

Abstract
A full account of concise, enantioselective syntheses of the anticancer agent (−)-salinosporamide A and derivatives, including (−)-homosalinosporamide, is described that was inspired by biosynthetic considerations. The brevity of the synthetic strategy stems from a key bis-cyclization of a (β)-keto tertiary amide, which retains optical purity enabled by A1,3-strain rendering slow epimerization relative to the rate of bis-cyclization. Optimization studies of the key bis-cyclization, enabled through by-product isolation and characterization, are described that ultimately allowed for a gram scale synthesis of a versatile bicyclic core structure with a high degree of stereoretention. An optimized procedure for zincate generation by the method of Knochel, generally useful for the synthesis of salino A derivatives, led to dramatic improvements in side-chain attachment and a novel diastereomer of salino A. The versatility of the described strategy is demonstrated by the synthesis of designed derivatives including (−)-homosalinosporamide A. Inhibition of the human 20S and 26S proteasome by these derivatives using an enzymatic assay are are also reported. The described total synthesis of salino A raises interesting questions regarding how biosynthetic enzymes leading to the salinosporamides proceeding via optically active β-keto secondary amides, are able to maintain the stereochemical integrity at the labile C2 stereocenter or if a dynamic kinetic resolution is operative.
Keywords: β-lactone, aldol-lactonization, biomimetic, organocatalysis
Graphical abstract
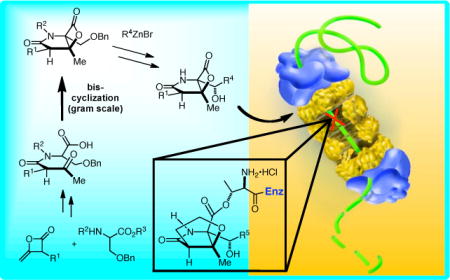
Introduction
The development of practical, scaleable total syntheses of complex, bioactive natural products has become of great interest in the recent past due to a desire to further explore biological activity or to facilitate development of structure-activity relationship profiles. Inevitably, one of the best ways to achieve such a practical synthesis involves consideration of potential biosynthetic strategies utilized by the producing organism to assemble such stereochemically complex structures.1
The potential of human 20S proteasome inhibitors continues to be of interest for anticancer chemotherapy and the recent FDA approval of bortezomib (Velcade) validates the proteasome as a target for cancer chemotherapy.2 Isolated by Fenical and coworkers3 from the marine actinomycete, Salinispora tropica, salinosporamides A and B (1a-b, salino A and B) are unique bicyclo [3.2.0] β-lactone-containing natural products (Figure 1). Phase I human clinical studies for multiple myeloma following the finding that salino A exhibited great potential in mouse models toward several cancers when administered intravenously despite the potentially labile β-lactone.4 The terrestrial metabolites, cinnabaramides (e.g. A, 2),5 lactacystin-β-lactone (omuralide, 3a) derived from the prodrug lactacystin (4),6 and the recently isolated mixed congener antiprotealide (3b)7 bear close structural similarities to salino A and congeners which have been targets for total8 and formal9 syntheses,10 structure-activity studies,11 biosynthetic engineering,12 and crystallographic studies with the 20S proteasome.13
Figure 1.
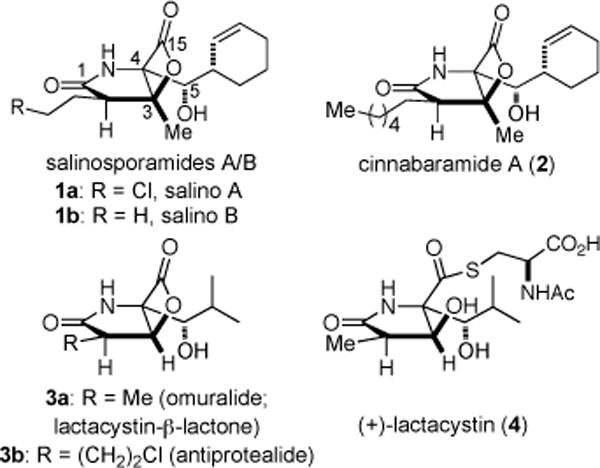
Structures of Naturally-Occuring Bicyclic-β-Lactone Proteasome Inhibitors
Our synthetic strategy to salino A and derivatives utilizes a nucleophile-catalyzed aldol-lactonization (NCAL) process, a reaction we previously developed for the synthesis of carbocycle-fused β-lactones (Figure 1).14 Indeed, salino A and related natural products were the initial inspiration for the development of the NCAL methodology in our group (Figure 2). In particular, the study of an intramolecular aldol-lactonization was inspired by biosynthetic considerations of this class of proteaseome inhibitors and this general biosynthetic pathway has recently been supported by detailed biosynthetic studies.12 We previously reported racemic syntheses of cinnabaramide A and salino A8e and more recently an enantioselective synthesis of (−)-salino A and (−)-homosalino A based on these strategies.8k Retention of optical purity during the bis-cyclization process was made possible by the A1,3-strain of (β)-keto tertiary amides.15 Herein, we disclose a full account of our bioinspired syntheses of salino A and derivatives including optimization of the key bis-cyclization process that simultaneously forms the γ-lactam and fused β-lactone core. Furthermore, the bioactivity of these derivatives was assayed against the chymotripsin-like, caspase-like, and trypsin-like activities of the human proteasome.
Figure 2.
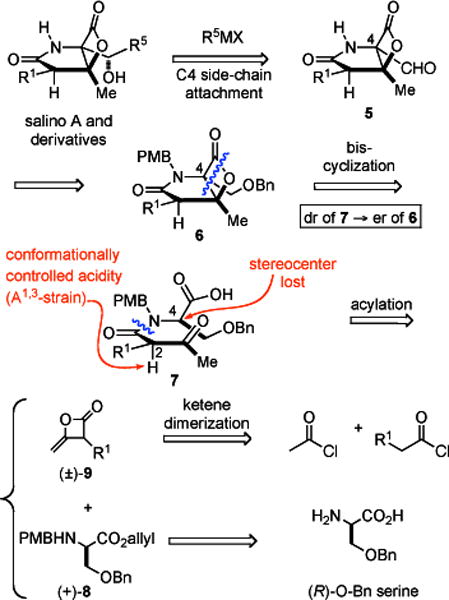
Enantioselective Strategy to Salino A and Derivatives
Our primary retrosynthetic disconnections, namely the bis-cyclization (7→6) and amide coupling steps (8+9→7), which were inspired by biosynthetic considerations, are key steps in our retrosynthetic analysis. The optical purity of β-lactone 6 would reflect the diastereomeric purity of β-ketoamide 7 derived from acylation of serine derivatives with racemic ketene dimer 9. The C4 stereocenter in β-ketoamide 7 is lost during the bis-cyclization process due to formation of an ammonium enolate intermediate, thus the integrity of the C2 stereocenter primarily dictates the optical purity of β-lactone 6 which we proposed would be preserved by A1,3 strain.15 Introduction of the C4-sidechain initially relied on the strategy developed by Corey8a and in this report we describe the use of Knochel’s procedure for zincate formation,16 however the success of this process and subsequent manipulations was initially not guaranteed given the potential reactivity of the β-lactone.17
Results and Discussion
Initial Studies of the Bis-cyclization Leading to C4-Unsubstituted Bicyclic β-Lactones
We first targeted simple C4-unsubstituted substrates for the bis-cyclization. Ketene homodimers 12/13 and subsequently employed (vide infra) heterodimers 14/15 were prepared by dimerization of acid chlorides 10/11 by the method of Sauer (Scheme 1).8e,k,18 As expected, homodimers 12/13 were obtained in moderate to good yields (43–65%) following silica gel purification, however heterodimers 14/15 provided the expected statistical mixtures and further reductions in yields were also noted during purification leading to low absolute yields (5–13% yield, 20–52% based on a statistical, theoretical yield of 25%). Importantly, these dimerizations are readily run on multigram scale from inexpensive, commercially available acid chlorides and heterodimers 14/15, despite low absolute yields, can also be obtained in multigram quantities following silica gel purification.
Scheme 1.
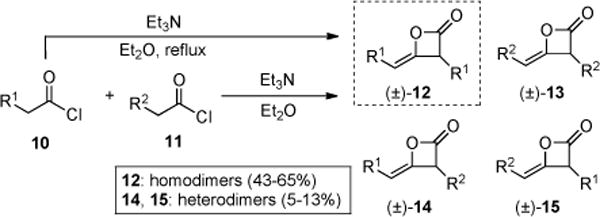
Synthesis of Ketene Homodimers 12/13 and Heterodimers 14/15
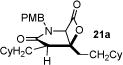
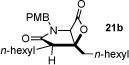
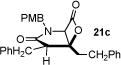
With racemic ketene homodimers 12a–e in hand, these were coupled with N-PMB-glycine benzyl ester (16) by the method of Calter,19 which proceeded efficiently to provide keto acid substrates 19a-e following hydrogenolysis (Table 1). Bis-cyclization of these ketoacids was achieved using conditions similar to those developed for carbocycles, with 4-pyrrolidinopyridine (4-PPY) as nucleophilic promoter at 0 °C and proceeded efficiently (70–93%) in most cases to give bicyclic-β-lactones 21a-e with moderate diastereoselectivity (dr, 2.2–4:1). Ketoacid 19c gave high diastereoselectivity (>19:1) but in reduced yield (54%; Table 1, entry 3), however monitoring this reaction by 1H NMR at 25 °C indicated that the high selectivity was due to selective degradation of the minor diastereomer by prolonged reaction times. The C2-unsubstituted ketoacid 19d gave only 25% yield (Table 1, entry 4) which may be due to facile enolization due to the absence of the C2-substitutent (R1), which likely leads to diminished rates of the initial aldol step. Importantly, ketoacid 19e bearing two primary chlorides thus mimicking the substrate required for the proposed salino A synthesis also proceeded efficiently (Table 1, entry 5).
Table 1.
Synthesis of Simplified, C4-Unsubstituted Salino A Derivatives 21a-e from Homodimers 12a-e
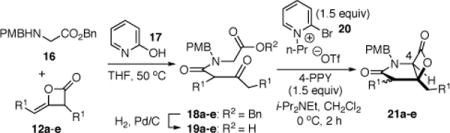
| ||||
|---|---|---|---|---|
| entry | % yield (19)a,b |
% yield (21)b |
β-lactone | drc |
| 1 | 84 (19a) |
93 (21a) |
|
2.2:1 |
| 2 | 80 (19b) |
90 (21b) |
|
2.2:1 |
| 3 | 72 (19c) |
85 (21c) |
|
2.5:1 (>19:1)d |
| 4 | 77 (19d) |
25 (21d) |
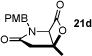
|
– |
| 5 | 81 (19e) |
70 (21e) |
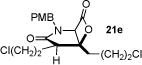
|
4:1 |
Yield is for 2 steps.
Yields refer to isolated, purified (SiO2) product.
Determined by 1H NMR analysis of crude reaction mixtures.
Observed diastereomeric ratio (dr) if reaction is allowed to proceed at 25 °C for 36 h (54% yield).
PMB = p-methoxybenzyl, 4-PPY = 4-pyrrolidinopyridine.
Total Synthesis of rac-Cinnabaramide A (2)
Encouraged by the results with C4-unsubstituted keto acid substrates, we next studied more sterically demanding substrates (i.e. bearing C4-substititution) by targeting the synthesis of cinnabaramide A (2). The synthesis commenced with reductive amination of commercially available (S)-O-benzylserine ((S)-22) with p-anisaldehyde (Scheme 2). Subsequent esterification provided the protected serine derivative 23a in 58% overall yield (2 steps). The required unsymmetrical ketene dimer 14a, derived from heterodimerization of acetyl and octanoyl chlorides (5% yield),18a was coupled with serine derivative 23a to provide diastereomeric esters 24 (dr 1:1) and a Sn-mediated hydrolysis20 provided ketoacid 25. The key bis-cyclization provided diastereomeric β-lactones 26/27 in 45% yield with moderate diastereoselectivity (dr 3.3:1), and nOe analysis confirmed that the major diastereomer corresponded to that found in the cinnabaramides.21 Hydrogenolysis of the benzyl ether facilitated separation of the major alcohol diastereomer 28 (79%, dr >19:1) and this was followed by Parikh-Doering oxidation22 to give an intermediate aldehyde, which was used directly in the next step. Applying the method developed by Corey with zinc reagent 29 gave alcohol 30 in 57% yield (2 steps, dr 4.7:1). Oxidative PMB deprotection with ceric ammonium nitrate gave rac-cinnabaramide A((±)-2), which could be isolated diastereomerically pure in 48% yield. Spectral data for the synthetic material correlated with the published data and further verification of the relative stereochemistry was possible by X-ray analysis.21 This synthesis demonstrated that several reactions could be performed in the presence of the (β)-lactone including organozinc additions to aldehydes.
Scheme 2.
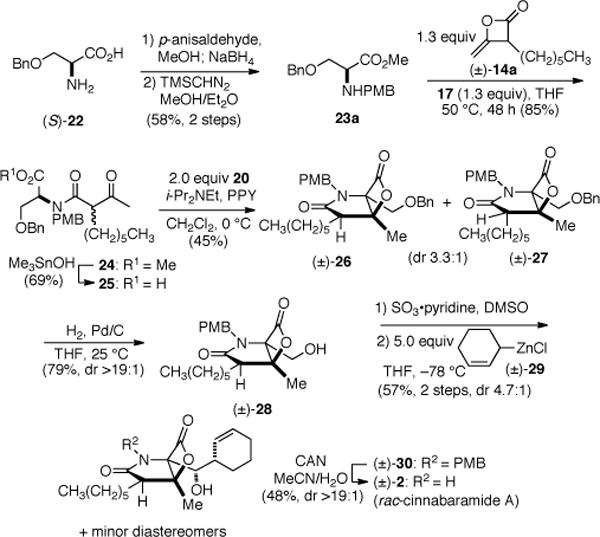
Total Synthesis of rac-Cinnabaramide A ((±)-2)
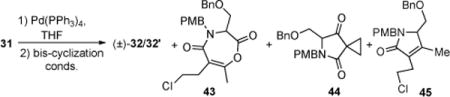
Total Synthesis of rac-Salinosporamide A ((±)-1a)
In a highly analogous manner to that described for cinnabaramide A, the synthesis of racemic salino A was accomplished, and thus only key reactions are highlighted (Scheme 3). The ketoacid substrate 31 was obtained from coupling of heteroketene dimer 14b, from heterodimerization of acetyl chloride and commercially available 4-chlorobutanoyl chloride on multigram scale by the method of Sauer,18 and serine derivative 23b bearing an allylester, which aided subsequent deprotection. In this case, the unoptimized bis-cyclization proceeded to give β-lactone 32 in 25–35% yield (dr 2–3:1), however the relative stereochemistry of the major diastereomer corresponded to salino A as confirmed by subsequent conversion to the natural product. Bicyclic β-lactone 32 was processed to racemic salino A (1a) using an identical sequence to that described for cinnabaramide A (cf. Scheme 2). Once again, it was gratifying to determine that the potentially labile β-lactone and primary chloride were compatible with several final reaction steps in the synthesis.
Scheme 3.
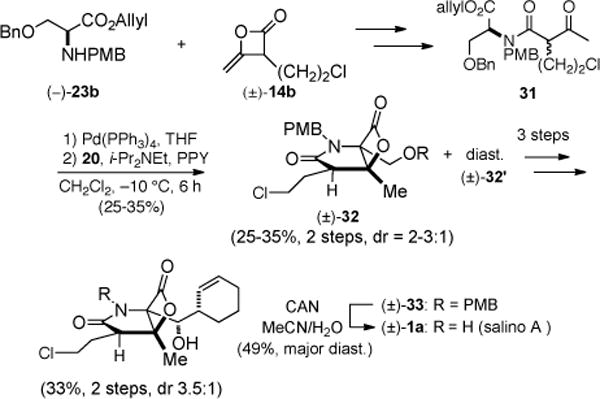
Total Synthesis of rac-Salino A ((±)-1a)
Optimization Studies of the Bis-Cyclization
Our racemic syntheses of cinnabaramide A and salino A demonstrated the viability of our synthetic approach. However, with an ultimate goal of developing an efficient, enantioselective route to a versatile salino A core (i.e. β-lactone 32) amenable to the synthesis of derivatives, we set out to identify reaction conditions that would minimize formation of observed byproducts and most critically, epimerization of the C2 stereocenter of (β)-ketoacid 34 during the bis-cyclization. Our working mechanism for this bis-cyclization invokes a nucleophile catalyzed aldol-lactonization process and follows from our previous studies with carbocycle-fused p-lactones (Scheme 4).14 Activation of the carboxylic acid is followed by transacylation with 4-PPY to provide acyl ammonium 36, and deprotonation provides ammonium enolate 37. Equilibration to the reactive conformer 37” enables subsequent aldol reaction leading to aldolate 38. Lactonization, which may occur via a ‘SN2’ process,14c,23 then delivers μ-lactone 39. While a [2+2] cycloaddition mechanism via an intermediate ketene has not been excluded,24 evidence to date including reduced conversions upon varying nucleophilic promoters (vide infra) is suggestive of nucleophile involvement in the ratedetermining or prior step. Indirect evidence comes from previously reported highly enantioselective bis-cyclization routes to carbocycle-fused β-lactones employing chiral nucleophiles.14a,c
Scheme 4.
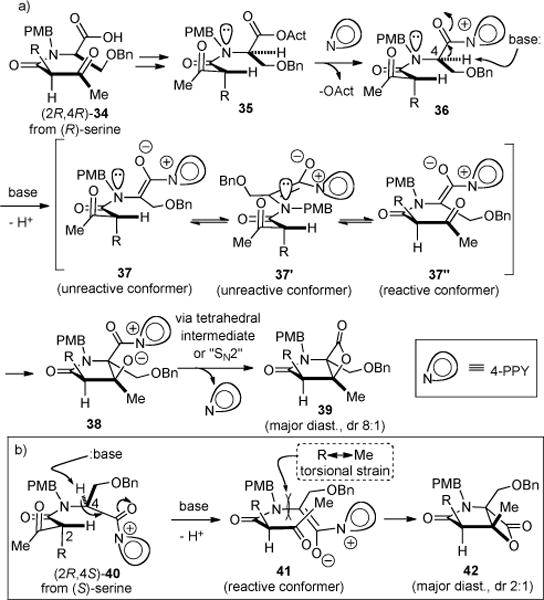
Proposed Mechanism for the bis-Cyclization with Two Diastereomeric Ketoacid Substrates (a) (2R, 4R)-34 from (R)-Serine (b) (2R, 4S)-40 from (S)-Serine
At the outset of our studies, we recognized that several energetically favored conformers dictated by A1,3-strain e.g. conformers 37 and 37′, while unproductive for aldol-lactonization since the ketone is not proximal to the ammonium enolate,25 would preserve the enantiopurity of the single stereocenter (C2). This would be possible since the α-proton is nearly in plane with the amide carbonyl in these conformations due to A1,3-strain (Scheme 4a).15 Furthermore, an expectedly higher energy conformation such as that represented by 37” is required to achieve bis-cyclization and, due to the proximity and favorable formation of a 5-membered lactam ring, aldol reaction might be expected to be facile. What we did not anticipate was the impact of the C4 absolute stereochemistry on the diastereoselectivity of the bis-cyclization. In our early optimization studies, we determined that diastereoselectivity was highly dependent on whether (S) or (R)-serine derived keto acids were employed leading to different diastereoselectivities, 1:2 vs 8:1 respectively. We suggest that the moderate diastereoselectivity observed for the bis-cyclization is governed by developing torsional strain between the methyl group (C3) of the ketone and the C2 substituent (cf. 41, R⇔Me) during the initial aldol reaction, leading to the preferred relative stereochemistry following lactonization (cf. 39). Note that only a moderate preference for this conformation (cf. 37″) over the alternative (cf. 41) is expected26 and this is reflected in the maximum observed diastereoselectivity for the bis-cyclization (dr 4-7:1, vide infra). Regarding the C4 absolute stereochemistry dependence, this is likely a result of required conformations for deprotonation imposed by the N-C4 bond rotamer of the two diastereomeric, acyl ammonium species 36 and 40 leading to ammonium enolates 37 and 41 and suggests the intriguing possibility of a memory of chirality effect which we are currently studying in related systems. With acyl ammonium 36 derived from (R)-serine, deprotonation leads directly to a N-C4 conformer that can access reactive conformer 37″ leading to major diastereomer 39. Alternatively, deprotonation of the diastereomeric acyl ammonium 40 derived from (S)-serine leads directly to reactive conformer 41 that delivers the diastereomeric β-lactone 42. This analysis assumes a rapid bis-cyclization since enantiopurity could indeed be maintained in a non-Curtin-Hammett situation, if once the higher energy reactive conformation is achieved, bis-cyclization immediately takes place. For these reasons, we utilized (R)-serine for the enantioselective synthesis (vide infra).
In early optimization studies of the bis-cyclization of ketoacid 31 directed toward salino A, we isolated and identified three by-products; enol lactone 43, cyclopropyl ketoamide 44, and γ-lactam 45 (Table 1). Only the former two by-products were observed when both 4-PPY and Hünig’s base were employed accompanied by low yields of (β)-lactones 32,32′ (Table 1, entries 1 and 2). Enol lactone 43 is likely derived from intramolecular acylation of enol 46 of the (β)-ketoamide with the activated ester (Scheme 5). Cyclopropyl ketoamide 44 may be derived from a condensation reaction of enol 46 onto the activated acid leading to (β)-diketone 47 followed by a retro-Claisen process induced by 4-PPY via 48, and finally cyclization via enol 49 leading to the cyclopropane (Scheme 5, red mechanism arrows). We next studied the use of 4-PPY (7.0 equiv) as both base and nucleophilic promoter, which reduced the amount of by-products 43 and 44 with attendant dramatic improvements in yield of β-lactone but formation of a new by-product, unsaturated γ-lactam 45 (Table 2, entry 3). Consideration of possible mechanisms for formation of this by-product suggested control experiments to determine if lactam 45 was derived from decomposition of β-lactones 32, 32′ via a decarboxylation pathway. Subjecting β-lactones 32/32′ to 4-PPY at 23 °C indeed led to lactam 45 (31%) and interestingly recovered β-lactones 32, 32′ with improved diastereomeric ratio (dr, 5:1→9:1, Scheme 6). Decomposition was much slower at (→)5 → −10 °C and was not observed if only Hünig’s base was added to β-lactones 32, 32′. The increased diastereomeric ratio suggests a differential rate of decomposition for the two diastereomers. This may be explained by a lower barrier syn-elimination (endothermic, late transition state) for β-lactone 32′ due to a more accessible proton and release of torsional strain (R⇔Me) which is absent in diastereomeric β-lactone 32. Finally, decarboxylation and protonation leads to the observed unsaturated γ-lactam 45.
Scheme 5.
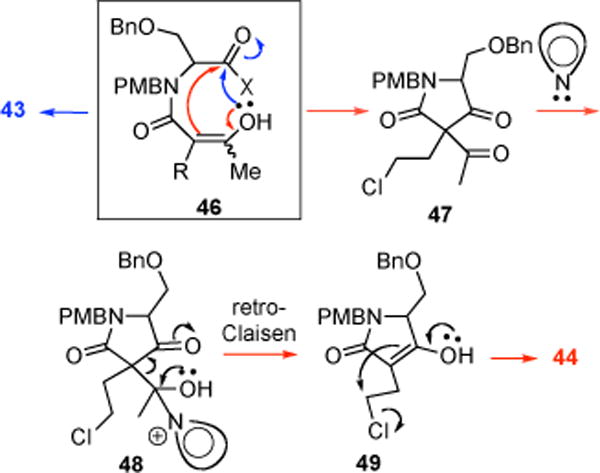
Proposed Mechanisms for Formation of By-Products 43 and 44
Table 2.
Optimization of the Bis-cyclization Leading to Bicyclic-β-Lactone (±)−32
| ||||||
|---|---|---|---|---|---|---|
| reaction conditionsa
|
% yield 32/32′d | % yield 43–45e | ||||
| entry | act. agentb (equiv) | base (equiv) | nuc.c (equiv) | solvent | ||
| 1 | 20 (1.5) | i-Pr2NEt (3.0) | 4-PPY (1.5) | CH2Cl2 | 10 | 23 (43/44) |
| 2 | 20 (1.5) | i-Pr2NEt (1.0) | 4-PPY (4.0) | CH2Cl2 | 33 | 25 (43/44) |
| 3 | 20 (1.5) | – | 4-PPY (7.0) | CH2Cl2 | 42 | 19 (43/45) |
| 4 | MsCl (1.5) | – | 4-PPY (3.5) | PhMe | 61 | 10f (rec. SM 31) |
| 5 | MsCl (1.5 | – | DMAP (3.5) | PhMe | 54 | 8f (rec. SM 31) |
All reactions were performed at −5 → −10 °C except entry 1, performed at 23 °C.
Carboxylic acid activating agent.
Nucleophilic promoter.
Yields are given for 2 steps (bis-cyclization/benzyl deprotection) and refer to isolated, purified (SiO2) products.
By-products were not easily separated and thus approximate yields are provided based on 1H NMR (500 MHz) integration.
Recovered starting material, ketoacid 31.
Scheme 6.
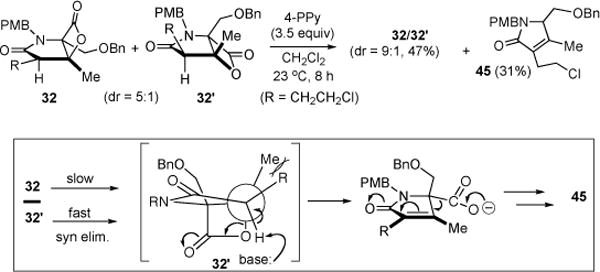
Differential Decomposition of β-Lactones 32/32′ Leading to Unsaturated γ-Lactam 45
These studies highlighted the importance of minimizing reaction time to avoid decomposition of β-lactones and minimizing C2-deprotonation to avoid these by-products and ultimately to maintain optical purity during the bis-cyclization process. Dramatic improvements were therefore realized when alternative activating agents and most importantly a less polar solvent (e.g. toluene), which may slow C2-deprotonation, was employed. Use of MsCl in toluene avoided formation of all by-products and yields of the (β)-lactones 32, 32′ increased to 61% (Table 2, entry 4). Use of the less nucleophilic dimethylaminopyridine (DMAP) also led to (β)-lactone 32, 32′ but in slightly reduced yields (54%) under similar conditions suggestive of nucleophile involvement in the rate-determining or prior step (Table 2, entry 5).
The impact of the chlorine atom on the acidity of the C2-proton was investigated under the original bis-cyclization conditions with a dual purpose of studying inductive effects on conversion and by-product formation and demonstrating the versatility of this synthetic strategy for C2-derivative synthesis. Ketoamide substrate 50a was prepared from a β-ketothioester while 50b was prepared from heteroketenedimer 14c (not shown).21 Bis-cyclization of ketoacid derived from allyl ester 50a bearing a γ-siloxy group (Table 3, entry 2) led to similar results as from ketoamide 31 bearing a γ-chloro substituent (salino core shown for comparison, Table 3, entry 1), however the δ-chloro ketoacid derived from allylester 50b led to a significant increase in yield (Table 3, entry 3). The yields obtained appear to correlate with the 1H NMR chemical shifts of the C2 protons of β-ketoamides 32, 51a-b (Table 3, entries 1–3). Comparison with the salino A core (Table 3, entry 1) shows a Δδ of 0.4 between the γ- and δ-heteroatom substituted substrates, pointing to the inductive effects and attendant increased acidity leading to by-product formation imposed by the heteroatoms. Inductive effects leading to reduced yields (70–85%) and greater by-product formation was also observed in simpler C4-unsubstituted substrates bearing α-phenyl and β-chloro C2 substituents (Table 1, entries 3 and 5) in comparison to other C2-alkyl substrates in that series (90–93% yield).
Table 3.
Variation of the C2-Side Chain: Inductive Effects on Efficiency of Bis-cyclization
| ||||
|---|---|---|---|---|
| entry | R1 | β-lact. | % yielda,b | δ Ha, Hbc |
| 1 | (CH2)2Cl | 32/32′ | 40 | 3.92, 3.94 |
| 2 | (CH2)2OTBS | 51a/51a′ | 42 | 3.92, 3.95 |
| 3 | (CH2)3Cl | 51b/51b′ | 62 | 3.52, 3.57 |
Yield for 2 steps (deprotection and bis-cyclization).
Yields refer to isolated, purified β-lactones.
Chemical shift of C2 protons (Ha/b) of diastereomeric ketoamide esters 31, 50a-b.
Total Synthesis of (−)-Salinosporamide A
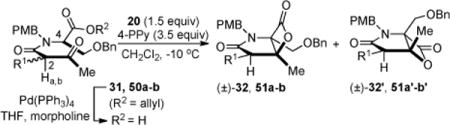
Having identified bis-cyclization conditions that improved yields and diastereoselectivity, we turned our attention to the asymmetric version of the bis-cyclization. The synthesis of salino A began with reductive amination of commercially available (R)-O-benzyl serine ((R)-22) (99% ee) with p-anisaldehyde and subsequent esterification provided allyl ester (+)-23b (Scheme 7). After extensive optimization, the desired ester (+)-23b could be obtained with minimal racemization (98% ee) in 79% yield (2 steps) by careful control of reaction temperature and duration.27 Acylation of serine derivative (+)-23b with unsymmetrical ketene dimer (±)-14b under microwave conditions gave diastereomeric β-ketoamides 31/31′ (dr 1:1) in 80% yield. Separation of the diastereomers by MPLC on mulitgram scale (3–4 g/run) provided the required (R,R)-β-ketoamide 31 (dr 30:1, 98% ee, 46%). Under acidic conditions, the undesired diastereomer (2S, 4R)-31′ could be transformed to a 1:1 mixture of diastereomers via epimerization of the C2 but not the C4 stereocenter (verified by chiral HPLC) thus achieving an effective resolution of ketene dimer (±)-14b28 and greater material throughput.29 Optimization of the Pd(0)-mediated allyl deprotection of ester (R, R)-31 was also required to minimize epimerization of the C2 stereocenter and ultimately delivered keto acid 52 with neglibile erosion of the diastereomeric purity.
Scheme 7.
9-Step Enantioselective Synthesis of (–)-Salino A (1a) from (R)-OBn Serine ((+)-22)
At the outset of the studies toward an asymmetric process, it was unclear whether the A1,3-strain induced conformational bias in β-keto tertiary amides (~4 kcal/mol) would be sufficient to avoid epimerization of this center in the time frame and under the basic conditions of the bis-cyclization. In our initial studies, application of bis-cyclization conditions developed for the racemic series8e to keto acid 52 (dr ~29:1, 98% ee) using modified Mukaiyama’s reagent but without Hünig’s base led to similar yields and diastereoselectivity (40%, dr 2.5:1) obtained previously, with an attendant erosion of optical purity of (β)-lactone 32 (85% ee). As described in our optimization studies above, we ultimately found that mesyl chloride at lower temperature with less polar solvents led to increased diastereoselectivity. When these conditions were utilized, the diasteroeselectivity improved (7:1) and minimal epimerization (~3%) was observed providing bicyclic β-lactone 32 in 35% yield and 92% ee. Further improvements in yield up to 60% were realized with longer reaction times under these same conditions, however this led to reduced diastereoselectivity and enantiopurity (dr 4:1, 88% ee). Importantly, the bis-cyclization could be performed on gram scale with comparable diastereoselectivity and retention of enantiopurity (52% over 2 steps, dr 5:1, 90% ee).
Completion of the salino A synthesis entailed hydrogenolysis to provide alcohol (−)-53 which could be separated at this stage from the minor diastereomer produced during the bis-cyclization process (75% yield). Modified Moffatt oxidation30 and addition of the zinc reagent 29 derived from the reaction of n-butyllithium with cyclohexenyl tributyltin and ZnCl2 following the procedure of Corey8a gave a mixture of C5,C6-diastereomeric alcohols (dr 11:3:1:1) in 62% yield (2 steps, Scheme 7, Procedure A ‘Proc. A’) and the desired diastereomer 33 was the major adduct. Alternately, zinc reagent 29′ could be prepared directly from commercially available 3-bromocyclohexene and activated zinc by the method of Knochel,16 which greatly simplified product purification since tin by-products were avoided (Scheme 7, Proc. B). Most importantly, this protocol gave only two diastereomers (dr 4:1), improved the yield to 74%, and avoided the use of toxic tin reagents. This procedure also enabled isolation of a novel salino A diastereomer 1a′ (C5, C6-bis-epi-salino A, not shown) whose relative stereochemistry was verified by X-ray analysis.21 Finally, deprotection of PMB-protected lactam (−)-33 led to pure (−)-salino A (1a) which correlated well with spectroscopic data reported previously (syn. [α]23D −71.3; lit. [α]25D −72.9).
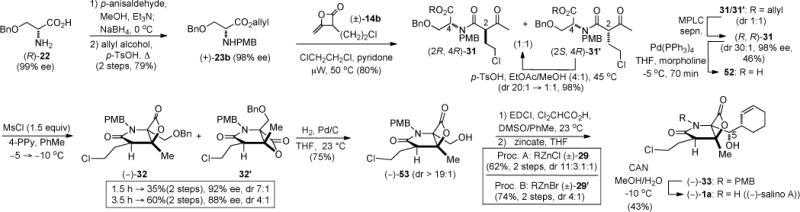
Total Synthesis of (−)-Homosalinosporamide A (Homosalino A)
In our optimization studies described above, we altered the C2-side chain with the dual purpose of studying inductive effects (cf. Table 3) and providing the one carbon homologated side-chain that could ultimately lead to (−)-homosalino A (54). Groll and coworkers have proposed that acylation of the proteasome by the β-lactone of salino A at the catalytic N-terminus (Thr 1) is followed by cyclization of the incipient alcohol with the pendant chloroethyl group leading to the formation of a tetrahydrofuran as verified by X-ray analysis (Scheme 8).13a This led us to consider the synthesis of homosalino A (54) to determine if a tetrahydropyran (cf. 57, n = 2) could be formed at the active site of the proteasome in analogy to the known formation of a tetrahydrofuran.
Scheme 8.
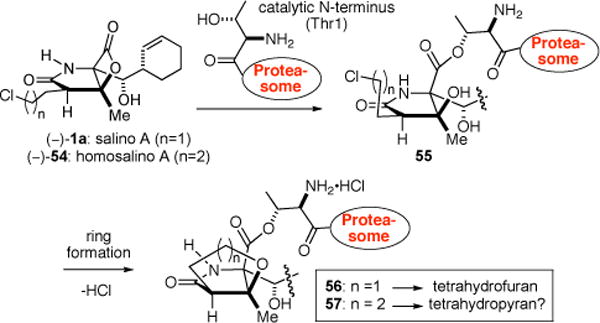
Mechanism of Inhibition of the Proteasome (CT Site) with Salino A (1a, ref. 13a) and Proposed for Homosalino A (56)
The synthesis followed an identical route as for salino A but employed heteroketene dimer (±)-14c with serine derivative (+)-23b to provide the homologous ketoester (−)-58 (Scheme 9). Bis-cyclization following ester deprotection provided the bicyclic-β-lactone (−)-59 in 60% yield (dr 3.5:1). Using an identical sequence as described for salino A, the β-lactone core was converted to (−)-homosalino A ((−)-54, 88% ee).21
Scheme 9.
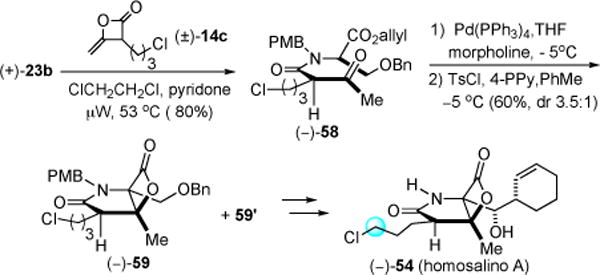
Varying the C2-Sidechain: Synthesis of (−)-Homosalino A (54)
Variation of the C4-Side-Chain of the Salino Core
Our synthetic strategy also enables facile variation of the C4-side chain by addition of organometallic reagents to the versatile aldehyde obtained by oxidation of alcohol (±)-53 (Scheme 10). Addition of Grignard reagents (dr ~3:1) followed by PMB deprotection provided the known phenyl12c and novel dimethylphenyl salino derivatives (±)-61 and (±)-63, respectively. The relative stereochemistry of the major diastereomer of phenyl derivative (±)-61 was verified by single crystal X-ray analysis.21
Scheme 10.
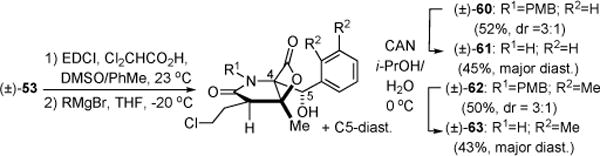
Varying the C4-Sidechain of Salino A
Biological Studies
The salino A derivatives synthesized were analyzed for their ability to inhibit the chymotrypsin-like (CT-L), caspase-like (C-L), and trypsin-like (T-L) activities of the 20S and 26S human proteasome in a luminogenic enzymatic assay (Table 4). Both velcade (entry 6) and synthetic salino A (92% ee, entry 2) were included as controls and data obtained correlates well with previous analyses.13 As expected based on known structure-activity relationships,7,13a changes made to the C5-sidechain in general led to lower activity with drastic loss of activity observed for the rac-dimethylphenyl derivative 63. The potency of the racemic phenyl derivative against the human 20A proteasome was much greater than that reported previously for the optically active phenyl derivative against the yeast 20S proteasome.12c However, as previously noted, changes in the C2-sidechain are more tolerable as indicated by (−)-homosalino A (54, Table 4, entry 5) and indeed this derivative exhibited very similar activity to the natural product with the exception of decreased inhibition of the trypsin-like activity of the 20S proteasome (~3X decrease).
Table 4.
IC50 Values for Inhibition of the Human 20S and 26S Proteasome by Salino A Derivatives in a Luminogenic Enzymatic Assaya
| IC50
|
||||
|---|---|---|---|---|
| entry | compound | CT-L activity 20S/26S (nM) | C-L activity 20S/26S (nM) | T-L activity 20S/26S (nM) |
| 1 | rac-cinnabaramide A, (±)−2 | 2.8±0.6/2.2±0.04 | 125±12b/230+24b | 260±29b/412±83 |
| 2 | (−)-salino A (1a) (92% ee) | 0.8±0.08/2.5±1.3 | 111±22/156±18 | 39±7/37±16 |
| 3 | rac-phenyl deriv.(61) | 29±0.7/57±20 | 788±50/1585±633 | 702±235/589±120 |
| 4 | rac-diMePh (63) | inact.b,c | inact.b,c | inact.b,c |
| 5 | (−)-homosalino (54) (88% ee) | 0.7±0.04/2.3±1.1 | 144±12/188±3 | 118±28/125±30 |
| 6 | velcade | 2.6 ±0.7/7.4±1.9 | 25±7/72±17 | 254±24/680±150 |
IC50 values are the mean ± standard deviation of 4 or more experiments.
The average of 2 experiments.
Essentially inactive at >99,000 nM.
(CT-L: chymotrypsin-like activity; C-L: caspase-like activity; T-L: trypsin-like activity)
In summary, we developed a concise, 9-step enantioselective route to (−)-salino A and derivatives from O-benzyl serine. The key bis-cyclization of a β-ketoamide, amenable to gram scale, constructs both the γ-lactam and the fused-β-lactone in one operation leading to the brevity of the synthesis. The flexibility of the described strategy derives from the versatility of the bicyclic core (−)-32 enabling attachment of various C4-sidechains, even in the presence of the β-lactone, and the ability to use alternative ketene dimers 14 to vary the C2 side chain. Several derivatives including ((β))-homosalino A were synthesized and their activity toward proteasome inhibition was measured. The ability of the described (β)-keto tertiary amide substrates to maintain stereochemical integrity by virtue of A1,3 strain raises interesting questions regarding how such integrity is maintained with (β)-keto secondary amides, known salino A precursors,4c or if a dynamic kinetic resolution is operative during related biosynthetic aldol-lactonizations.31 The synthesis of additional hypothesis-driven salino derivatives by the described strategy, their bioactivity, and crystallographic studies of their interaction with the proteasome will be reported in due course.
Experimental Section
(2R, 4R)-(β)-Ketoacid, 52
To a solution of ketoamide (2R, 4R)-31 (2.30 g, 4.55 mmol, ~ 32:1 dr) in THF (91.0 mL) at −5 °C (ice and saturated NaCl solution) was added Pd(PPh3)4 (526 mg, 0.455 mmol), followed by immediate addition of morpholine (0.475 ml, 5.46 mmol). The reaction mixture was stirred at −5 °C for 70 min and diluted with ice cold Et2O (800 mL). A 0.02 N HCl solution was added until the pH was measured to be ~3. The layers were separated and the organic layer was washed with brine (400 mL), dried over MgSO4 and concentrated. The crude ketoacid (2R, 4R)-52 (~ 29:1 dr according to 500 MHz 1H NMR) was used directly in the subsequent step without further purification. (Note: longer reaction times led to epimerization).
Gram-scale bis-cyclization leading to benzyloxy-β-lactone, (−)-32/32′
To a solution of 4-pyrrolidinopyridine (2.60 g, 18.2 mmol, 4.0 equiv) in toluene (84 mL) at −5 °C (ice and saturated NaCl solution) was added MsCl (0.53 mL, 6.83 mmol, 1.5 equiv). Immediately, a solution of the freshly prepared ketoacid (R,R)-5a (total amount from above, ~4.55 mmol) in toluene (25 mL) was added to the resulting suspension via syringe pump over 45 min and 5 mL of additional toluene were used to ensure complete transfer. After 3 h, the reaction mixture was diluted with ice cold Et2O (700 mL) and washed with 20% CuSO4 solution (500 mL) to remove most of the 4-pyrrolidinopyridine, saturated NH4Cl (500 mL), and then washed with water (2 × 500 mL). The organic layer was dried over MgSO4, filtered, and concentrated. The residue was purified by flash chromatography (1:9→3:7 EtOAc/hexanes) to give a mixture of two inseparable β-lactones (−)-32/32′ (1.05 g, 52%, dr 5:1, 500 MHz 1H NMR) as a yellow oil and recovered ketoacid (10 %, 4:1 dr). Rf = 0.36 (20% EtOAc/hexanes). Enantiomeric excess of (β)-lactone 32 was determined to be 90%. The diasteromeric (β)-lactones were carried directly forward to the deprotection at which point they could be separated. Data for ((β))-32: IR (neat) 1830, 1703 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.32–7.36 (m, 3H), 7.13–7.15 (m, 4H), 6.80 (d, J = 8.5 Hz, 2H), 4.73 (d, J = 15.5 Hz, 1H), 4.31 (d, J = 15.5 Hz, 1H), 4.17 (d, J = 12.0 Hz, 1H), 4.13 (d, J = 11.5 Hz, 1H), 4.01 (ddd, J = 5.0, 7.5, 12.5 Hz, 1H), 3.77–3.81 (m, 1H), 3.77 (s, 3H), 3.73 (d, J = 11.5 Hz, 1H), 3.57 (d, J = 11.5 Hz, 1H), 2.91 (t, J = 7.5 Hz, 1H), 2.31–2.38 (m, 1H), 2.10–2.16 (m, 1H), 1.72 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 173.8, 166.1, 159.2, 136.4, 129.2(2C), 128.6, 128.5(2C), 128.2, 128.0(2C), 113.9(2C), 83.4, 79.3, 73.5, 61.6, 55.2, 45.0, 44.3, 42.5, 28.4, 19.2; LRMS (ESI) Calcd. for C24H27ClNO5 [M+H] 444, found 444. Enantiomeric excess of (−)-32 was determined to be 92% by chiral HPLC (CHIRALPAK IA, 250 × 4.6 mm (L × I.D.), solvent (isocratic) 87:13 hexanes/2-propanol, flow rate 1.0 mL/min, λ = 230 nm). Retention times: (−)-32: 13.68 min; (+)-32: 16.12 min.
Hydroxy β-lactone (−)-53
To a mixture of β-lactones (−)-32 and 32′ (260 mg, 0.586 mmol, dr 7:1) in THF was added palladium on carbon (52 mg, 20 wt%). After evacuating twice by aspirator vacuum, and refilling with H2, a balloon of H2 was attached to the flask and the heterogenous solution was stirred vigorously at 23 °C for 12 h. The reaction mixture was then diluted with Et2O, and dried over MgSO4. The organics were filtered through a pad of Celite, concentrated, and purified by MPLC (SiO2, 5:95 EtOAc/CH2Cl2) to give the desired alcohol (−)-53 in 75% yield (155 mg, dr >19:1, 92% ee) as a waxy solid. Rf = 0.29 (5:95 EtOAc/CH2Cl2); [λ]23D = − 67.0 (c = 0.95, CHCl3). Data for (−)-53: IR (neat) 3449, 1831, 1687 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.30 (d, J = 8.5 Hz, 2H), 6.89 (d, J = 8.5 Hz, 2H), 5.13 (d, J = 15.0 Hz, 1H), 4.06 (d, J = 15.5 Hz, 1H), 4.03 (ddd, J = 5.5, 7.5, 12.5 Hz, 1H), 3.92 (dd, J = 9.0, 13.5 Hz, 1H), 3.85 (dd, J = 4.5, 13.5 Hz, 1H), 3.80 (s, 3H), 3.78–3.82 (m, 1H), 2.94 (t, J = 7.0 Hz, 1H), 2.32–2.38 (m, 1H), 2.01–2.18 (m, 1H), 1.77 (s, 3H), 0.86 (dd, J = 5.0, 9.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) 6 174.2, 166.7, 159.6, 129.0(2C), 128.7, 114.7(2C), 83.6, 80.2, 55.3, 55.1, 44.9, 44.1, 42.4, 28.4, 19.1; LRMS (ESI) Calcd. for C17H21ClNO5 [M+H] 354, found 354.
Representative Procedure for Modified Moffatt Oxidation and Zincate Formation/Addition as Described for N-PMB-salino A, (−)-33
To a solution of alcohol (−)-53 (42 mg, 0.119 mmol, dr >19:1) in DMSO/toluene (0.9 mL/0.9 mL) was added EDCI (0.114mg, 0.595mmol), and dichloroacetic acid (5 μL, 0.060 mmol) at 23 °C. The reaction mixture was stirred for 6 h, and diluted with EtOAc (100 mL). The reaction mixture was acidified using 0.1 N HCl to pH 3. The organic layer was then washed with brine (60 mL), dried over MgSO4, filtered, and concentrated. The crude aldehyde was used for the next step without further purification due to some instability of the aldehyde observed upon flash column chromatography.
Preparation of a stock solution of zincate 29′: Zinc dust (1.64 g, 25.0 mmol, Baker, 20 mesh) was activated by the addition of an HCl solution (5 mL, 1.0 M) in a 25 mL round-bottomed flask. After stirring vigorously for 15 min at 23 °C, the acid solution was decanted and the zinc was repeatedly washed with dry THF (3 × 5 mL). the zinc was then dried under high vacuum for 2 h. Addition of dry THF (5 mL) was followed by slow addition of 3-bromo-cyclohexene (0.806 g, 5.0 mmol, Acros) over 15 min. The resulting mixture was stirred at 23 °C for 18 h, and then transferred via syringe to a dry centrifuge tube to remove the excess zinc suspension (10 min, 80 rpm). The resulting clear solution was then utilized directly, following titration as described below, being careful not to disturb the pellet at the bottom during transfers. The concentration of the zincate 29′ solution was determined as follows: A stock solution of iodine in THF was prepared by addition of iodine (254 mg, 1.0 mmol) in 10 mL of THF leading to a 100 μM solution. A portion (500 μL) of this iodine solution was placed into a dry 5 mL round-bottomed flask equipped with a magnetic stir bar. The zincate solution, after being centrifuged, was added dropwise into the iodine stock solution until the red color disappeared. Using this procedure, the prepared zincate solution was determined to be ~200 μM and was used (in excess) directly in the following reaction.
To a solution of the crude aldehyde (~0.119 mmol) in THF (1.0 mL) at −78 °C was added ~1.2 mL (0.238 mmol, ~200 pM in THF, ~2.0 equiv) of zincate 29′ via syringe pump over 30 min. The resulting mixture was slowly warmed up to −20 °C for 1 h, quenched with water and diluted with Et2O (120 mL). Saturated NH4Cl was added until a pH of 7 was achieved. The layers were separated and then the organic layer was washed with brine (80 mL). The filtrate was dried over MgSO4, filtered, and concentrated. The residue was purified by flash chromatography (95:5 → 85:5 EtOAc/hexanes) to give a mixture of two inseparable diastereomers of N-PMB salino A (−)-33 and C5, C6-bis-epi-N-PMB salino A (−)-33′ (38 mg, 74%, dr = 4:1 according to 500 MHz 1H NMR) as a colorless oil, which was carried directly to the next step without further purification.
Salino A ((−)-1a) and C5, C6-bis-epi-Salino A ((−)-la’)
To a methanolic solution (0.8 mL) containing the mixture of alcohols (−)-33/33′ (38 mg, 0.088 mmol) was added an aqueous solution of CAN (0.240 g, 0.44 mmol) in H2O (0.2 mL) at 0 °C dropwise. After stirring at 0 °C for 6 h, the reaction mixture was diluted with EtOAc (100 mL), washed with saturated solution of NaHCO3, and brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was purified by flash chromatography (5:95 → 15:85 EtOAc/CH2Cl2) to give salino A (−)-1a (14 mg, 51 %, dr > 19:1, 500 MHz 1H NMR) as a white solid (previously described, see ref. 8k) in addition to a novel diastereomeric salino A 1a′ (3.3 mg, 12%, dr > 19:1, 500 MHz 1H NMR). Data for C5, C6-bis-epi-salino A (1a′): Rf = 0.56 (33 % EtOAc/hexanes); IR (neat) 2931, 1830, 1702 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.28 (s, 1H), 6.10 – 6.14 (m, 1H), 5.85 – 5.88 (m, 1H), 3.99 (ddd, J = 5.0, 7.5, 12.5 Hz, 1H), 3.83 (dd, J = 7.5, 9.0 Hz, 1H), 3.78 (ddd, J = 5.0, 7.5, 12.5 Hz, 1H), 2.83 (t, J = 7.5 Hz, 1H), 2.48–2.50 (m, 1H), 2.27–2.34 (m, 1H), 2.11 – 2.18 (m, 1H), 2.06–2.08 (m, 2H), 1.98 (d, J = 9 Hz, 1H), 1.80 – 1.91 (m, 2H), 1.87 (s, 3H), 1.60 – 1.67 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 175.8, 165.0, 135.2, 123.5, 85.4, 78.7, 71.8, 44.9, 42.7, 38.4, 28.5 26.6, 25.2, 21.8, 19.4; HRMS (MALDI) Calcd. for C15H20ClNO4Na [M+Na] 336.0973, found 336.0957.
Proteasome Inhibition Assay
The Proteasome-Glo Assay (Promega) was used, which is a homogeneous coupled-enzyme system that utilizes luminogenic substrates to measure the three proteolytic activities (chymotrypsin-like(CT-L), trypsin-like (T-L), and caspase-like (C-L)) associated with the proteasome. Purified human 20S and 26S proteasome (BioMol/Enzo Life Sciences, Inc) at 0.5 – 2 μg/mL was incubated in 10 μL of assay buffer (10 mM Hepes, pH 7.6) at room temperature for 2030 min in the presence of the salino A derivatives at various concentrations or equivalent volume of solvent DMSO as a control along with salino A and Velcade.® The reaction was initiated by addition of 10 μL of 2× solution of various substrates. Reactions were performed at room temperature. After 1 h, inhibition of each proteasomal activity was measured on a multilabel microtiter plate reader.
Supplementary Material
Acknowledgments
We thank the NIH (GM069784) and the Welch Foundation (A-1280) for support. We thank Drs. Joe Reibenspies and Nattamai Bhuvanesh (TAMU) for X-ray analysis. We thank Mr. Nicholas Krudy (TAMU NSF REU, CHE-0755207) for technical assistance. The U.S. Department of Energy Genomics: GTL Program, http://doegenomestolife.org is acknowledged for the proteasome graphic used in the TOC entry.
Footnotes
Supporting Information Available. General procedures for synthesis and the human proteasome inhibiton assay and characterization data including 1H and 13C NMR spectra (for compounds 12e, 14b, 21e, (+)-23b, 31, 44, 45, 61, 63) and X-ray analysis (61, 1a′). This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Sorensen EJ, Theodorakis E. Tetrahedron. 2006;62 5159 and papers following this Preface to the Tetrahedron Symposia-In-Print “Nature-Inspired Approaches to Chemical Synthesis.”. [Google Scholar]
- 2.(a) Voorhees PM, Dees EC, O’Neil B, Orlowski RZ. Clin Cancer Res. 2003;9:6316. [PubMed] [Google Scholar]; (b) Rajkumar SV, Richardson PG, Hideshima T, Anderson KC. J Clin Oncol. 2005;23:630. doi: 10.1200/JCO.2005.11.030. [DOI] [PubMed] [Google Scholar]; (c) Joazeiro CAP, Anderson KC, Hunter T. Cancer Res. 2006;66:7840. doi: 10.1158/0008-5472.CAN-06-2033. [DOI] [PubMed] [Google Scholar]
- 3.Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical WF. Angew Chem Int Ed. 2003;42:355. doi: 10.1002/anie.200390115. [DOI] [PubMed] [Google Scholar]
- 4.For a review describing the development of salinosporamide A as an anticancer agent, see:; Fenical W, Jensen PR, Palladino MA, Lam KS, Lloyd GK, Potts BC. Bioorg Med Chem. 2009;17:2175. doi: 10.1016/j.bmc.2008.10.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stadler M, Bitzer J, Mayer-Bartschmid A, Muller H, Benet-Buchholz J, Gantner F, Tichy HV, Reinemer P, Bacon KB. J Nat Prod. 2007;70:246. doi: 10.1021/np060162u. [DOI] [PubMed] [Google Scholar]
- 6.Omura S, Fujimoto T, Otoguro K, Matsuzaki K, Moriguchi R, Tanaka H, Sasaki Y. J Antibiot. 1991;44:117. doi: 10.7164/antibiotics.44.113. [DOI] [PubMed] [Google Scholar]
- 7.Manam RR, Macherla VR, Tsueng G, Dring CW, Weiss J, Neuteboom STC, Lam KS, Potts BC. J Nat Prod. 2009;72:295. doi: 10.1021/np800578e. [DOI] [PubMed] [Google Scholar]
- 8.(a) Reddy LR, Saravanan P, Corey EJ. J Am Chem Soc. 2004;126:6230. doi: 10.1021/ja048613p. [DOI] [PubMed] [Google Scholar]; (b) Reddy LR, Fournier JF, Reddy BVS, Corey EJ. Org Lett. 2005;7:2699. doi: 10.1021/ol0508734. [DOI] [PubMed] [Google Scholar]; (c) Endo A, Danishefsky SJ. J Am Chem Soc. 2005;127:8298. doi: 10.1021/ja0522783. [DOI] [PubMed] [Google Scholar]; (d) Mulholland NP, Pattenden G, Walters IAS. Org Biomol Chem. 2006;4:2845. doi: 10.1039/b607109k. [DOI] [PubMed] [Google Scholar]; (e) Ma G, Nguyen H, Romo D. Org Lett. 2007;9:2143. doi: 10.1021/ol070616u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Ling T, Macheria VR, Manam RR, McArthur KA, Potts BCM. Org Lett. 2007;9:2289. doi: 10.1021/ol0706051. [DOI] [PubMed] [Google Scholar]; (g) Mulholland NP, Pattenden G, Walters IAS. Org Biomol Chem. 2008;6:2782. doi: 10.1039/b803818j. [DOI] [PubMed] [Google Scholar]; (h) Takahashi K, Midori M, Kawano K, Ishihara J, Hatakeyama S. Angew Chem Int Ed. 2008;47:6244. doi: 10.1002/anie.200801967. [DOI] [PubMed] [Google Scholar]; (i) Fukuda T, Sugiyama K, Arima S, Harigaya Y, Nagamitsu T, Omura S. Org Lett. 2008;10:4239. doi: 10.1021/ol8016066. [DOI] [PubMed] [Google Scholar]; (j) Mosey RA, Tepe JJ. Tetrahedron Lett. 2009;50:295. [Google Scholar]; (k) Nguyen H, Ma G, Romo D. Chem Comm. 2010;46:4803. doi: 10.1039/c0cc00607f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Caubert V, Masse J, Retailleau P, Langlois N. Tetrahedron Lett. 2007;48:381. [Google Scholar]; (b) Villanueva MI, Rupnicki L, Lam HW. Tetrahedron. 2008;64:7896. [Google Scholar]; (c) Momose T, Kaiya Y, Hasegawa J, Sato T, Chida N. Synthesis. 2009;17:2983. [Google Scholar]; (d) Struble JR, Bode JW. Tetrahedron. 2009;65:4957. doi: 10.1016/j.tet.2009.03.103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ling T, Potts BC, Macherla VR. J Org Chem. 2010;75:3882. doi: 10.1021/jo100432g. [DOI] [PubMed] [Google Scholar]
- 10.For a recent review on syntheses of lactacystin and salino A, see:; Shibasaki M, Kanai M, Fukuda N. Chem Asian J. 2007;2:20. doi: 10.1002/asia.200600310. [DOI] [PubMed] [Google Scholar]
- 11.(a) Macherla VR, Mitchell SS, Rama Rao Manam RR, Reed KA, Chao T-H, Nicholson B, Deyanat-Yazdi G, Mai B, Jensen PR, Fenical WF, Neuteboom STC, Lam KS, Palladino MA, Potts BM. J Med Chem. 2005;48:3864. doi: 10.1021/jm048995+. [DOI] [PubMed] [Google Scholar]; (b) Hogan PC, Corey EJ. J Am Chem Soc. 2005;127:15386. doi: 10.1021/ja056284a. [DOI] [PubMed] [Google Scholar]
- 12.(a) Beer LL, Moore BS. Org Lett. 2007;9:845. doi: 10.1021/ol063102o. [DOI] [PubMed] [Google Scholar]; (b) Eustaquio AS, Moore BS. Angew Chem Int Ed. 2008;47:3936. doi: 10.1002/anie.200800177. [DOI] [PubMed] [Google Scholar]; (c) Nett M, Gulder Tobias AM, Kale Andrew J, Hughes CC, Moore BS. J Med Chem. 2009;52:6163. doi: 10.1021/jm901098m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Liu Y, Hazzard C, Eustaquio AS, Reynolds KA, Moore BS. J Am Chem Soc. 2009;131:10376. doi: 10.1021/ja9042824. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Eusthquio AS, McGlinchey RP, Liu Y, Hazzard C, Beer LL, Florova G, Alhamadsheh MM, Lechner A, Kale AJ, Kobayashi Y, Reynolds KA, Moore BS. Proc Nat Acad Sci USA. 2009;106:12295. doi: 10.1073/pnas.0901237106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Groll M, Huber R, Potts BCM. J Am Chem Soc. 2006;128:5136. doi: 10.1021/ja058320b. [DOI] [PubMed] [Google Scholar]; (b) Mosey RA, Tepe JJ. Tetrahedron Lett. 2009;50:295. [Google Scholar]; (c) Groll M, McArthur KA, Macherla VR, Manam RR, Potts BC. J Med Chem. 2009;52:5420. doi: 10.1021/jm900559x. [DOI] [PubMed] [Google Scholar]; (d) Manam R, McArthur KA, Chao TH, Weiss J, Ali JA, Palombella VJ, Groll M, Lloyd GK, Palladino MA, Neuteboom STC, Macherla VR, Potts BCM. J Med Chem. 2008;51:6711. doi: 10.1021/jm800548b. [DOI] [PubMed] [Google Scholar]
- 14.(a) Cortez GS, Tennyson R, Romo D. J Am Chem Soc. 2001;123:7945. doi: 10.1021/ja016134+. [DOI] [PubMed] [Google Scholar]; (b) Oh SH, Cortez GS, Romo D. J Org Chem. 2005;70:2835. doi: 10.1021/jo050024u. [DOI] [PubMed] [Google Scholar]; (c) Henry-Riyad H, Lee CS, Purohit VC, Romo D. Org Lett. 2006;8:4363. doi: 10.1021/ol061816t. [DOI] [PubMed] [Google Scholar]; (d) Purohit VC, Matla AS, Romo D. J Am Chem Soc. 2008;130:10478. doi: 10.1021/ja803579z. [DOI] [PubMed] [Google Scholar]
- 15.Evans DA, Ennis MD, Le T, Mandel N, Mandel G. J Am Chem Soc. 1984;106:1154. [Google Scholar]
- 16.Ren H, Dunet G, Mayer P, Knochel P. J Am Chem Soc. 2007;129:5376. doi: 10.1021/ja071380s. [DOI] [PubMed] [Google Scholar]
- 17.Jacobsen and coworkers had previously demonstrated the stability of a related spiro-β-lactone in their studies toward omuralide, see:; Balskus EP, Jacobsen EN. J Am Chem Soc. 2006;128:6810. doi: 10.1021/ja061970a. [DOI] [PubMed] [Google Scholar]
- 18.(a) Sauer JC. J Am Chem Soc. 1947;69:2444. [Google Scholar]; b) Purohit VC, Richardson RD, Smith JW, Romo D. J Org Chem. 2006;71:4549. doi: 10.1021/jo060392d. [DOI] [PubMed] [Google Scholar]; c) Duffy RJ, Morris KA, Romo D. J Am Chem Soc. 2005;127:16754. doi: 10.1021/ja053478h. [DOI] [PubMed] [Google Scholar]
- 19.Calter MA, Orr RK, Song W. Org Lett. 2003;5:4745. doi: 10.1021/ol0359517. [DOI] [PubMed] [Google Scholar]
- 20.Furlám RLE, Emesto G, Mata EG, Masearetti OA. Tetrahedron. 1998;54:13023. [Google Scholar]
- 21.See supporting information for experimental details.
- 22.Parikh JP, Doering WE. J Am Chem Soc. 1967;89:5505. [Google Scholar]
- 23.For lead references to this mechanistic possibility, see:; (a) Adler M, Adler S, Boche G. J Phys Org Chem. 2005;18:193. [Google Scholar]; (b) Fox JM, Dmitrenko O, Liao L, Bach RD. J Org Chem. 2004;69:7317. doi: 10.1021/jo049494z. [DOI] [PubMed] [Google Scholar]
- 24.For previous reports of β-lactones from keto acid derivatives via proposed [2+2] mechanisms, see:; a) Boswell GA, Dauben WG, Ourisson G, Rull T. Bull Soc Chim Fr. 1958:1598. [Google Scholar]; b) Kagan HB, Jacques J. Bull Soc Chim Fr. 1958:1600. [Google Scholar]; c) Brady WT, Gu YQ. J Org Chem. 1988;53:1353. [Google Scholar]; d) Reddy LR, Corey EJ. Org Lett. 2006;8:1717. doi: 10.1021/ol060464n. [DOI] [PubMed] [Google Scholar]; For a previous report of an aldol-lactonzation pathway, see:; e) Merlic CA, Marlog BC. J Org Chem. 2003;68:6056. doi: 10.1021/jo034476n. [DOI] [PubMed] [Google Scholar]; For an anionic version, see:; (f) Shindo M, Matsumoto K, Sato Y, Shishido K. Org Lett. 2001;3:2029. doi: 10.1021/ol0159928. [DOI] [PubMed] [Google Scholar]
- 25.The alternate N-C1 amide rotamer, which is also accessible energetically but also unproductive for aldol-lactonization, is not considered in this analysis.
- 26.The preferred conformation of 2-butanone places the methyl and carbonyl oxygen nearly in plane (eclipsed conformation). However, the energy difference is low (~1 kcal/mol), see:; Eliel EL, Wilen SH. Stereochemistry of Oganic Compounds. Wiley & Sons; New York: 1994. p. 616617. [Google Scholar]
- 27.Optical purities were determined by chiral HPLC analysis (ref. 21).
- 28.Attempts to prepare the required optically active heteroketene dimer 14b by the method of Calter (ref. 19) led to very low yields.
- 29.Chiral HPLC analysis verified that epimerization only occurred at C2 within limits of detection (ref. 21).
- 30.(a) Pfitzner KE, Moffatt JG. J Am Chem Soc. 1965;87:5661. [Google Scholar]; (b) Coulton S, Southgate R. J Chem Soc, Perkin Trans. 1992;1:961. [Google Scholar]
- 31.The C2-epimer of salino A has been isolated, see ref. 11.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.