

Abstract
Cyclopropanations of alkene-containing natural products that proceed under mild conditions are reported for simultaneous arming and structure-activity relationship studies. An alkynyl diazo ester under Rh(II) catalysis is employed for cyclopropanations of electron-rich olefins while an alkynyl sulfonium ylide is used for electron-poor olefins. This approach enables simultaneous natural product derivatization for SAR studies and arming (i.e. via alkyne attachment) for subsequent conjugation with reporter tags (e.g. biotin, fluorophores, photoaffinity labels) for mechanism of action studies including cellular target identification and proteome profiling experiments.
Graphical Abstract
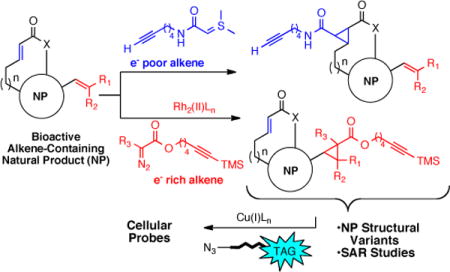
Natural products are an enduring class of privileged structures that exhibit an incredible range of structural complexity and functional diversity. In addition, they display a plethora of biologically and medicinally important activities that include antibiotic, antifungal, antiparasitic, anticancer, and immunosuppressive properties. Historically, natural product extracts provided the first medicines to humankind and today account for roughly 50% of all drugs in clinical use, either directly or indirectly.1 Natural products are also a highly productive source of leads for the development of new therapeutics,2 and are often used as invaluable tools for the study of biological processes and the discovery of protein targets for therapeutic intervention.3 A challenge to fully utilizing natural products in drug discovery programs is the lack of methods to enable direct modification of native natural products to tailor their properties. For this reason, we set out to develop chemo- and site selective methods to alter the structure of native natural products for structure-activity relationship (SAR) studies, generate new natural product-like structures, and to rapidly synthesize natural product-based cellular probes including biotin, fluorophore, and photoaffinity natural product conjugates.4 To date, we have described methods for simultaneous arming and SAR studies of natural products employing mild Rh(II)-catalyzed O-H insertions with alkynyl diazo acetates of alcohol-containing natural products5 and In(OTf)3-catalyzed iodination of arene-containing natural products.6 To expand these derivatizations to other commonly found functional groups, we now report the development of mild and selective cyclopropanation reactions of both electron-rich and electron-poor olefins. The utility of this strategy including the scope and functional group compatibility of these derivatizations is demonstrated by application to several commercially available natural products.
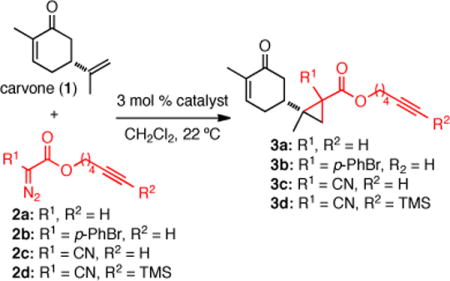
We initiated our studies with carvone (1) as a simple model system for the development of chemoselective cyclopropanations, since it contains both an electron-rich and electron-deficient alkene. For cyclopropanation of electron-rich alkenes we explored the reaction of carvone (1) and hexynyl diazo acetate 2a7 catalyzed by various transition metals (Table 1). The length of the tether between the diazo-bearing carbon and the alkyne were chosen to minimize undesired intramolecular cyclopropenation.8 While it is well known that several metals are capable of catalyzing this process, several reaction conditions require elevated temperatures for cyclopropanations to proceed at reasonable rates.9 With a goal of applying this strategy to the derivatization of complex and often sensitive natural products, we sought to develop the most mild and practical cyclopropanation conditions that proceeded at ambient temperature (22 °C). In our initial screening, we found that Rh(II) catalysts were the most efficient at promoting the site-selective cyclopropanation of the isopropylidene group of carvone (1) employing diazo acetate 2a at 22 °C. Both copper and palladium catalysts proved ineffective under these reaction conditions (Table 1, entries 1–3), while Rh2(OCOCF3)4, Rh2(OAc)4 and the Du Bois-Espino catalyst, Rh2(esp)2,10 provided the desired cyclopropanes in 20, 28 and 45% yield, respectively (Table 1, entries 4–6). These reactions led to complete site selectivity but a mixture of only two diastereomeric cyclopropanes (dr 2:1, R1 = H). Diazo esters 2b–2c7 led to cyclopropanation, however, the chemical yields remained modest (19 and 55%, Table 1, entries 7–8). Careful analysis of side-products revealed that the alkyne on the side-chain was in fact reacting competitively with the metallocarbenoid intra- and intermolecularly leading to the corresponding cyclopropenes. To circumvent this side reaction, a TMS-protected alkynyl diazo reagent 2d was studied8a,c and found to completely supress cyclopropenations as evidenced by a dramatic increase in the desired diastereomeric cyclopropanes to 91% (dr 1:1:1:1, Table 1, entry 9). Under these optimized conditions, only 1.2 equivalents of the diazo reagent were needed for complete conversion of carvone.11
Table 1.
Screening of Catalysts and Diazo Reagents for the Cyclopropanation of Carvone (1).a
| ||||
|---|---|---|---|---|
|
| ||||
| entry | diazo | catalyst | % yieldb | drc |
| 1 | 2a | Cu(acac)2 | 0 | – |
| 2 | 2a | IprCuCl | trace | – |
| 3 | 2a | PdCl2(CH3CN)2 | 0 | – |
| 4 | 2a | Rh2(OCOCF3)4 | 20% | 2:1 |
| 5 | 2a | Rh2 (OAc)4 | 28% | 2:1 |
| 6 | 2a | Rh2 (esp)2 | 45% | 2:1 |
| 7 | 2b | Rh2 (esp)2 | 19% | 2:2:1:1 |
| 8 | 2c | Rh2(esp)2 | 55% | 1:1:1:1 |
| 9d | 2d | Rh2 (esp)2 | 91% | 1:1:1:1 |
Reaction of carvone (1) and diazo esters 2a–c, (4 equiv) at 22 °C.
Values refer to isolated yields.
Ratios were determined by 1H NMR (500 MHz); the relative stereochemistry was not determined.
Only 1.2 equiv of 2d were used. acac = acetylacetonate; IPr = [1,3-bis(diisopropylphenyl)imidazole-2-ylidene]; esp = α,α,α′,α′-tetramethyl-1,3-benzenedipropionate.
The scope of this reaction was assessed with several commercially available alkene-containing natural products (Scheme 1) using the established optimized conditions (Table 1, entry 9). Cyclopropanation of caryophyllene oxide provided the desired cyclopropanes 4 in 94% yield with high facial selectivity and with 3:1 diastereoselectivity at the newly generated quaternary carbon. The antineoplastic agent bexarotene methyl ester was also an excellent substrate delivering racemic cyclopropanes 5 in 93% yield and 8:1 dr. The insecticide rotenone gave all four possible diastereomeric cyclopropanes 6 in 88% yield and 3:3:1:1 ratio. Trisubstituted alkenes participate under these conditions as demonstrated with vitamin K1, providing four diastereomeric cyclopropanes 7 in 84% yield. The reaction of more sterically encumbered alkenes was more challenging as evidenced by attempted cyclopropanation of forskolin which required 3 equiv of diazo ester 2d for complete conversion to deliver cyclopropanes 8 in 81% yield. To prevent OH insertion5 with accessible free alcohols, mild global silylation was performed prior to cyclopropanation of forskolin and gibberellic acid methyl ester. However, in the case of forskolin, the hindered tertiary C6 and secondary C9 alcohols are not protected under these conditions and also did not undergo OH insertion. With the accessible alcohols protected, cyclopropanation proceeded smoothly to give cyclopropyl forskolin derivative 8 (81% yield, dr 1:1) and in the case of gibberellic acid methyl ester which contains two electron-rich olefins, only the less hindered 1,1-disubstituted olefin undergoes cyclopropanation to provide derivative 9 in 79% yield (dr 3:1). The TMS groups are readily removed simultaneouly during subsequent Sharpless-Huisgen cycloaddition (vide infra).
Scheme 1. Rh2(Esp)2-Catalyzed Cyclopropanations of Natural Products Bearing Electron-Rich Alkenes with Alkynyl Diazo Ester 2d.a.
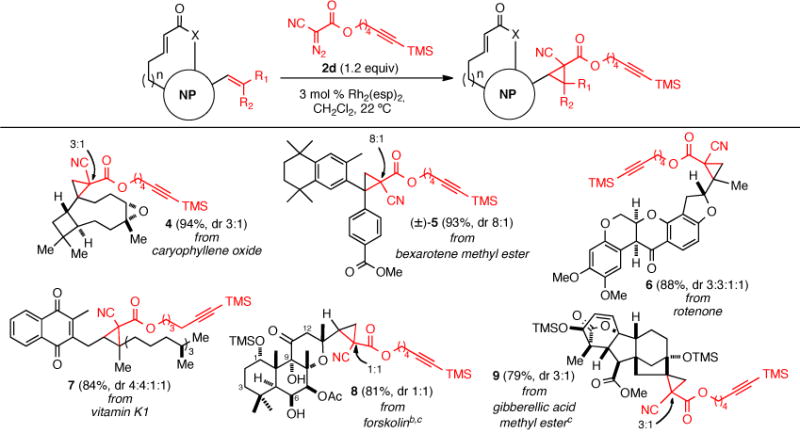
aDiastereomeric ratios were determined by 1H NMR (500 MHz); the relative stereochemistry of the major diastereomers was not determined since in some cases they were inseparable (see Supporting Information). b3.0 equiv of diazo ester 2d were employed. cSilylation was performed prior to cyclopropanation (TMSCl, DMAP, Et3N, CH2Cl2, 22 °C).
For cyclopropanation of electron-deficient alkenes,12 we prepared the alkynyl amide-containing sulfonium salt 10.13 Addition of this salt to a solution of t-BuOK in DMSO generated the corresponding sulfur ylide which reacted selectively with electron-poor alkenes in several natural products (Scheme 2). Carvone (1) provided the desired cyclopropane 11 in good yield (94%) and diasteroselectivity (8:1 dr). Brefeldin A reacted under these conditions affording the cyclopropane 12 albeit in low yield (32%) and diastereoselectivity (2.5:1). Similarly, phorbol dibutyrate gave cyclopropane 14 in modest yield (24%) and low diastereoselectivity. In the case of parthenin and achillin, this reaction exhibited high chemosite (chemo- and site) selectivity providing monocyclopropane 13 in 32% yield (dr 6:1) and cyclopropane 15 in 72 % yield (dr >19:1), respectively. Parthenin reacted selectively at the more reactive exo-methylene and the structure of this adduct was verified by X-ray crystallography (Scheme 2, inset).
Scheme 2. Scope of the Cyclopropanation of Natural Products Bearing Electron-Poor Alkenes with Alkynyl Sulfonium 10.a.
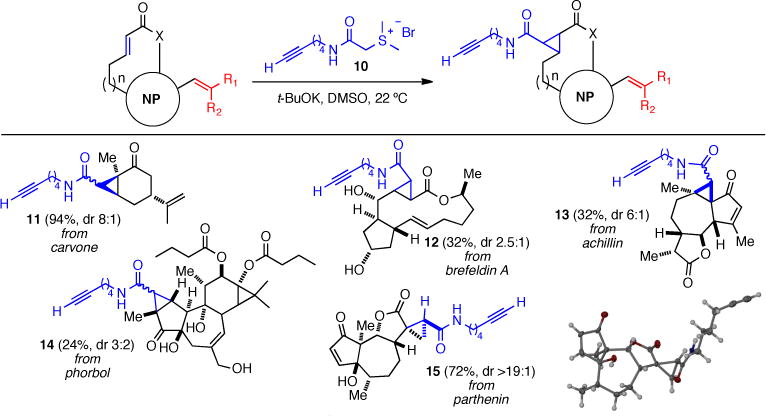
aDiastereomeric ratios were determined by 1H NMR (500 MHz); the relative stereochemistry was not confirmed (with exception of 15) but where indicated is based on conformational considerations of the natural product substrate.
The need for global silylation prior to cyclopropanation to prevent OH insertion was readily accomodated since a procedure for in situ desilylation of TMS-alkynes and subsequent Sharpless-Huisgen cycloaddition was recently reported in the presence of tetrabutylammonium fluoride (TBAF).14 The utility of this process was demonstrated by the synthesis of a biotin conjugate from the TMS-protected forskolin cyclopropane 8 (Scheme 3). This tandem desilylation/cycloaddition proceeded in good yield (76%) to give the deprotected, forskolin-biotin conjuate 17.
Scheme 3.
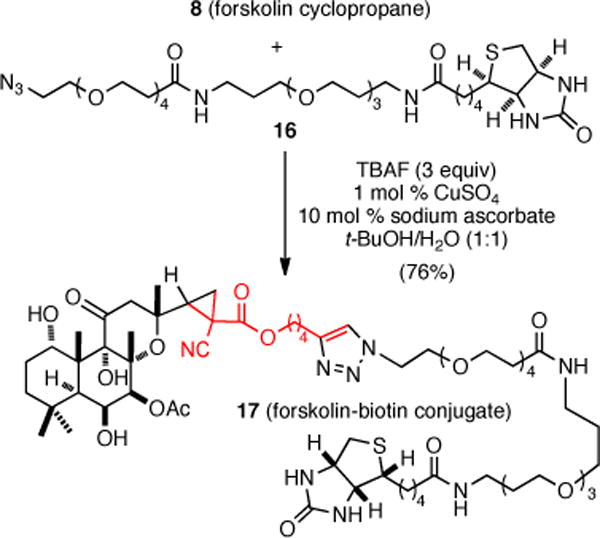
Desilylation/Conjugation of Cyclopropyl-Forskolin 8 with a Biotin Tag via a Sharpless-Huisgen Cycloaddition.
We also demonstrated that the described Rh(II)-catalyzed cyclopropanation/conjugation strategy is amenable to microscale,15 as demonstrated by the derivatization of 1 mg of bexarotene methyl ester (18). The two step sequence provided the bexarotene-biotin conjugate 19 cleanly and in sufficient quantities to be characterized by 1H NMR (500 MHz) and HRMS (Scheme 4).
Scheme 4.
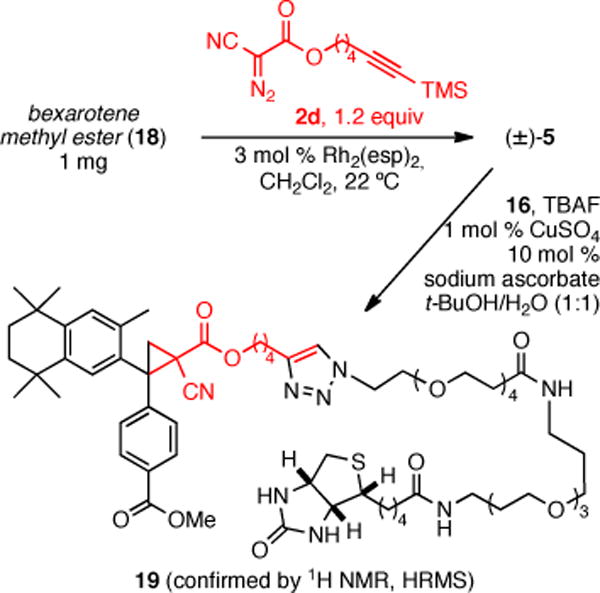
Microscale Cyclopropanation/Conjugation of Bexarotene Methyl Ester (18).
In summary, we have developed reagents for selective cyclopropanations that enable simultaneous derivatization and arming of both electron rich and electron poor olefin-containing natural products under mild conditions. We anticipate that use of chiral Rh(II)-catalysts and chiral sulfoximines could alter observed diastereoselectivity and most importantly could alter site selectivity in natural products with multiple electron rich and electron poor alkenes, respectively.9 The described methods enable SAR studies of native or transiently protected natural products in addition to subsequent conjugation, via the appended alkyne, with reporter tags for cellular target identification, mechanism of action studies and the synthesis of activity-based molecular probes for global protein profiling.
Supplementary Material
Acknowledgments
We thank the NIH (GM 086307) and the Welch Foundation (A-1280) for financial support of this work. We also thank Dr. Chun-Xiao Xu (TAMU) for helpful suggestions on the utility of TMS-protected diazo reagent 2d. We thank Dr. Joseph Reibenspies (TAMU) for solving the X-ray crystal structure of 15.
Footnotes
Supporting Information Available: Detailed experimental procedures and characterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Harvey AL. Drug Discov Today. 2008;13:894. doi: 10.1016/j.drudis.2008.07.004. [DOI] [PubMed] [Google Scholar]; (b) Newman DJ, Cragg GM. J Nat Prod. 2007;70:461. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 2.(a) Welsch ME, Snyder SA, Stockwell BR. Curr Opin Chem Biol. 2010;14:347. doi: 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ganesan A. Curr Opin Chem Biol. 2008;12:306. doi: 10.1016/j.cbpa.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 3.(a) Carlson EE. ACS Chem Biol. 2010;5:639. doi: 10.1021/cb100105c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jung HL, Kwon HJ. J Microbiol Biotechnol. 2006;16:651. [Google Scholar]; (c) Stockwell BR. Nature. 2004;432:846. doi: 10.1038/nature03196. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Burdine L, Kodadek T. Chem Biol. 2004;11:593. doi: 10.1016/j.chembiol.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 4.(a) Tang P, Furuya T, Ritter T. J Am Chem Soc. 2010;132:12150. doi: 10.1021/ja105834t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen MS, White MC. Science. 2007;318:783. doi: 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]; (c) Lewis CA, Miller SJ. Angew Chem Int Ed. 2006;45:5616. doi: 10.1002/anie.200601490. [DOI] [PubMed] [Google Scholar]
- 5.(a) Peddibhotla S, Dang Y, Liu JO, Romo D. J Am Chem Soc. 2007;129:12222. doi: 10.1021/ja0733686. [DOI] [PubMed] [Google Scholar]; (b) Chamni S, He QL, Dang Y, Bhat S, Liu JO, Romo D. ACS Chem Biol. 2011;6:1175. doi: 10.1021/cb2002686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou CY, Li J, Peddibhotla S, Romo D. Org Lett. 2010;12:2104. doi: 10.1021/ol100587j. [DOI] [PubMed] [Google Scholar]
- 7.For the synthesis of diazo compounds 2a-c, see ref. 5a and 5c. For the synthesis of 2d see Supporting Information.
- 8.(a) Briones JF, Davis HML. Org Lett. 2011;13:3984. doi: 10.1021/ol201503j. [DOI] [PubMed] [Google Scholar]; (b) Briones JF, Hansen J, Hardcastle KI, Autschbach J, Davis HML. J Am Chem Soc. 2010;132:17211. doi: 10.1021/ja106509b. [DOI] [PubMed] [Google Scholar]; (c) Davis HML, Lee GH. Org Lett. 2004;6:1233. doi: 10.1021/ol049928c. [DOI] [PubMed] [Google Scholar]; (d) Lou Y, Horikawa M, Kloster RA, Hawryluk NA, Corey EJ. J Am Chem Soc. 2004;126:8916. doi: 10.1021/ja047064k. [DOI] [PubMed] [Google Scholar]; (e) Doyle M, Protopopova M, Müller P, Ene D, Shapiro E. J Am Chem Soc. 1994;116:8492. [Google Scholar]; (f) Protopopova M, Doyle M, Müller P, Ene D. J Am Chem Soc. 1992;114:2755. [Google Scholar]; (g) Müller P, Imogaï H. Tetrahedron: Asymmetry. 1998;9:4419. [Google Scholar]
- 9.For a comprehensive overview, see:; (a) Davis HML, Antoulinakis EG. Organic Reactions; New York. 2001;57:1–326. [Google Scholar]; (b) Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds. Wiley; New York: 1998. [Google Scholar]; (b) Lebel H, Marcoux JF, Molinaro C, Charette AB. Chem Rev. 2003;103:977. doi: 10.1021/cr010007e. [DOI] [PubMed] [Google Scholar]
- 10.Espino GCF, K W, Kim M, Du Bois J. J Am Chem Soc. 2004;126:15378. doi: 10.1021/ja0446294. [DOI] [PubMed] [Google Scholar]
- 11.Hydrolysis experiments established that ester 3d was resistant to hydrolysis after 72 h at pH 10 and 13. See supporting Information for details.
- 12.(a) Appel R, Hartmann N, Mayr H. J Am Chem Soc. 2010;132:17894. doi: 10.1021/ja1084749. [DOI] [PubMed] [Google Scholar]; (b) Collado I, Domínguez C, Ezquerra J, Pedregal C, Monn JA. Tetrahedron Lett. 1997;38:2133. [Google Scholar]; (c) Payne GB. J Org Chem. 1967;32:3351. [Google Scholar]
- 13.For the synthesis of compound 10, see Supporting Information.
- 14.Maisonneuve S, Fang Q, Xie J. Tetrahedron. 2008;64:8716. [Google Scholar]
- 15.Lewis CA, Longcore KA, Miller SJ, Wender PA. J Nat Prod. 2009;72:1864. doi: 10.1021/np9004932. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.