermore, a myriad of cellular stresses, including exposure to various chemotherapeutics, leads to global inhibition of protein synthesis and remodeling of the cancer proteome (de Haro et al., 1996; Walter and Ron, 2011).
A powerful new tool to measure gene-specific regulation of translation is ribosome profiling, the deep sequencing of mRNA fragments protected by actively translating ribosomes (Ingolia et al., 2009, 2011; Michel and Baranov, 2013). A central assumption of ribosome profiling is that indirect measurement of ribosome footprint occupancy on transcripts is directly reflective of true protein synthesis. While this assumption has been shown to be largely true in bacteria (Li et al., 2014a), the relationship between footprint occupancy and protein synthesis remains less clear in the more complex translational system of eukaryotes (Liu et al., 2016). Furthermore, using standard ribosome profiling approaches it can be difficult to capture global cellular changes in translational capacity (Ingolia, 2016), such as those which occur in response to drug therapy in cancer.
A potential orthogonal method to measure gene-specific translational regulation is pulsed-SILAC (pSILAC) proteomics. In this approach, stable isotope-labeled amino acids are added to the cellular media and subsequently incorporated into all newly synthesized proteins. pSILAC has been used to directly monitor the synthesis of new proteins in various systems using shotgun proteomic approaches (Jovanovic et al., 2015; Schwanhausser et al., 2011). Therefore, combining pSILAC with high-accuracy quantitative proteomics may offer an alternate approach to assess gene-specific translational regulation through the direct measurement of newly synthesized proteins.
Here, as a test-bed for comparing ribosomal footprint profiling and pSILAC, we monitor the effects of low-dose bortezomib therapy on translation in multiple myeloma cells using both methods. Bortezomib is a first-line therapy for this hematologic cancer, and proteasomal blockade by this drug is known to lead to ER stress due to accumulation of unfolded and misfolded proteins (Obeng et al., 2006; Vincenz et al., 2013). This stress triggers downstream signaling pathways that inhibit the translation of the large majority of mRNAs (Walter and Ron, 2011). We demonstrate that before the onset of bortezomib-mediated translational repression, ribosomal footprint profiling and pSILAC measurements were well correlated, providing important support for the assumption that ribosome footprint density is quantitatively reflective of protein synthesis in eukaryotes. However, we observed that under conditions of proteasomal blockade this correlation breaks down in characteristic ways. Using a quantitative statistical model to describe protein synthetic rates, we found that pSILAC methods were able to directly detect global alterations of translation not identified by standard ribosome profiling approaches, including dynamic, protein-level responses to different levels of stress-induced translational inhibition. These findings underscore the utility of pSILAC proteomics as a complementary method in studies of translational regulation to ribosome profiling, particularly under conditions of cellular stress. Given that similar, global perturbations of translation occur during heat shock, DNA damage, and oxidative stress (Duncan and Hershey, 1984; Powley et al., 2009; Shenton et al., 2006), we suggest that this approach may be widely applicable.
RESULTS
In the first portion of this study, we directly compared ribosomal footprint profiling and pSILAC in MM1.S multiple myeloma cells treated with a low dose (0.5 nM) of bortezomib (Figure S1A). This dose is well below the half-maximal effective concentration (EC50 ~ 8 nM) for inhibition of proteasomal catalytic activity (Chauhan et al., 2005). We previously studied MM1.S response to a high dose of bortezomib (20 nM), but found that due to wides-pread mRNA degradation during rapid apoptosis we could not establish a quantitative relationship between ribosome footprint occupancy and protein synthesis (Wiita et al., 2013). The low dose of bortezomib used here is able to suppress cell growth over 48 hr, indicating some degree of drug-induced stress, but does not lead to appreciable apoptosis (Figures S1B–S1E).
At each of six time points over 48 hr after 0.5 nM bortezomib treatment, we harvested cells for mRNA sequencing (mRNA-seq), ribosome profiling, and quantitative proteomics (Figure S1A). mRNA-seq and ribosome profiling were performed as described previously (Wiita et al., 2013). For high-accuracy quantification of newly synthesized proteins, we designed quantitative selected reaction monitoring (SRM) assays (Picotti and Aebersold, 2012) measuring synthesis (“heavy” channel) and degradation (“light” channel) of 272 proteins in this cellular system. This analysis included monitoring at least two unique sequence peptides per protein, in technical duplicate, in both the light and heavy channels by SRM (Figure 1A). SRM data were normalized across time points using the total intensity (light + heavy channel intensity) of a panel of “housekeeping” proteins that remain unchanged at the transcript level (STAR Methods). While SRM has the advantage of consistent quantification of targeted peptides across all time points, a main drawback is the lower throughput compared with “shotgun” proteomic methods. Therefore, our analysis is necessarily limited to a subset of expressed proteins.
Figure 1. Direct Monitoring of Protein Synthesis by Targeted pSILAC Mass Spectrometry with Simultaneous Measurement of Transcript.

(A) Example time-course SRM data for peptides from PROF1 and DDX5. Red traces, “light” channel intensity (degraded from baseline); blue traces, “heavy” channel intensity (newly synthesized post-stable isotope pulse). Each trace represents added intensity of all monitored SRM transitions (four per peptide per channel). Inset: intensity values for each channel plotted over time; error bars reflect ±SD from replicate assays.
(B) Relative mRNA abundance and ribosome footprint read density (ratio versus 0 hr [0h], in RPKM) move together over the time course. Changes in protein abundance are not as prominent as transcript-level changes. SRM measurements of total protein here refers to the sum of all peptide intensities in light and heavy channels.
Overall Changes over the Time Course
We first compared the relative read density of transcripts identified by mRNA-seq and ribosome profiling across the time course to those found at baseline (untreated cells at 0 hr). We found a strong correlation between mRNA-seq and ribosome footprint density at the baseline and 6 hr time points (Figures S1F–S1H). As expected, we also found that relative ribosome footprint density generally moves in concert with relative transcript abundance (Figures S2A and S2B). The biological effects of low-dose proteasome inhibition were similar to those seen previously at high dose (Wiita et al., 2013), with prominent upregulation of proteasomal subunits and downregulation of ribosomal subunits (Data S2). However, this general correlation between mRNA abundance and ribosome footprint density does not capture dynamics in this system at the protein level. For example, when comparing the relative fold change to 0 hr of total abundance of the 272 proteins monitored by SRM across the time course with that of mRNA-seq and ribosome footprint read density on the corresponding transcript, we observed that while relative increases in mRNA indeed drove increases in protein abundance, most protein-level increases were less prominent than transcript-level increases (Figure 1B). Furthermore, downregulated transcripts did not lead to detectable decreases in protein abundance over 48 hr (Figure 1B). This finding is consistent with those of others (Jovanovic et al., 2015; Schwanhausser et al., 2011) suggesting that high-abundance proteins, as we primarily monitored here, typically have long half-lives. These half-lives may be further extended by partial blockade of proteasomal degradation by bortezomib treatment (Figure S4E).
To further investigate the quantitative relationship between these two orthogonal methods to measure protein synthesis, we compared the amount of protein synthesis inferred from ribosome profiling with that directly measured by SRM for individual proteins. For these comparative measurements we first required an estimate of the absolute copy number of newly synthesized proteins per cell. We therefore used the intensity Based Absolute Quantification (iBAQ) approach (Schwanhausser et al., 2011) to estimate baseline protein copy number per cell in untreated MM1.S cells. This estimate is based on measured peptide intensities in label-free shotgun proteomic analyses of biological duplicate samples (Figures S2F and S2G).
Compared with iBAQ-estimated absolute protein abundance across the MM1.S proteome, we found a stronger correlation with ribosome footprint read density (R = 0.76) than with mRNA-seq read density (R = 0.62) (Figures S1I and S1J), consistent with prior studies (Ingolia et al., 2009). To ensure that these baseline copy numbers were of the correct order of magnitude, we verified protein copy number per cell for three representative proteins, spanning the range of estimated copy numbers per cell (~105 to ~107) for the majority of proteins included in the SRM assay, using quantitative western blotting (Figures S2D and S2E). Using the heavy-channel SRM intensity, which represents newly synthesized proteins, and extrapolating from baseline protein copies per cell, we estimated the number of protein copies per cell synthesized between the 0 hr and 12 hr time points, when cellular protein synthesis appears largely unaffected by drug treatment (Figures S2H, S2I, and 3A). We compared these data with the average ribosome footprint density (in reads per kilobase million [RPKM]) across the 0, 6, and 12 hr time points (Figure 2A). We found a good correlation between ribosome footprint density and protein synthesis (Pearson’s R = 0.80 on log-transformed data). A linear best fit to these data on a log scale resulted in a slope of 0.97 (95% confidence interval 0.86–1.06). This strong correlation and linear fit with slope near unity in this eukaryotic system suggests that indirect measurement of synthesis via ribosome footprint occupancy for any gene indeed appears to quantitatively reflect absolute protein synthesis. However, the observed correlation is not perfect, requiring further exploration of potential causes of divergence between these two orthogonal measurements.
Figure 3. Biochemical Measurements of Protein Synthesis and Translational Efficiency Are Consistent with Targeted pSILAC and Allow for Simulations of Protein Synthesis Based on Experimental Data.

(A) Puromycin incorporation into nascent protein chains (1 hr pulse of puromycin added at each time point prior to cell harvest; western blot with anti-puromycin antibody) demonstrates decreased global protein synthesis at later time points.
(B) Simulation of protein abundance using the differential equation we proposed with the derived mRNA abundance Mg(t), degradation rate constant and translational rate parameter . At each sampling time τ we simulated the protein abundance after 1 hr with the initial conditions that the protein abundance at τ = 0 was 0. The heatmaps were normalized to the first column to show the relative abundance.
(C) Relative intensity in puromycin incorporation shows a slight increase at 6 hr and 12 hr and then a decrease in synthesis at 36 hr and 48 hr. The line represents fits using degree-3 orthogonal natural cubic splines with four knots and linearity constraints (see STAR Methods).
(D) The ratio of total ribosome footprint reads in each sample to the total number of ChrM-mapping reads, an independent measurement of global protein synthesis, also shows a very similar decrease across the time course. Here, the total number of ChrM-mapping reads is the sum of the individual genes in Figure S2C. The line represents the orthogonal natural cubic spline fits.
(E) The computationally estimated changes in global protein synthetic capacity Gr(t) (STAR Methods) demonstrates a very similar pattern to both the puromycin incorporation measurements (C) and the ratio of total ribosome footprint reads to the total number of ChrM-mapping reads (D).
(F) The total mRNA nucleotide abundance Gm(t) per cell is obtained from the biochemical data shown in Figure S1D. The line represents the orthogonal natural cubic spline fits.
(G) The ratio of the relative intensity in puromycin incorporation (C) to the total mRNA nucleotide abundance (F) is used to represent bulk translational efficiency from biochemistry experiments. This ratio shows a global translational repression over the time course. The line represents the orthogonal natural cubic spline fits.
(H) The average normalized translational rate parameter derived from pSILAC proteomics also shows a global translational repression, similar to the pattern observed for the independent biochemical measurements in (G). The line represents the orthogonal natural cubic spline fits.
Figure 2. Measuring and Modeling Translational Rate by pSILAC Proteomics and Ribosome Profiling.

(A) Early in the time course, before any biochemical evidence of translational inhibition (Figure S2), comparison of average ribosome footprint density on transcript coding sequence and newly synthesized proteins per cell, measured by SRM intensity and extrapolated based on iBAQ estimate of total protein copies per cell, show a strong correlation (Pearson’s R = 0.80 on log-transformed data). Red line is line of best fit, with slope = 0.97 (95% confidence interval 0.86–1.06). Molecules synthesized per cell by SRM represent “heavy” protein copies at 12 hr (12h) minus “heavy” copies at 0 hr (see STAR Methods); footprint data are average RPKM from the 6, 12, and 24 hr time points.
(B) Example plots from proteins EIF6 and YWHAE show proteomic data. Lines represent fits from model fitting based on orthogonal splines with linearity constraints (see STAR Methods). Solid lines, total protein; short dashed lines, degradation; long dashed lines, synthesis.
(C) Differential rate equation model describes protein abundance as a function of protein synthesis and degradation as measured from proteomic experiments and mRNA-seq. The primary free fitting parameter describes the number of proteins synthesized per transcript per unit time, is abundance of “light” proteins, is protein degradation rate constant derived from single-exponential fit to light channel data, Mg is absolute transcript abundance as derived from mRNA-seq data, and is abundance of newly synthesized “heavy” protein. The definition of translational efficiency from ribosome profiling literature is denoted by TE per gene: ribosome footprint read density rg(t) divided by mRNA-seq read density mg(t) (both in RPKM). The normalized translational efficiency is denoted by : ribosome footprint read density normalized to mitochondrial footprints Rg(t) divided by absolute transcript abundance Mg(t).
(D) Baseline comparison of TE and show that they are correlated, but imperfectly (Pearson’s R = 0.58 on log-transformed data).
(E–G) TE (E), (F), and plotted across all 272 proteins across the time course, as a ratio to the value at 0 hr (0h), shows no change in TE by standard analysis but similar changes in translational rate as measured by proteomics (F) and mitochondrial-corrected ribosome footprints (G).
To generate hypotheses as to the causes of this divergence, we turned to mathematical modeling. We explored the dynamics of protein degradation and production by using a system of differential equations. For each protein, we fitted the estimated number of “heavy” and “light” protein copies per cell, as well as the total protein abundance based on the addition of these two SRM intensities (Figure 2B), using orthogonal natural cubic splines with linearity constraints to obtain functional forms, denoted by , and , respectively (STAR Methods). This enabled us to describe changes of protein abundance ( and ) in terms of protein synthesis and degradation (Figure 2C). For each corresponding transcript we also monitored the mRNA-seq read density (Mortazavi et al., 2008) and ribosome footprint density in RPKM.
For estimating the degradation rate constant for each gene g, we found that a single-exponential fit well described protein degradation for the included proteins. Other proteomic and deep-sequencing data were fitted using the same approach described above (see STAR Methods). The primary gene-specific free parameter in this model is , the translation rate parameter at time t for gene g describing the number of protein molecules produced per transcript per unit time, which provides a proteomic-based measure of translational efficiency for each gene. Notably, our model allows us to determine changes in as a function of time, as needed after cellular perturbation, unlike in prior approaches describing a static term in steady-state cells (Schwanhausser et al., 2011).
With our model in hand, we directly compared with a measure of translational efficiency (TE) used in the ribosome profiling literature, where TE is defined as the ratio of the relative ribosome footprint read density to the relative mRNA-seq read density (Ingolia et al., 2009, 2011). Using standard ribosome profiling analysis methods (see STAR Methods), we observed little change in TE (Figure 2E) over time. This finding is in surprising contrast to changes found in as measured by proteomics, where comparisons to 0 hr indicate a reduction in proteins synthesized per transcript across the time course (Figure 2F).
We reasoned that our proteomic methods may be directly detecting global decreases in protein synthesis induced by bortezomib treatment not captured by standard ribosome profiling approaches. In agreement with this notion, polysome analysis by sucrose gradient centrifugation (Figure S2H), incorporation of puromycin into nascent proteins (Figure 3A), and dephosphorylation of the translation initiation factor eIF4E-binding protein 1 (4EBP1) (Figure S2I) all supported a diminishment of global translational capacity at time points after 12 hr, despite little decrease in global mRNA levels compared with baseline (Figure S1D). We also computed the ratio of bulk puromycin incorporation (Figure 3C) to the total mRNA nucleotide abundance, Gm(t), (Figure 3F) to represent bulk translational efficiency from biochemistry experiments (Figure 3G). This showed a response over time of decreasing translational capacity similar to that of the average normalized translational rate parameter, (Figure 3H).
The inability of standard ribosome profiling approaches to detect global changes in translational capacity has been previously recognized (Ingolia, 2016). To address this issue, we used a recently described method of normalization incorporating ribosome footprints mapping to mitochondria-encoded genes (ChrM), which are proposed to remain constant despite inhibition of cytosolic translation (Iwasaki et al., 2016). The normalized translational efficiency from ribosome profiling is critical for assessing global changes in translational capacity under conditions of cellular stress. This normalized TE, denoted by (see STAR Methods), demonstrates qualitative agreement between changes in global translational capacity as measured by both ribosome profiling and proteomics across the time course (Figures 2F and 2G). At 48 hr we measure a median ~40% decrease in translational efficiency of measured transcripts by both methods, albeit with greater variance in the proteomic measurement. However, we note that the normalization method above may be limited by the low number of ribosome footprint reads mapping to ChrM (Figure S2C), the long duration of low-dose bortezomib treatment, or other alterations in mitochondrial ribosome dynamics in this system. Therefore, while this comparison is supportive of our proteomic data, we cannot exclude the possibility that this concordance is coincidental. Importantly, however, the overall decrease in translational efficiency we measure by pSILAC also appears consistent with biochemical measurements indicating decreased global translational efficiency (Figures S2H, S2I, and 3). This finding highlights that monitoring protein synthesis by mass spectrometry can directly confirm global changes in translational capacity independent of ribosome profiling data.
Given the potential limitations of the mitochondrial footprint normalization approach, we also developed an algorithm to computationally estimate the changes in global protein synthetic capacity. A scaling function Gr(t) (common to all genes) was incorporated into the system of differential equations. This function Gr(t) was used to correct TE to reflect changes in the global synthetic capacity in the cell (blue curve in Figures 4A and 3E). We inferred a Gr(t) that optimizes a squared loss function based on proteomic data (STAR Methods). When our inferred Gr(t) was included in simulations of low-dose bortezomib treatment (Figure 3B), the resulting simulated protein synthesis dynamics (Figure 3E) were similar to those noted by puromycin incorporation (Figure 3A). Gr(t) further demonstrates a very similar pattern to both the puromycin incorporation measurements (Figure 3C) and the ratio of total ribosome footprint reads to the total number of ChrM-mapping reads (Figure 3D).
Figure 4. A Dynamic Model Predicts Absolute Protein Synthesis under Conditions of Cellular Stress.

(A) Simulation of absolute protein copies synthesized as a function of different levels of translational capacity (denoted by Gr(t)) in MM1.S, with inputs of iBAQ protein copy number, ribosome footprint density, mRNA-seq, β, and from MM1.S data. Three conditions are considered: Gr(t) being constant, as in untreated cells (black); Gr(t) varying as found in low-dose bortezomib (btz)-treated MM1.S (blue); and Gr(t) decreasing toward zero, as in high-dose bortezomib-treated MM1.S (green).
(B) The simulated heavy channel protein abundance normalized by the heavy channel protein abundance at 48 hr under constant Gr(t), denoted by in MM1.S. Under high-dose btz, protein synthesis is strongly curtailed, consistent with Wiita et al. (2013).
(C) The simulated total protein abundance normalized by the total protein abundance at 48 hr under constant Gr(t), denoted by . High-dose btz again shows a significant decrease in total protein abundance.
(D) Untreated Epstein-Barr virus-immortalized B cells also show a strong correlation (Pearson’s R = 0.84 on log-transformed data) between absolute protein synthesis and footprint RPKM as in Figure 2A. Red line is line of best fit, with slope = 0.93 (95% confidence interval 0.84–1.02).
(E) SRM pSILAC-measured absolute protein copy number synthesis per B cell (left) compared with simulated estimates of total absolute proteins synthesized in untreated B-cells over 48 hr using quantitative model (right), with inputs of iBAQ protein copy number, ribosome footprint density, and mRNA-seq measured in B cells and β and from MM1.S data, show good agreement.
As a further comparison between ribosome profiling and pSILAC proteomics, we also investigated the correlation at baseline between translational efficiency and across proteins (Figure 2D). We first estimated a multiplicative constant, β that, when applied to all genes, largely reconciles the TE measured from ribosome profiling with estimated from proteomic experiments (STAR Methods). A major question is whether the discrepancy of the fit between TE and for different genes represents real biology (i.e., gene-specific translational regulation at the post-translational level, only detectable by proteomics) or systematic biases in one or both methods. Notably, our quantitative model relies on absolute protein copy-number estimates from the iBAQ method (Schwanhausser et al., 2011). While the proteins included in our targeted SRM assay on MM1.S showed high reproducibility by iBAQ (Figure S2G), simulations suggest that even this limited iBAQ replicate error could account for over one-third of the residual variance of the correlation presented in Figure 2A (Figure S3B). To evaluate further potential sources of error, we examined whether accounting for annotated transcript isoforms could improve the correlation between footprint and proteomic data, but found only minor improvements (Figure S3D).
Extensions of the Model: Quantitative Predictions of Protein Synthesis
One direct application of our model is predicting absolute protein synthesis and abundance under different global levels of translational inhibition. Using our estimated values of in combination with baseline mRNA-seq and ribosome profiling data, we predicted proteome remodeling under three different scenarios of Gr(t) in MM1.S cells (Figure 4A): untreated cells, low-dose bortezomib, and high-dose bortezomib. Our model predicted significantly reduced absolute protein synthesis under conditions of strong translation inhibition by high-dose bortezomib (Figures 4B and 4C), consistent with that found in our prior study (Wiita et al., 2013).
The model can also be used to define protein synthesis rates in other systems. For example, we obtained a similar dataset of pSILAC proteomics paired with baseline mRNA-seq and ribosome profiling in untreated Epstein-Barr virus (EBV)-immortalized B cells (Figures 4D and S4). Importantly, the relationship between absolute protein synthesis and footprint RPKM (Figure 4D) is also strong in this setting (Pearson’s R = 0.84 on log-transformed data). A linear fit to these data on a log scale results in a slope of 0.93 (95% confidence interval 0.84–1.02). Again, this strong correlation and linear relationship indicates that ribosome footprint occupancy is quantitatively reflective of absolute protein synthesis in a different cell type and without any drug perturbation.
Using our mathematical model, we predicted the “heavy” (newly synthesized) protein copy number in these B cells at later times (measured using SRM), using inputs of iBAQ protein copy number, ribosome footprint density, and mRNA-seq measured in untreated B cells at baseline, and β and estimated from MM1.S data (Figure 4E, right). In our prediction model, we assumed that ribosome footprint read density, mRNA-seq read density, Gm(t), and Gr(t) remain constant over time in this untreated scenario. Indeed, the values predicted by our model were similar to the experimental pSILAC measurements of heavy protein copy number (Figure 4E, left). Our results from Figures 2A and 4D further support the notion that ribosome profiling data, in combination with biochemical knowledge of global translational inhibition, may be sufficient to predict changes in the proteome using our quantitative model.
DISCUSSION
Here, we directly measured protein synthesis and ribosome footprint density in the setting of cancer therapy. Ribosome profiling has become a widespread technique to assess translational regulation and protein synthesis. One important question about this technique, however, is whether the resulting data truly reflect protein synthesis and translational rate. A recent study in Escherichia coli demonstrated that when compared with previously published absolute copy numbers per cell, extrapolated synthesis rates based on ribosome footprint density correlated very well (R = 0.98) (Li et al., 2014a). In our work, we also find a strong positive correlation between ribosome footprint density and absolute protein synthesis as measured by targeted time-resolved pSILAC (Figures 2A [R = 0.80] and 4D [R = 0.84]), supporting the notion that ribosome footprint density, as measured by ribosome profiling, is directly reflective of absolute protein synthesis, even in the more complex translational system of eukaryotes (Jackson et al., 2010; Kozak, 1999).
Others have compared the capture and analysis of nascently translated proteins by mass spectrometry with ribosome profiling data and have found weaker correlations (R = 0.66) (Zur et al., 2016). However, this “Punch-P” approach has significant disadvantages as an orthogonal quantitative validation of ribosome profiling data as it relies on incorporation of a chain-terminating puromycin analog for enrichment. Such truncated polypeptides will likely be rapidly degraded, skewing abundances in the captured cohort. Furthermore, enrichment-based methods suffer from biases in differential protein capture on streptavidin beads and artifacts from non-specific binding. These limitations make it difficult to quantitatively compare ribosome profiling with Punch-P. In contrast, the pSILAC approach we take here, combined with high-accuracy targeted quantification, allows us to directly measure protein synthesis in a complex system in an unbiased fashion.
With these data, we find that noise in baseline absolute protein abundance using the iBAQ method (Li et al., 2014b; Wilhelm et al., 2014) strongly affects the correlation between proteomic and ribosome profiling data. Other sources of error in our comparison that remain to be investigated may relate to ribosome footprint sample preparation methods (Weinberg et al., 2016) or splice isoform-specific translational control (Floor and Doudna, 2016). Due to these limitations, we cannot exclude the possibility that for some genes there is a divergence between ribosome footprint occupancy and true protein synthesis, despite their overall strong correlation across the monitored genes.
The quantitative model we develop also allows us to determine a measure of translational efficiency ( ) using proteomic data and compare this with translational efficiency as measured by ribosome profiling. Our results, with a Pearson’s R = 0.58 on log-transformed data at baseline (Figure 2D), are in line with that of a recent extensive time-course study of protein synthesis in murine dendritic cells, in which Jovanovic et al. (2015) performed ribosome profiling at the baseline time point alone and also found a similar correlation (R = 0.5) between TE from ribosome profiling and measured from shotgun proteomic data. Given our findings (Figures 2E–2G), it appears that while proteomics may be able to broadly detect global changes in translational capacity, ribosome profiling may be more sensitive in determining translational efficiency changes for individual genes.
In the context of cancer therapy, our results here underscore that standard measurements of ribosome footprint density may not reflect absolute protein synthesis when global changes in translational capacity (i.e., the number of actively translating ribosomes) are present, whereas proteomics can more directly detect these changes (Figures 2E and 2F). pSILAC combined with targeted mass spectrometry may therefore be an important method to orthogonally validate quantitative changes in translational rate found by normalization of ribosome profiling data under conditions of cellular stress (Andreev et al., 2015; Ingolia, 2016).
Furthermore, we develop a new quantitative model that can capture and predict dynamic changes in protein synthesis during cancer therapy. As we find a linear relationship between ribosome footprint density and absolute protein synthesis across genes, we suggest that with inputs of untreated mRNA-seq, ribosome profiling, and absolute protein abundance estimates, in conjunction with biochemical data to describe the degree of translational inhibition, our model will provide a new window to predict the remodeling of the cancer proteome in response to therapeutic perturbation, even in the absence of a full pSILAC dataset. We suggest that this quantitative framework can readily be applied to any human cellular system exposed to cellular stress impinging on the translational machinery.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-GAPDH | Cell Signaling Technology | Cat# 2118L; RRID: AB_561053 |
| Rabbit monoclonal anti-vimentin | Cell Signaling Technology | Cat# 5741P; RRID: AB_10695459 |
| Rabbit polyclonal anti-Bid | Cell Signaling Technology | Cat# 2002P; RRID: AB_10830065 |
| Mouse monoclonal anit-puromycin | KeraFast | Cat# 3RH11 |
| Rabbit monoclonal anti-4E-BP1 | Cell Signaling Technology | Cat# 53H11; RRID: AB_10691384 |
| Rabbit monoclonal anti-phospho-4E-BP1 (T37/46) | Cell Signaling Technology | Cat# 236B4; RRID: AB_10695878 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Bortezomib | LC Laboratories | B-1408 |
| Cycloheximide | Sigma-Aldrich | Cat# C4859; CAS: 66-81-9 |
| L-Lysine 4,4,5,5-D4 (Lys4) | Cambridge Isotope Laboratories | Cat# DLM-2640 |
| L-Arginine 13C6 (Arg6) | Cambridge Isotope Laboratories | Cat# CLM-2265-H |
| L-Lysine 13C615N2 (Lys8) | Cambridge Isotope Laboratories | Cat# CNLM-291-H |
| L-Lysine 13C615N4 (Arg10) | Cambridge Isotope Laboratories | Cat# CNLM-593-H |
| L-Lysine | Sigma-Aldrich | Cat# 8662; CAS: 657-27-2 |
| L-Arginine | Sigma-Aldrich | Cat# A6969; CAS: 1119-34-2 |
| TRIzol® reagent | Life Technologies | Cat# 15596026 |
| HALT protease and phosphatase inhibitor single use cocktail | Thermo Fisher | Cat# 78443 |
| Sequencing Grade Modified Trypsin | Promega | Cat# V5111 |
| GAPDH recombinant protein | Abcam | Cat# ab82633 |
| Vimentin recombinant protein | PeptroTech | Cat# 110-10 |
| Bid recombinant protein | Sino Biological | Cat# 10468-HNCE-59 |
| Critical Commercial Assays | ||
| Rneasy mini kit | QIAgen | Cat# 74104 |
| Quantifluor RNA assay | Promega | Cat# E3310 |
| Oligo(dT)25 Magnetic Beads kit | New England BioLabs | Cat# S1419S |
| Pierce™ BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| SepPak C18 columns | Waters | Cat# WAT020515 |
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat# G7570 |
| Caspase-Glo® 3/7 Assay Systems | Promega | Cat# G8090 |
| Invitrogen MyOne streptavidin C1 dynabeads | Thermo Fisher | Cat# 65001 |
| Deposited Data | ||
| Raw genetic sequencing data | This paper | http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=chohoqacvbyffwf&acc=GSE69047 |
| Raw SRM data | This paper | https://panoramaweb.org/labkey/translation_model_2015.url |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| HMCL66_Transcript_Expression_FPKM | Keats Lab (Translational Genomics Research Institute, USA) | http://www.keatslab.org/data-repository |
| Experimental Models: Cell Lines | ||
| MM.1S | ATCC | Cat# CRL-2974 |
| EBV-transformed B-cell | Markus Muschen (University of California San Francisco, USA) | N/A |
| Sequence-Based Reagents | ||
| miRNA Cloning Linker 1 DNA oligo: 5′ AppCTGTAGGCACCATCAAT/3ddC 3′ | IDT | N/A |
| rRNA subtraction DNA oligo oNTI309: (biotin)-TCCTCCCGGGGCTACGCCTGTCTGAGCGTCGCT | IDT | N/A |
| rRNA subtraction DNA oligo oNTI301r: (biotin)-GGGCCTCGATCAGAAGGACTTGGGCCCCCCACGA | IDT | N/A |
| rRNA subtraction DNA oligo oNTI305r: (biotin)-GGCGAGACGGGCCGGTGGTGCGCCCTCGGCGGA | IDT | N/A |
| rRNA subtraction DNA oligo oNTI307hr: (biotin)-GCGGGGGACCGGCTATCCGAGGCCAACCGAGGCTC | IDT | N/A |
| rRNA subtraction DNA oligo oNTI298r: (biotin)-TGATCTGATAAATGCACGCATCCCCCC | IDT | N/A |
| rRNA subtraction DNA oligo oNTI303hr: (biotin)-CGCGCCGTGGGAGGGGTGGCCCGGCCCC | IDT | N/A |
| Rev Transcription DNA oligo oNTI225-Link1 (DNA): 5′/5Phos/GATCGTCGGACTGTAGAACTCTGAACCTGTCGGTGGTCGCCGTATCATT/iSp18/CACTCA/iSp18/CAAGCAGAAGACGGCATACGAATTGATGGTGCCTACAG 3′ | IDT | N/A |
| Amplification DNA primer oNTI230 (DNA): 5′-AATGATACGGCGACCACCGA | IDT | N/A |
| Amplification DNA primer oNTI231 (DNA): 5′-CAAGCAGAAGACGGCATACGA | IDT | N/A |
| Software and Algorithms | ||
| MaxQuant v1.5.1.2 | Cox and Mann, 2008 | http://www.biochem.mpg.de/5111795/maxquant |
| Skyline v2.5 | MacLean et al., 2010 | https://skyline.ms/project/home/software/Skyline/begin.view |
| Ribomap | Wang et al., 2016 | https://github.com/Kingsford-Group/ribomap |
| Star v.2.4.0j | Dobin et al., 2012 | https://github.com/alexdobin/STAR/releases/tag/STAR_2.4.0j |
| Cluster 3.0 | de Hoon et al., 2004 | http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm |
| Java TreeView | Saldanha, 2004 | https://sourcefourge.net/projects/jtreeview/ |
| DAVID Bioinformatics Resource | Huang et al., 2009 | http://david.ncifcrf.gov |
| MATLAB R2013a | Mathworks | http://www.mathworks.com/products/matlab/ |
| MATLAB spline fit script | This paper | https://sourceforge.net/projects/ncspline/ |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and request for reagents may be directed to and will be fulfilled by Lead Contact Arun Wiita (arun.wiita@ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
MM1.S cells were acquired from ATCC with cell line identity and female gender confirmed by karyotyping and DNA microarray. EBV-immortalized human B-cells derived from normal donor cord blood were a kind gift of Dr. Markus Müschen (UCSF Dept. of Laboratory Medicine) with normal female diploid genome confirmed by karyotyping.
METHOD DETAILS
Cell Culture and Drug Treatment
MM1.S cells were grown in suspension to 1×106 cells/ml in RPMI-1640 media with 10% FBS. EBV-immortalized B-cells were grown in suspension to 1 × 106 cells/mL in RPMI-1640 media with 20% FBS. For initial MM1.S experiments (Figure S1), bortezomib (LC Laboratories, Woburn, MA, USA) 20 μM stock solution in sterile-filtered phosphate buffered saline (PBS) was simultaneously added to a final concentration of 0.5 nM to flasks each containing 90 × 106 cells (PBS only added to control sample). At the indicated time point cells were separated into aliquots for each experimental approach (15×106 cells induplicate for each of mRNAseq, proteomics, and ribosome profiling). Cells for ribosome profiling alone were incubated at 37°C for 1 min with 100 μg/mL cycloheximide (Sigma-Aldrich, St. Louis, MO, USA). All cells were pelleted by centrifugation, washed in PBS (PBS + 100 μg/ml cycloheximide for ribosome footprint samples), pelleted by centrifugation again, and flash frozen in liquid N2, then stored at −80°C. Cell viability and caspase activity were assessed by Cell-Titer Glo and Caspase-Glo (Promega, Madison, WI, USA) assays per manufacturer protocol, respectively. For pulsed-SILAC experiments, MM1.S cells were grown for 6 cell doublings in SILAC RPMI media depleted of arginine and lysine (Thermo Fisher Scientific, Waltham, MA, USA), dialyzed FBS (Life Technologies, Carlsbad, CA, USA) supplemented with unlabeled L-lysine (70 mg/L) and L-arginine (40mg/L) (Sigma-Aldrich). EBV-immortalized B-cells were similarly grown for 6 cell doublings in SILAC RPMI media supplemented with “medium” (4,4,5,5-D4) lysine and 13C6 arginine. For both cell lines, at time 0h, 100 × 106 cells at a density of 1 × 106 cells/ml were pelleted by centrifugation and resuspended in SILAC RPMI media supplemented with “heavy” 13C6-15N2 lysine (70 mg/L) and 13C6-15N4 arginine (40 mg/L) (Cambridge Isotope Laboratories, Andover, MA, USA). Cells were harvested at the indicated time points, washed once in PBS, and stored at −80°C until sample analysis. For MM1.S cells, ribosome profiling, mRNA-seq, and proteomics were performed at each time point. For B-cell analysis, proteomics were performed at each time point, while ribosome profiling and mRNA-seq analysis were performed in biological duplicate on the baseline sample alone as the cells are not perturbed during the time course.
Measurement of Total RNA, mRNA, and Protein
Total RNA was extracted either by Trizol (Life Technologies) per manufacturer protocol or using QIAgen RNeasy kit (QIAgen, Germantown, MD, USA). mRNA was further purified from isolated total RNA by poly(A) separation using Oligo (dT)25 Magnetic Beads kit (New England BioLabs, Ipswich, MA, USA) per manufacturer protocol. Total RNA and mRNA concentration was measured either by NanoDrop ND-1000 UV-Vis spectrophotometer (Thermo Fisher) or QuantiFluor RNA assay (Promega, Santa Clara, CA, USA). Total protein was isolated by lysis in either 8M Urea buffer for proteomics (see below) or RIPA buffer (EMD Millipore, Billerica, MA, USA) for immunoblotting and concentration measured using BCA assay (Thermo Fisher Scientific).
Ribosome Profiling and mRNA-Seq
Ribosome profiling and mRNA-seq samples were prepared and analyzed as in our prior study (Wiita et al., 2013). Briefly, harvested cell pellets for ribosome profiling were suspended and lysed in 500 μl ice-cold polysome lysis buffer (20 mM Tris, pH 7.4, 250 mM NaCl, 15 mM MgCl2, 1 mM dithiothreitol, 0.5% Triton X-100, 24 U/ml Turbo DNase (Ambion, Austin, TX, USA), and 100 μg/ml cycloheximide). Lysate was clarified by centrifugation and RNaseI 100 U/μl (Ambion) was added to digest polysomes to monosomes. Digested samples were then loaded onto a 1 M sucrose cushion and pelleted by centrifugation for 4 hr at 70,000 rpm. The pellet was resuspended in Trizol and RNA isolated per manufacturer protocol. RNA was separated by gel electrophoresis on a 15% TBE-Urea gel (Life Technologies) and gel fragments extracted corresponding to ~25–35 nt in size. RNA was extracted from gel as in (Ingolia et al., 2011), by disrupting gel slices with centrifugation through a needle hole between 0.5 mL microfuge tube nested in a 1.5 mL microfuge tube. The gel was extracted in RNase-free water for 10 min at 70°C. The eluate was recovered by loading slurry onto a Spin-X column (Corning 8160) and centrifuging to recover eluate in collection tube. RNA was then precipitated from the filtered eluate by adding sodium acetate to a final concentration of 300 mM as a coprecipitant, followed by at least one volume of isopropanol. Precipitation occurred at −20°C overnight, RNA was pelleted and centrifuged for 45 min at 20,000 × g, 4°C. The supernatant was discarded and the RNA pellet was air dried, then resuspended in 10 μl 10 mM Tris (pH 7.0).
Harvested cell pellets for mRNA-seq were isolated by Trizol and total RNA isolated per manufacturer protocol. Poly(A) mRNA was purified from the total RNA sample using poly-dT magnetic beads (as above) per manufacturer protocol. mRNA was fragmented in high pH buffer (50 mM NaCO3, pH 9.2) for 20 min at 95°C, then precipitated and separated by gel electrophoresis as above. mRNA fragments of 50–90 nt were extracted.
Both poly(A)-selected and ribosome footprint size-selected RNA samples were dephosphorylated, ligated to linker, and separated by gel electrophoresis as described previously (Ingolia et al., 2011). RNA was dephosphorylated with T4 DNA polynucleotide kinase (NEB M0201S), by resuspending RNA in 25ul 10mM Tris (pH 8.0), denaturing the fragments for 2 min at 75°C then equilibrating at 37°C and brought to a volume of 50 μl in 1X T4 polynucleotide kinase reaction buffer with 25 U T4 polynucleotide kinase (NEB M0201S) and 12.5 U Superasein (Thermo Fisher AM2694). This dephosyporylation reaction was incubated for 1 hr at 37°C and enzyme heat inactivated at 70°C for 10 min, then purified by precipitation as described above. Linker was ligated in a 20 μl reaction with dephosphorylated RNA, 12.5% w/v PEG 8000, 10% DMSO, 1X T4 RNA Ligase 2, truncated (NEB M0242L) reaction buffer, 20 U Superasein, 500 ng preadenylated miRNA cloning linker 1 (IDT), 200 U T4 RNA Ligase 2 (tr). This ligation was incubated at 37°C for 2.5 hr and the products were separated by gel electrophoresis and extraction as described above. Reverse transcription and cDNA library preparation were completed as in (Ingolia et al., 2011). Reverse transcription was carried out by preparing a reaction with RNA in 18 μl SuperScriptIII (Thermo Fisher 18080044) and 50 pmol oNTI-225 link1 primer (see Key Resources Table). Reactions were denatured for 5 min at 65°C, equilibrated at 48°C with 2.0μl 1N NaOH and incubating 20 min at 98°C. Products were purified by gel electrophoresis and extracted as described above. Reverse transcription products were circularized with 20 μl CircLigase (Epicentre CL4111K) reaction. After circularization, subtraction of rRNA sequences was performed by subtractive hybridization using biotinylated oligos that reverse complement overabundant rRNA contaminants (see Key Resources Table: oNTI309, 301r, 305r, 397hr, 298r, 303hr), by being suspended in 30 μl 2X SSC with 250 pmol total biotinylated subtraction oligos. The sample was denatured for 2 min at 70°C and transferred to 37°C. Hybridization was incubated for 30 min at 37°C. Biotinylated oligos were removed by MyOne streptavidin C1 dynabeads (see Key Resources Table), using 1mg magnetic beads. The rRNA-subtracted, circularized cDNA was used as a template for PCR amplication (see Key Resources Table for amplification primers) using Phusion polymerase (NEB M0530S). Reaction products were then separated by gel electrophoresis as described above and DNA extracted using same procedure as above, with NaCl substituted as a co-precipitant. Extracted DNA was resuspended in 10 μl 10 mM Tris (pH 8.0) and expected library size verified using an Agilent Bioanalyzer 2100.
Sequencing was performed on an Illumina HiSeq 2500 using single end, 50-bp reads at the UCSF Center for Advanced Technology. Before alignment, linker sequences were computationally removed from the 3′ ends of raw sequencing reads. STAR_2.4.0j was used to perform the alignments with up to one mismatch allowed. For footprint data only reads of length 25–36 nt (footprint length with cycloheximide (Ingolia et al., 2011)) were used for alignment. Reads were first aligned vs mitochondrially-translated genes; aligned reads were filtered. Next, all remaining reads were aligned vs human non-coding RNA and tRNA sequences; aligned reads were discarded. Finally remaining reads were aligned to human transcriptome reference (downloaded from http://www.gencodegenes.org/18.html Sep 2016) on reference genome GRCh37.
Software Ribomap (Wang et al., 2016) was used with default settings to assign multi-mapped reads to isoforms according to the mRNA abundance of each isoform. mRNA and footprint read density were calculated in units of reads per kilobase million (RPKM) to normalize for gene length and total reads per sequencing run.
Unsupervised hierarchical clustering was performed using complete linkage across mRNA, footprint, and translational efficiency data with uncentered correlation in Cluster 3.0 and visualized in TreeView. Where indicated, gene lists were analyzed by NIH DAVID resource (Huang et al., 2009) using default settings for included genes and interaction networks and “human” selected as species.
The RPKM of genes monitored by ribosome profiling and mRNA seq and the DAVID analysis results are available in Data S2.
Selected Reaction Monitoring Proteomics
Frozen cell pellets were lysed by probe-tip sonication in buffer containing 8M Urea, 50 mM NaCl, and 100 mM Tris pH 8.0 supplemented with 1x HALT protease and phosphatase inhibitor cocktail (Thermo Fisher). Lysates were cleared by centrifugation at 16,500 × g for 10 min and protein concentration measured using the BCA assay. Lysate containing ~500 ug protein was diluted to 200 uL with lysis buffer. Disulfide bonds were reduced with 5 mM dithiothreitol and cysteines alkylated with 10 mM iodoacetimide. Lysate was diluted 1:6 with trypsin dilution buffer (100 mM Tris pH 8.0, 1 mM CaCl2, 75 mM NaCl). Sequencing grade modified trypsin (see Key Resources Table) was added at an enzyme:substrate ratio of 1:25. Proteins were trypsin digested overnight with agitation at room temperature. Samples were adjusted to pH<3 with trifluoracetic acid and precipitate removed by centrifugation at 16,500 × g for 10 min. Tryptic peptides were desalted on SepPak C18 columns (Waters, Milford, MA, USA), evaporated to dryness on a vacuum concentrator, and stored at −80°C. For mass spectrometry analysis, peptides were resuspended in 0.1%formic acid to a final concentration of ~0.2 μg/μL.
We previously developed targeted, label-free Selected Reaction Monitoring (SRM) assays for 152 proteins in our prior work in MM1.s myeloma cells treated with 20 nM bortezomib (Wiita et al., 2013). We applied this same assay to our samples as shown in Figure S1 (SRM transitions and intensities available in Data S1). Here we further developed new SRM assays to measure relative protein quantification in both the light and heavy SILAC channels (as in Figure 1). For method development, data-dependent (or “shotgun”) proteomic data acquired on an LTQ Orbitrap Velos (Thermo Fisher) in HCD mode in our prior study was imported into the open-source software Skyline (v. 2.5) (MacLean et al., 2010) to build targeted assays consisting of parent ion and fragment ion “transitions” based on MS2 sequencing data. For heavy channel analysis, we used calculated increases in m/z of primarily y-ion fragments to develop targeted methods. Two to four peptides per protein were targeted in initial method development. Proteins for analysis were primarily chosen based on high MS2 fragment intensity in “shotgun” data. Peptides were chosen having unique sequence identity for the targeted protein based on canonical sequence in Uniprot database.
All SRM analysis was carried out on a QTRAP 5500 (SCIEX, Framingham, MA) triple quadrupole mass spectrometer interfaced inline with a nanoAcquity UPLC system (Waters) identical to that on the LTQ Orbitrap Velos (Thermo Fisher) on which the spectral library was acquired (Analytical column: BEH130 (0.075×200 mm column, 1.7 μm; Waters)). LC buffer A = 0.1% formic acid in water; buffer B = 0.1% formic acid in acetonitrile. We injected ~1 μg of tryptic peptides from MM1.S cells onto the mass spectrometer with the following conditions: Direct sample loading at 3% B for 10 min after injection, a linear gradient from 3%–35% B over 80 min, an increase to 90% B over 5 min, then held for 5 min, then a decrease to 3% B for 10 min (total run time 110 min). Unit resolution was used at Q1 and Q3. A three second duty cycle time was used for all runs. For unscheduled runs a 10 ms acquisition time was used per transition. Multiple injections were used to test for all targeted peptides. Using data analysis in Skyline software, peptides were selected for further method development based on 1) the signal detection (above baseline) of at least 5 of 7 co-eluting transitions in both the light (0h sample used for method development) and heavy (48h sample used for method development) channels; 2) a retention time within 7 min of that acquired in the initial spectral library (acquired under the same chromatographic conditions); 3) fragment ion intensity of similar rank to that found in the initial spectral library.
Peptides chosen for further development were then limited to the four most intense transitions in both the light and heavy channels as found in unscheduled runs. A scheduled SRM method was developed with a retention time window of ±5 min. We then applied this scheduled method across multiple injections, with a minimum scan time per transition of 10 ms, at each time point, in technical duplicate. We ultimately chose to include in our pulsed SILAC analysis only those peptides which demonstrated detectable SRM intensity above background and at a consistent LC retention time at all time points in both the light and heavy channels (with the exception of heavy channel at 0h, where we expect to detect only background signal). Therefore, we ultimately included 733 peptides from 272 proteins in the analysis here for analysis of MM1.S cells. We applied this same method to EBV-immortalized B-cells and found that 165 of these proteins demonstrated sufficient signal-to-noise for analysis, likely based on differential protein expression between the two cell lines; these 165 proteins were used for all B-cell analysis.
Peptide intensity in each sample was measured as the sum of all transition peak areas for that peptide in each of the light and heavy channels (as measured by analysis in Skyline). Total peptide intensity was measured as the sum of the light and heavy channel intensities, and total protein intensity was measured as the sum of total intensities for all peptides from that protein. To normalize peptide concentration across samples, we used peptides derived from a set of high abundance proteins not expected to significantly change during the time course based on transcript-level data. We derived an index based on the geometric mean total intensity of peptides from these ‘housekeeping’ proteins (ENO1, KPYM, PPIA, FLNA, ACTB, TUBA1B) and scaled SRM intensity of all peptides in each channel based on the median value of this index. Corrected peptide intensity was averaged across injections for each sample.
Western Blots
For quantitative Western blots, 10 × 106 untreated MM1.S cells were counted by taking the average of measurements from both manual hemocytmeter and automated cell counting using a Sceptre instrument (EMD Millipore). Cells were pelleted, washed 1x in cold PBS, and lysed in RIPA buffer (EMD Millipore) supplemented with 1x HALT protease and phosphatase inhibitors (Thermo Fisher) by probe-tip sonification. Protein concentration in lysate was measured by BCA assay. Recombinant proteins (GAPDH, Abcam; Vimentin, PeproTech; Bid, Sino Biological, see Key Resources Table) at the manufacturer’s indicated concentration were used to generate a standard curve. Lysate and recombinant protein were separated on Mini-PROTEAN any Kd TGXgels (Bio-Rad, Hercules, CA, USA) and transferred to 0.45 μm PVDF membrane (EMD Millipore). Membranes were blocked using Odyssey blocking buffer (LI-COR, Lincoln, NE, USA) and probed with anti-GAPDH rabbit monoclonal antibody, anti-vimentin rabbit monoclonal, and anti-Bid rabbit polyclonal (Cell Signaling Technology, see Key Resources Table) diluted at 1:1000 in Odyssey blocking buffer. Membranes were washed and blotted with infrared reporter-conjugated secondary antibodies and imaged on a LI-COR Odyssey system. LI-COR Image Studio software was used to quantify standard curve and lysate band intensity. Western blots for phospho- and total 4EBP-1were performed as previously described with identical reagents (Wiita et al., 2013, see Key Resources Table). All blots were completed in biological duplicate.
Puromycin Incorporation
The bortezomib treatment time course was performed in biological duplicate. One hour prior to each time point, 1 μM puromycin (Sigma-Aldrich) was added to 4.5 × 106 cells at 1.0 × 106 cells/mL. Cells from were allowed to incorporate puromycin for one hour, pelleted, washed in PBS, and lysed in RIPA buffer as above. 25 μg of lysate was separated by gel electrophoresis and transferred to PVDF membrane as above. Membranes were probed with a mouse anti-puromycin monoclonal antibody (KeraFast, see Key Resources Table) and imaged on LI-COR Odyssey system.
Sucrose Density Gradient
10 × 106 MM1.S cells at each time point were lysed in 500 μL buffer containing 20 mM Tris pH 7.5, 50 mM NaCl, 5 mM MgCl2, 30% glycerol, 1% Triton X-100, 20 U/mL SuperASEin (Ambion), 1 mM DTT, and 0.1 mg/mL cycloheximide. Lysate was loaded over a 10%–50% sucrose gradient, centrifuged at 35,000 × g for 3 hrs, and polysomes analyzed using a Density Gradient Fractionation System, model 160 (Teledyne Isco, Lincoln, NE) with absorbance measured at 254 nm and recorded with Logger Pro 3.6.1 software (Vernier, Beaverton, OR).
Estimation of Absolute Protein Copy Number at Baseline
We previously used the iBAQ method (first described in (Schwanhausser et al., 2011)) implemented in MaxQuant (Cox and Mann, 2008) to estimate protein copy number per cell at baseline in MM1.S cells. Here we identically analyzed unlabeled trypic peptides from a biological replicate of untreated MM1.S cells on an LTQ Orbitrap Velos mass spectrometer (Wiita et al., 2013). iBAQ values are available in Data S3. We estimated protein copy number per cell ρg based on the ion current assigned to each protein group iBAQg and scaled by a constant σ, such that ρg = σ[iBAQg]. By incorporating the molecular mass μg of each protein, the total mass of protein per cell μtotal, and , we derived the scaling constant σ, and thereby estimated the protein copy number per cell ρg. For incorporation into the quantitative model, we used the mean of the absolute protein copy number per cell from two iBAQ replicate analysis, denoted as ρ̄g, as the “0h” value for copies per cell.
This baseline quantity is related to the background corrected SRM intensity by , in which is the background corrected intensity of the light SRM channel, is the background corrected intensity of the heavy channel, and ag is a gene specific constant. The background correction was conducted by subtracting the signal intensity in the heavy channel at 0h from all measurements, since no labeling had occurred, represented as and . We extracted ag using the background corrected SRM intensity and baseline absolute protein copy number per cell, and estimated protein copy number per cell at later time points in the heavy channel and light channel by and .
Estimation of Absolute mRNA Copy Number
We estimated mRNA copy number per cell Mg,i(t) from mRNA-seq data using a method proposed previously (Schwanhausser et al., 2011; Wiita et al., 2013). Let γg,i(t) represent the number of sequencing reads mapped to the transcript of gene g, isoform i, where i ∈ {1, …, Ig} indexes the Ig isoforms of gene g, lg,i represent the transcript length, and T (6.40 × 10−16 mol/cell) represent the total number of mRNA nucleotides per cell at baseline. Also let Gm(t) represent the relative total mRNA nucleotide abundance at time t, normalized so that Gm(0) = 1. Gm(t) is obtained by fitting the experimental data, shown in Figure S1D (see the section Measurement of total RNA, mRNA, and protein) using degree-3 orthogonal natural cubic splines with 4 knots and linearity constraints (see the section Exponential and orthogonal natural cubic spline fitting). The resulting function is shown in Figure 3F.
These quantities can be related to the absolute mRNA copy number,
Note that, up to a scale factor, the right hand side is the definition of relative mRNA abundance of gene g, isoform i (in RPKM), denoted by mg,i(t). Therefore, Mg,i(t) = c mg,i(t)Gm(t), where c is a constant. In what follows, we define , and .
Statistical Analysis of Experimental Data
In this study there was no blinding of any experimental data. SRM analyses were performed using random-block injections to prevent artifacts from carryover. There was no sample-size estimation used. All error bars for experimental data represent +/− S.D. for the number of replicates indicated in the Figure Legend. All correlations are performed using Pearson R on log-transformed data.
QUANTIFICATION AND STATISTICAL MODEL
Here we describe our quantitative model of protein synthesis dynamics and our inference procedure for parameter estimation. The corresponding data can be found in Data S4.
System of Differential Equations
We propose a system of differential equations to model the integrated longitudinal data of ribosome profiling, mRNA-seq, and pSILAC mass spectrometry. In what follows, we use the index g ∈ {1, 2, …, N} to denote the gene or transcript ID, and use and to denote, respectively, the newly synthesized protein abundance of gene g in the heavy channel and the degrading protein abundance of gene g in the light channel. The gene-specific degradation rate constant is denoted by , while the translational rate parameter for gene g is denoted by , which is allowed to vary over time. Let rg(t)Gr(t) denote the number of active ribosomes bound to each transcript of type g at time t, where denotes the relative ribosome footprint abundance for transcript g (in RPKM), rg,i(t) denotes the relative ribosome footprint abundance for transcript g isoform i (in RPKM), i ∈ {1, …, Ig} indexes the Ig isoforms of gene g, and Gr(t) denotes the total number of active ribosomes. Gr(t) is normalized so that Gr(0) = 1. Ingolia et al. (Ingolia et al., 2009) defined “translational efficiency” as , and we employ this definition.
In addition, we propose a new definition of translational efficiency that incorporates the ribosome footprints mapping to mitochondrially-translated genes. Let denote the normalized ribosome footprint abundance of gene g and isoform i, where i ∈ {1, …, Ig} indexes the Ig isoforms of gene g. It is normalized by the total number of reads that mapped to mitochondrially-translated genes, representing reads per kilo base per mitochondria read. And let denote the summation across the isoforms of gene g. The new definition of translational efficiency becomes . This definition incorporates the total cellular protein synthesis capacity Gr(t) and total mRNA nucleotide abundance Gm(t) into the calculation, which both can be time varying functions.
The proposed dynamical model of protein synthesis is a modification of the mass-action models for translation introduced earlier (Hargrove and Schmidt, 1989; Jovanovic et al., 2015; Schwanhausser et al., 2011; Wiita et al., 2013):
| (Equation 1) |
| (Equation 2) |
| (Equation 3–1) |
| (Equation 3–2) |
This model is a significant extension of our prior work (Wiita et al., 2013) in the following aspects: i) The pSILAC mass spectrometry technique enables us to extract the degradation rate constant and thereby disentangle the degradation process from the synthesis process. ii) The relative ribosome profiling footprints measured in RPKM units are not sufficient to quantify how the absolute amount of footprints for each transcript varies over time. To address this problem, we incorporate a global function Gr(t) (which we infer) that reflects the total cellular protein synthetic capacity (Equation 3–2). An alternative solution is to use the mitochondrial footprints normalized translational efficiency (Equation 3–1). iii) Our modified model treats the translational rate parameter as a time varying function that depends on the density of ribosomes on the transcript. The major difference between the above model and that of Jovanovic et al. (Jovanovic et al., 2015) is that whereas their model utilizes only pulsed SILAC mass spectrometry data, we also incorporate the ribosome profiling footprint information into our model via Equations 3–1 or 3–2.
Dilution Effect Resulted from Cell Growth
The number of cells in the low-dose bortezomib-treated MM1.S data remains constant over time. Hence, and represent respectively the heavy channel and light channel protein copies per cell. However, the untreated EBV-immortalized B-cells are growing, and the amount of light channel proteins per cell will be diluted due to cell division. We therefore scale the SRM intensities and by the growth function g(t), and obtain the heavy channel and light channel protein abundance as and , respectively. This allows us to model the light channel protein abundance as an exponential function, discussed in the next section.
Exponential and Orthogonal Natural Cubic Spline Fitting
The solution to Equation 1 is given by , so fitting the observed degrading protein abundance with an exponential function yields an estimate of the degradation rate constant . We employ the framework of functional data analysis in our study. In particular, measurements of , Mg(t), Rg(t) sampled at discrete time points are fitted with orthogonal natural cubic splines. Further details are provided below.
Spline is one of the widely used bases when approximating non-periodic functions (Ramsay and Silverman, 2005). We apply matrix factorization to the B-spline basis with degree 3 and three knots to construct an orthonormal basis , where t ∈ [T1, T2]. Let Σ be the Gram matrix of with the (i, j)-entry . By matrix factorization, one can find an invertible transformation Λ such that ΛTΛ = Σ−1 and B(t) = ΛB̃(t) forms an orthonormal basis (Redd, 2012). Let be a set of longitudinal data, where xi is the measurement, ti is the time when the measurement is observed, and n is the number of measurements. The data are approximated by a linear combination of the orthonormal basis functions , where α = (α1, …, αJ). We adopt the penalized least-squares estimator to fit the function f(t;α) and utilize generalized cross-validation to decide the penalty parameter (Ruppert, 2002). Since the behavior of polynomials fit beyond the boundaries can vary erratically, we impose additional constraints such that f(t;α) is linear at T1 and T2, as in natural cubic spline (Hastie et al., 2009). The problem can be formulated as
| (Equation 4) |
subject to f″(t; α) = 0 and f‴(t; α) =0 at t = T1 or T2, where λ is the penalty parameter, and f″ and f‴ denote the second and the third derivatives of f, respectively. This constrained optimization problem can be solved by the method of Lagrange multipliers. The Lagrangian of Equation 4 can be written as
where λ1, λ2, λ3, λ4 are the Lagrange multipliers. The solution should satisfy ∇α,λ1,λ2,λ3,λ4L(α, λ1, λ2, λ3, λ4) = 0. For convenience, define X ≜ [x1 x2 … xn]T,
and
This then leads to a system of linear equations
which can be solved by blockwise matrix inversion. Examples of the fitted functional form of and can be found in Figure 2B. Since the replicates of SRM measurements had high reproducibility, we use the average of them in the following analysis.
Solving for Parameters in the System of Differential Equations
We estimated the derivative of using its functional representation obtained from the mass spectrometry data, and fit using degree-3 orthogonal natural cubic splines with 4 knots. Let denote the resulting fit. Then, using Equations 2 and 3-1, we solved for β̃ that minimizes the objective function
Fit Rg(t) using degree-3 orthogonal natural cubic splines with 4 knots. Let denote the fit. Then the objective function becomes
Since the basis is orthonormal, the objective function is simplified as a least-squares regression problem: .
Similarly, using Equations 2 and 3-2, we solved for β and the total cellular protein synthetic capacity Gr(t) that minimizes the objective function
We achieved this by the following two steps:
Initialize β=1. Optimize with respect to
Prediction of Translational Rate Parameters and Protein Synthesis Rates Using Ribosome Profiling and RNA-seq Measurements
To test whether β is a universal factor that applies both in the absence of and during exposure to bortezomib, we carried out leave-one-out prediction tests as follows. In testing gene g, we estimated Gr(t) using the remaining N−1 genes and estimated β using the protein measurement for g at time 0 (i.e., prior to the application of bortezomib). These parameters were then used together with ribosome profiling and RNA-seq measurements to predict the translational rate parameter . Then, the protein synthesis rate was predicted as . We assessed the performance of our predictions using the data for 6–48 hours, i.e., during exposure to bortezomib. The average performance in terms of the Pearson correlation coefficient and the relative mean absolute error are reported in Data S4, which illustrates that the error of our estimation and prediction procedures is comparable to the variation observed in experimental replicates.
Of note, Jovanovic et al. (Jovanovic et al., 2015) introduced the concept of the “recycling rate” in SILAC mass spectrometry experiments. This rate describes the incorporation of unlabeled amino acids into newly synthesized proteins after SILAC pulse. However, in our targeted SRM measurements, we can only measure fully “light” and fully “heavy” peptides. We cannot readily measure the infrequent mixed peptides, with both light and heavy residues in the same peptide, necessary for measuring the recycling constant γ. Therefore, we made a “shotgun” measurement at the 12h time point sample with an LTQ Orbitrap mass spectrometer using previous instrument and LC parameters (Wiita et al., 2013). We analyzed the results with Protein Prospector (prospector.ucsf.edu) using 13C6-15N4 arginine and 13C6-15N2 lysine as variable modifications. Of 372 peptides with at least one missed tryptic site (i.e. multiple lysines and/or arginines), and at least one labeled residue, only 8 showed evidence of mixed labeling. This result leads to a γ(12h) of 0.012, which has a negligible impact on measurement of protein synthesis using the model of Jovanovic et al. (Jovanovic et al., 2015). This result is also in line with their results, finding the recycling constant to have minimal effects after 12h of SILAC pulse (Jovanovic et al., 2015). Therefore, our measurements of protein synthesis, going up to 48h, are likely unaffected by not explicitly including the recycling constant in our model.
Simulation of the Biochemical Experiment with Puromycin Incorporation
The biochemical experiment (Figure 3A) was conducted with 1 hour pulse of puromycin added at each time point prior to cell harvest. To test the accuracy of our proposed dynamical model, we performed a protein synthesis simulation using the parameters , mg(t), Gm(t), estimated from RNA-seq and mass spectrometry experiments. The initial condition was set such that , and the simulation was conducted to acquire the abundance after 1 hour, , where τ ∈ {0, 6, 12, 24, 36, 48}. The simulation results qualitatively showed a similar pattern as the biochemical experiment (Figure 3B).
Simulations of Under- or Over-Estimation in iBAQ
We examined how much variation in (Figures 2A and 4D) can be explained by noise in iBAQ by performing simulations to model under- or over-estimation in iBAQ values. Assume that the newly synthesized protein copies correlated perfectly with ribosome density, i.e. the red lines in Figures 2A and 4D. We randomly generated scaling factors sg according to the fitted normal distribution of the differences between the log-transformed iBAQ replicates (Figure S3A). Multiplying the ideal newly synthesized protein copies by these independent scaling factors, and repeating the process for 500 trials, we built a confidence interval for the abundance of newly synthesized protein copies (Figures S3B and S3C). Then, we compared the average vertical offsets from the red line (Figures S3B and S3C) in terms of MSE to the measured vertical offsets (Figures 2A and 4D). Results showed that the iBAQ noise explained 36% and 23% of the deviation from the linear regression fits for MM1.S and B-cells respectively.
We further examined how the presence of transcript isoforms affected our estimation of these iBAQ noise statistics. We partitioned all 272 protein-transcript pairs monitored in MM1.S cells into two groups (Figure S3D), using the criterion: one dominant transcript isoform (>80% of RNA-seq read density on a single isoform, per paired-end RNA-seq analysis at www.keatslab.org/data-repository:HMCL66_Transcript_Expression_FPKM). The group that has one dominant transcript isoform has almost half (47%) of the deviation from the linear regression fits explained by iBAQ noise, whereas the other group has 35% of the deviation explained by iBAQ noise. This suggests that some other sources of error may exist, e.g., the difficulty of footprint alignment in the presence of transcript isoforms, which remains an open question.
DATA AND SOFTWARE AVAILABILITY
Raw sequencing data is available in the GEO repository with accession number GEO: GSE69047 (reviewer access link: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=chohoqacvbyffwf&acc=GSE69047).
Raw SRM data is available via Skyline Panorama (https://panoramaweb.org/labkey/translation_model_2015.url).
MATLAB Software to enable use of the statistical model is available at https://sourceforge.net/projects/ncspline/
Supplementary Material
Highlights.
Confirm ribosome footprints reflect protein synthesis using targeted proteomics
Pulsed-SILAC captures alterations in global protein synthesis after chemotherapy
Alterations in global synthesis not seen by standard ribosome profiling analysis
Quantitative model predicts proteome remodeling under translational repression
Acknowledgments
This work was supported by a Post-doctoral Fellowship (DRG 11-112) and Dale Frey Breakthrough Award (DFS 14-15) from the Damon Runyon Cancer Research Foundation, NCI K08CA184116-01A1, and an American Cancer Society grant #IRG-97-150-13 (to A.P.W.); and NSF CAREER grant DBI-0846015, a Packard Fellowship for Science and Engineering, and Math + X Research grant from the Simons Foundation (to Y.S.S.). Mass spectrometry was performed at the UCSF NIH-NIGMS Mass Spectrometry Facility directed by Dr. Alma Burlingame supported by NIH grants P41RR001614 and S10RR026662. We thank Jessica Lund at the UCSF Center for Advanced Technology for assistance with HiSeq sequencing.
Footnotes
Supplemental Information includes four figure and four datasets and can be found with this article online at http://dx.doi.org/10.1016/j.cels.2017.05.001.
AUTHOR CONTRIBUTIONS
T.-Y.L., H.H.H., J.A.W., Y.S.S., and A.P.W. designed the experiments and analysis approach. H.H.H., D.W., Y.X., and A.P.W. performed experiments and analyzed data. T.-Y.L. and Y.S.S. analyzed data and built the quantitative model. T.-Y.L., H.H.H., Y.S.S., and A.P.W. wrote the paper.
References
- Andreev DE, O’Connor PB, Fahey C, Kenny EM, Terenin IM, Dmitriev SE, Cormican P, Morris DW, Shatsky IN, Baranov PV. Translation of 5′ leaders is pervasive in genes resistant to eIF2 repression. Elife. 2015;4:e03971. doi: 10.7554/eLife.03971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussemart L, Malka-Mahieu H, Girault I, Allard D, Hemmingsson O, Tomasic G, Thomas M, Basmadjian C, Ribeiro N, Thuaud F, et al. eIF4F is a nexus of resistance to anti-BRAF and anti-MEK cancer therapies. Nature. 2014;513:105–109. doi: 10.1038/nature13572. [DOI] [PubMed] [Google Scholar]
- Chauhan D, Catley L, Li G, Podar K, Hideshima T, Velankar M, Mitsiades C, Mitsiades N, Yasui H, Letai A, et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from bortezomib. Cancer Cell. 2005;8:407–419. doi: 10.1016/j.ccr.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- de Haro C, Mendez R, Santoyo J. The eIF-2alpha kinases and the control of protein synthesis. FASEB J. 1996;10:1378–1387. doi: 10.1096/fasebj.10.12.8903508. [DOI] [PubMed] [Google Scholar]
- de Hoon MJL, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20:1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gigeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2012;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan R, Hershey JW. Heat shock-induced translational alterations in HeLa cells. Initiation factor modifications and the inhibition of translation. J Biol Chem. 1984;259:11882–11889. [PubMed] [Google Scholar]
- Floor SN, Doudna JA. Tunable protein synthesis by transcript isoforms in human cells. Elife. 2016;5:e10921. doi: 10.7554/eLife.10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargrove JL, Schmidt FH. The role of mRNA and protein stability in gene expression. FASEB J. 1989;3:2360–2370. doi: 10.1096/fasebj.3.12.2676679. [DOI] [PubMed] [Google Scholar]
- Hastie T, Tibshirani R, Friedman J. The Elements of Statistical Learning. 1. Vol. 2. New York: Springer; 2009. [Google Scholar]
- Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Ingolia NT. Ribosome footprint profiling of translation throughout the genome. Cell. 2016;165:22–33. doi: 10.1016/j.cell.2016.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki S, Floor SN, Ingolia NT. Rocaglates convert DEAD-box protein eIF4A into a sequence-selective translational repressor. Nature. 2016;534:558–561. doi: 10.1038/nature17978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol. 2010;11:113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic M, Rooney MS, Mertins P, Przybylski D, Chevrier N, Satija R, Rodriguez EH, Fields AP, Schwartz S, Raychowdhury R, et al. Immunogenetics. Dynamic profiling of the protein life cycle in response to pathogens. Science. 2015;347:1259038. doi: 10.1126/science.1259038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. Initiation of translation in prokaryotes and eukaryotes. Gene. 1999;234:187–208. doi: 10.1016/s0378-1119(99)00210-3. [DOI] [PubMed] [Google Scholar]
- Li GW, Burkhardt D, Gross C, Weissman JS. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell. 2014a;157:624–635. doi: 10.1016/j.cell.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JJ, Bickel PJ, Biggin MD. System wide analyses have underestimated protein abundances and the importance of transcription in mammals. Peer J. 2014b;2:e270. doi: 10.7717/peerj.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Beyer A, Aebersold R. On the dependency of cellular protein levels on mRNA abundance. Cell. 2016;165:535–550. doi: 10.1016/j.cell.2016.03.014. [DOI] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AM, Baranov PV. Ribosome profiling: a Hi-Def monitor for protein synthesis at the genome-wide scale. Wiley Int Rev RNA. 2013;4:473–490. doi: 10.1002/wrna.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107:4907–4916. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picotti P, Aebersold R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat Methods. 2012;9:555–566. doi: 10.1038/nmeth.2015. [DOI] [PubMed] [Google Scholar]
- Powley IR, Kondrashov A, Young LA, Dobbyn HC, Hill K, Cannell IG, Stoneley M, Kong YW, Cotes JA, Smith GC, et al. Translational reprogramming following UVB irradiation is mediated by DNA-PKcs and allows selective recruitment to the polysomes of mRNAs encoding DNA repair enzymes. Genes Dev. 2009;23:1207–1220. doi: 10.1101/gad.516509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay JO, Silverman BW. Functional Data Analysis. New York: Springer; 2005. [Google Scholar]
- Redd A. A comment on the orthogonalization of B-spline basis functions and their derivatives. Stat Comput. 2012;22:251–257. [Google Scholar]
- Ruppert S. Selecting the number of knots for penalized splines. J Comp Graph Stat. 2002;11:735–757. [Google Scholar]
- Saldanha AJ. Java Treeview—extensible visualization of microarray data. Bioinformatics. 2004;20:3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Shenton D, Smirnova JB, Selley JN, Carroll K, Hubbard SJ, Pavitt GD, Ashe MP, Grant CM. Global translational responses to oxidative stress impact upon multiple levels of protein synthesis. J Biol Chem. 2006;281:29011–29021. doi: 10.1074/jbc.M601545200. [DOI] [PubMed] [Google Scholar]
- Vincenz L, Jager R, O’Dwyer M, Samali A. Endoplasmic reticulum stress and the unfolded protein response: targeting the Achilles heel of multiple myeloma. Mol Cancer Ther. 2013;12:831–843. doi: 10.1158/1535-7163.MCT-12-0782. [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang H, McManus J, Kingsford C. Isoform-level ribosome occupancy estimation guided by transcript abundance with Ribomap. Bioinformatics. 2016;32:1880–1882. doi: 10.1093/bioinformatics/btw085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg DE, Shah P, Eichhorn SW, Hussmann JA, Plotkin JB, Bartel DP. Improved ribosome-footprint and mRNA measurements provide insights into dynamics and regulation of yeast translation. Cell Rep. 2016;14:1787–1799. doi: 10.1016/j.celrep.2016.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiita AP, Ziv E, Wiita PJ, Urisman A, Julien O, Burlingame AL, Weissman JS, Wells JA. Global cellular response to chemotherapy-induced apoptosis. Elife. 2013;2:e01236. doi: 10.7554/eLife.01236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm M, Schlegl J, Hahne H, Moghaddas Gholami A, Lieberenz M, Savitski MM, Ziegler E, Butzmann L, Gessulat S, Marx H, et al. Mass-spectrometry-based draft of the human proteome. Nature. 2014;509:582–587. doi: 10.1038/nature13319. [DOI] [PubMed] [Google Scholar]
- Wolfe AL, Singh K, Zhong Y, Drewe P, Rajasekhar VK, Sanghvi VR, Mavrakis KJ, Jiang M, Roderick JE, Van der Meulen J, et al. RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature. 2014;513:65–70. doi: 10.1038/nature13485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zur H, Aviner R, Tuller T. Complementary post transcriptional regulatory information is detected by PUNCH-P and ribosome profiling. Sci Rep. 2016;6:21635. doi: 10.1038/srep21635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.