

Abstract
Nephropathy is a common and progressive complication of sickle cell anemia (SCA). In SCA mice, we found that hyperangiotensinemia in the absence of hypertension underlies nephropathy, and its downregulation by losartan, an angiotensin-II-receptor-1 blocker, reduced albuminuria and progression of nephropathy. Therefore, we performed a phase-2 trial of oral losartan, given for 6 months, to explore whether it reduced albuminuria in children and adults with SCA. Participants were allocated to groups defined by class of baseline urinary albumin-to-creatinine ratio (UACR): no albuminuria (NoA), microalbuminuria (MicroA), and macroalbuminuria (MacroA). The primary endpoint was a ≥25% reduction UACR from baseline. There were 32 evaluable participants (mean age 24 years; NoA=14, MicroA=12, MacroA=6). The primary endpoint was met in 83% of the MacroA group (P<0.0001) and 58% of the MicroA group (P<0.0001). Median fold-change in UACR was −0.74 for MacroA and −0.46 for MicroA. In MacroA and MicroA, UACR classification improved in 50% but worsened in 11%. Urine osmolality and estimated glomerular filtration rate (eGFR) did not change significantly. Losartan was discontinued in 3 participants [leg cramps, N=1; decline in eGFR >25% (142→104 mL/minute/1.73 m2), N=1; rise in serum creatinine >50% (0.2→0.3 mg/dL), N=1]. Albuminuria was associated with diastolic dysfunction and impaired functional capacity, although cardiopulmonary status was unchanged after 6 months of losartan therapy. In summary, losartan decreased urinary albumin excretion in most participants with albuminuria. Those with macroalbuminuria had the greatest benefit. This study forms the basis for a phase-3, randomized, placebo-controlled trial of losartan for the nephropathy of SCA.
Keywords: sickle cell disease, kidney, nephropathy, losartan, albuminuria, microalbuminuria, proteinuria, angiotensin receptor blocker
Introduction
Sickle cell anemia (SCA) affects the kidney in many ways.1,2 Young children with SCA have a urinary concentrating defect, increased glomerular filtration rate (GFR), supranormal proximal tubular function, and an impaired ability to excrete potassium. Medullary tubulointerstitial damage can also cause hematuria. With increasing age, GFR declines3 and glomerulopathy develops, which manifests initially as microalbuminuria (MicroA).4,5 Glomerulopathy is found in 20–25% of children and 20–70% of adults with SCA.4–12 Some studies suggest that the development of proteinuria or macroalbuminuria (MacroA) may precede the development of chronic kidney disease and end-stage renal disease (ESRD).13,14 ESRD is associated with a high mortality in SCA.14–16
Vaso-occlusive ischemia and infarction and anemia-related hyperfiltration are thought to be the main causes of sickle cell nephropathy,1,2 but the downstream, molecular pathogenesis, especially of the glomerulopathy, is poorly understood. We recently reported that mice and humans with SCA have hyperangiotensinemia without hypertension.17 In mice, we found that excessive activation of angiotensin-II receptor 1 (AT1R) contributes to glomerulopathy.18 Pharmacotherapy to interfere with pathological angiotensin signaling, therefore, may be beneficial for the nephropathy of SCA. Indeed, angiotensin converting enzyme (ACE) inhibition with captopril or enalapril has been reported to decrease albuminuria in humans with SCA,12,19–22 although there is no conclusive evidence of efficacy.23
Angiotensin-II is produced by the action of ACE on angiotensin-I. Angiotensin-II signals through two receptors, AT1R and angiotensin-II receptor 2 (AT2R). Therefore, ACE inhibitors decrease signaling through both AT1R and AT2R. While AT1R signaling contributes to glomerulopathy,18 AT2R signaling may be renoprotective.24–26 Consequently, ACE inhibitors block both pathological (AT1R) and potentially renoprotective (ATR2) signaling. In contrast, angiotensin receptor blockers (ARBs), like losartan and valsartan, only inhibit signaling via AT1R, leaving the potentially renoprotective and cardioprotective effects of AT2R signaling unopposed. 18,24–26 ARBs are also less likely to cause cough than ACE inhibitors,27 so ARBs may be better tolerated by people with SCA, in whom asthma and airway hyper-reactivity are so common.28
Given the different mechanism of action and side-effect profile of ARBs, compared to ACE inhibitors, we performed a phase-2, open-label trial of losartan in children and adults with SCA to explore its effects on sickle cell nephropathy. Our primary hypothesis was that losartan therapy would decrease the degree of albuminuria by ≥25% in participants with microalbuminuria (MicroA). We chose albuminuria as the primary outcome measure because it is a sensitive and early marker of glomerulopathy that portends renal failure.4,5,29 Reduction in urinary albumin excretion is also a therapeutic goal in other forms of kidney disease, such as diabetic nephropathy.30 We also measured the effects of losartan on other aspects of sickle cell nephropathy, urinary concentrating ability and GFR, and explored its effects on the cardiac phenotype of SCA.
Methods
Study Overview
This was a multicenter, phase 2, open-label study of losartan for sickle cell nephropathy. Participants were enrolled at 9 centers in the United States between 2012 and 2015. Institutional review boards at each site and a central data and safety monitoring board (DSMB) approved and monitored the study. Adult participants provided written informed consent. Minors provided written assent, when applicable, and their parents or legal guardians provided written permission. The study was conducted according to guidelines of the Declaration of Helsinki and the International Conference on Harmonisation and registered at clinicaltrials.gov (NCT01479439).
Selection of Participants
Complete selection criteria are provided in Supplemental Table 1. The main inclusion criteria were: homozygous sickle cell anemia or sickle-β0-thalassaemia; age ≥6 years (and <21 years for the no albuminuria group) and a stable dose of hydroxyurea in the 3 months preceding enrollment (if taking hydroxyurea). The main exclusion criteria were: estimated glomerular filtration rate by creatinine clearance (eGFR-CrCl) <60 mL/minute/1.73 m2; gross hematuria; hyperkalemia (K≥5.5 mEq/L); hypersensitivity to ARBs; and chronic transfusion therapy. Treatment with an ACE inhibitor or ARB in the 2 weeks preceding enrollment was exclusionary, but a controlled washout period could allow individuals being treated with these agents to participate.
Allocation of Participants
Participants were allocated into 3 pre-specified groups defined by baseline urinary albumin-to-creatinine ratio (UACR): no albuminuria (<30 mg/g; NoA); microalbuminuria (30–300 mg/g; MicroA); and macroalbuminuria (>300 mg/g; MacroA). UACR measurements were performed on first-morning-urine samples. We included the NoA group to explore a secondary hypothesis that losartan would prevent conversion to a classification of MicroA or MacroA during the study. Enrollment into each group stopped when its pre-specified sample size was reached.
Study Intervention
All participants received open-label losartan potassium by mouth as tablets (25 mg, 50 mg, or 100 mg) or an oral suspension (2.5 mg/mL). Complete dosing specifications are provided in Supplemental Table 2. Briefly, the starting dose was determined by age group (6–16 or >16 years). A lower starting dose was specified for concomitant diuretic therapy, volume depletion, or hepatic impairment. Participants in the baseline NoA group remained on the fixed (starting) dose, unless toxicity necessitated a reduction in dose. Those in the baseline MicroA or MacroA groups had up to 2 increases in dose in the first 12 weeks of the study. The dose could be increased if there was no improvement in class of albuminuria after at least 2 weeks at a particular dose. Reduction in dose or cessation of losartan due to toxicity could occur at any time.
Concomitant Treatments
Concomitant treatment with hydroxyurea was allowed, but the dose must have been stable in the 3 months preceding enrollment. Clinically indicated changes in dose of hydroxyurea therapy (e.g., for change in weight or toxicity) were permitted during the study. Chronic transfusion therapy was an exclusion criterion, but episodic transfusions of blood could be given for clinical indications during the study. Treatment with ACE inhibitors, other ARBs, or potassium-sparing diuretics was not allowed.
Withdrawal of Participants
Withdrawal criteria included: pregnancy; HIV positivity; initiation of chronic transfusion therapy; initiation of hydroxyurea therapy; significant decline in eGFR-CysC; significant increase in serum creatinine; serum potassium levels ≥5.5 mEq/L despite a low potassium diet and a reduction in dose; or blurred vision that did not improve with a reduction in losartan dose. Complete withdrawal specifications are provided in Supplemental Table 3. SCA-related events were not criteria for withdrawal from study, unless the administration of losartan interfered with medical care, in which case the participant was withdrawn if losartan had to be stopped for >4 consecutive weeks. We pre-specified that participants were to be withdrawn for non-adherence with one-third or more of study procedures.
Definitions, Measurements and Outcomes
Supplemental Table 4 provides the complete schedule of study assessments. The baseline value for all outcomes was defined as the average of the two pre-losartan measurements (obtained at screening visit and visit 1). In the original version of the protocol (before enrollment began), we specified that the UACR at the screening visit was considered the baseline value; however, because some participants changed class of albuminuria between the screening visit and visit 1 (Supplemental Figure 1), we re-defined the baseline value to be the average of the UACR for these two visits before accrual to the study was complete. The final value for all outcomes was defined as the visit 10 (end-of-study) measurement, or, if a participant exited the study early, the last recorded value. We pre-specified that a minimum of 3 months’ therapy with losartan was required for a participant to be considered evaluable.
The primary outcome measure was a ≥25% reduction in UACR (comparing the final to the baseline UACR). The main secondary outcomes included change in classification of UACR (NoA, MicroA, MacroA), UACR on a continuous scale, eGFR, urine osmolality, echocardiography, and 6-minute walk distance (6MWD). Two methods were used to calculate eGFR: creatinine clearance by 24-hour urine collection (eGFR-CrCl) and measurement of cystatin C (eGFR-CysC). Accommodations were made for participants with nocturnal enuresis for timed urine collections. First morning voids were used for measurement of UACR and urine osmolality. Participants were instructed not to drink any anything after 8 p.m. the night before the measurement of urine osmolality. Blood pressure was measured at all study visits according to a standard protocol for all sites. Blood pressure was measured twice with the participant sitting. A third measurement was taken if there was a >10% difference between these sequential values. The mean of these values was used for analysis. Transthoracic echocardiography was performed at study sites and analyzed centrally. 6MWD was measured at study sites per American Thoracic Society guidelines.
The renal structural injury biomarkers, N-acetyl-β-D-glucosaminidase (NAG) and kidney injury molecule-1 (KIM-1) were measured by commercial kits: a colorimetric assay for NAG (Roche) and an enzyme-linked immunosorbent assay for KIM-1 (R&D Systems). Arginine and related metabolites, monomethylarginine (MMA), asymmetric dimethylarginine (ADMA), and symmetric dimethylarginine (SDMA), were measured by high-pressure liquid chromatography.31
Sample Size and Statistical Analyses
The total planned sample size was 36 participants, distributed across groups defined by class of baseline albuminuria (NoA, N=14; MicroA, N=14; MacroA, N=8). To test the primary hypothesis, the sample size was calculated to detect the primary endpoint (≥25% reduction in UACR) in ≥30% of the MicroA group using a two-sided binominal test (>80% power, α=0.05), assuming that the proportion of significant reduction (≥25%) in UACR without losartan was ≤5%. Baseline differences across groups defined by class of albuminuria (NoA, MicroA, and MacroA) were tested by one-way ANOVA. The related-samples Wilcoxon signed rank test was used to assess treatment effects on outcomes (paired pre- and post-losartan comparisons). UACR data were analyzed longitudinally using a linear mixed effects model. The natural log fold change from baseline was used as the dependent variable to better suit normality assumptions. The dependent variables were group (MaA, MiA and NoA), week of measurement (2, 4, 8, 12, and 26) and their interaction. Group variances were allowed to differ in the fitted model. An autoregressive with lag 1 error structure was assumed for measurements within the same subject. Tests for the difference between the natural log fold change over baseline not equal to 0 were carried out using Wald tests for each time point. A P-value <0.05 was considered statistically significant, and no corrections were made for multiple comparisons.
Results
Participants
There were 36 participants (mean age 24.1 years; 53% female). Flow of participants is shown in Supplemental Figure 1. Four participants were not evaluable based on pre-specified criteria [<3 months of losartan therapy due to toxicity (N=1) or non-compliance (N=3)]. Baseline characteristics of the 32 evaluable participants are shown in Table 1. Each participant in the NoA group had detectable urinary albumin, but UACR was <30 mg/g by definition. Mean age was higher with increasing class of baseline albuminuria (P=0.005). There were no statistically significant differences in hemoglobin (Hb) concentration (P=0.14) across groups; however, participants with albuminuria (MicroA and MacroA combined) had lower Hb concentration compared to participants with NoA (83.3 vs. 92.2 g/L; P=0.049). There were no differences in systolic blood pressure by class of baseline albuminuria, but the NoA group had lower diastolic blood pressure than the MicroA and MacroA groups.
Table 1.
Baseline characteristics of all participants and by class of baseline albuminuria.
| Characteristic | All participants (N or mean ± SEM) | No albuminuria (N or mean ± SEM) | Micro-albuminuria (N or mean ± SEM) | Macro-albuminuria (N or mean ± SEM) | P* |
|---|---|---|---|---|---|
| Number | 32 | 14 | 12 | 6 | |
| Sex (male/female) | 14/18 | 8/6 | 4/8 | 2/4 | |
| Age (years) | 24.4 ± 15.7 | 14.6 ± 1.3 | 28.6 ± 4.8 | 38.8 ± 7.0 | <0.01 |
| Hb (g/dL) | 8.7 ± 2.3 | 9.2 ± 3.5 | 8.3 ± 3.6 | 8.4 ± 4.7 | 0.14 |
| UACR (mg/g) | 202 ± 59 | 8.5 ± 1.3 | 121 ± 25 | 815 ± 137 | <0.01 |
| eGFR-CrCl (mL/min/1.73m2) | 150 ± 10 | 146 ± 61 | 172 ± 59 | 108 ± 19 | 0.02 |
| eGFR-CysC (mL/min/1.73m2) | 134 ± 6.4 | 132 ± 5.0 | 155 ± 11.9 | 96 ± 9.6 | <0.01 |
| Osmolality (mosm/kg H20) | 393 ± 12 | 430 ± 21 | 383 ± 10 | 328 ± 21 | <0.01 |
| SBP (mm Hg) | 116 ± 2.1 | 116 ± 3.4 | 114 ± 3.5 | 120 ± 4.3 | 0.60 |
| DBP (mm Hg) | 65 ± 1.2 | 60 ± 1.3 | 68 ± 1.8 | 67 ± 2.7 | <0.01 |
Independent samples Kruskal-Wallis test of the distribution of the continuous variables across the three classes of baseline albuminuria.
Abbreviations: DBP, diastolic blood pressure; eGFR-CrCl, estimated glomerular filtration rate by creatinine clearance from 24-hour urine collection; eGFR-CysC, estimated glomerular filtration rate by measurement of cystatin C; Hb, hemoglobin concentration; SBP, systolic blood pressure; SEM, standard error of the mean; UACR, urinary albumin-to-creatinine ratio.
Dose of Losartan
As specified in the protocol, participants in the baseline NoA group (N=14) remained on the fixed (starting) dose of losartan, and none had a reduction in dose for toxicity. Dose escalation was permitted in the MicroA and MacroA groups based on UACR. Among the MicroA group (N=12), 3 remained on the starting dose, 6 had a single dose escalation (e.g., 50 mg to 75 mg), 2 had two dose escalations (e.g., 50 mg to 75 mg to 100 mg), and 1 had a dose reduction because of toxicity. Among the MacroA group (N=6), 2 remained on the starting dose, 3 had a single dose escalation (e.g., 50 mg to 75 mg), 1 had two dose escalations (50 mg to 75 mg to 100 mg), and 1 had a dose reduction because of initiation of diuretic therapy. The dose changes for individual MicroA and MacroA participants are indicated in Supplemental Figure 2.
UACR
The primary endpoint (≥25% reduction in UACR) was met in 83% (5/6) of the MacroA group (P<0.0001), 58% (7/12) of the MicroA group (P<0.0001), and 7% (1/14) of the NoA group (P=0.51) (Figure 1). Considering MacroA and MicroA participants together, 67% (12/18) met the primary endpoint (P<0.0001). The median fold-change in UACR was −0.74, −0.46, and 0.08 for the MacroA, MicroA, and NoA groups, respectively (Supplemental Table 5; Figure 2, Panel A). The difference in fold-change across groups was statistically significant (P=0.032), consistent with a greater therapeutic response in participants with albuminuria (MacroA > MicroA). Longitudinal analysis of UACR also demonstrated decreases in UACR in participants with albuminuria (MacroA > MicroA) but not in the NoA group (Figure 3). Most MacroA and MicroA participants had 0.5 to 1-fold decreases in UACR (12/18), but those who did not respond to losartan had relatively large increases in UACR (Supplemental Figure 2). In MacroA and MicroA participants, UACR classification improved (e.g., MacroA to MicroA) with losartan therapy in 50% but worsened in 11%. Only 1 (7%) of the NoA participants had worsened class (NoA to MicroA) at the end of the study, while 13/14 (93%) remained as NoA. Participants who had concomitant treatment with hydroxyurea had numerically greater reduction in UACR than those not treated with hydroxyurea (median fold change −0.08 vs 0.46, P=0.14), but this was not a statistically significant difference.
Figure 1. Percent of participants meeting the primary endpoint by baseline class of albuminuria.
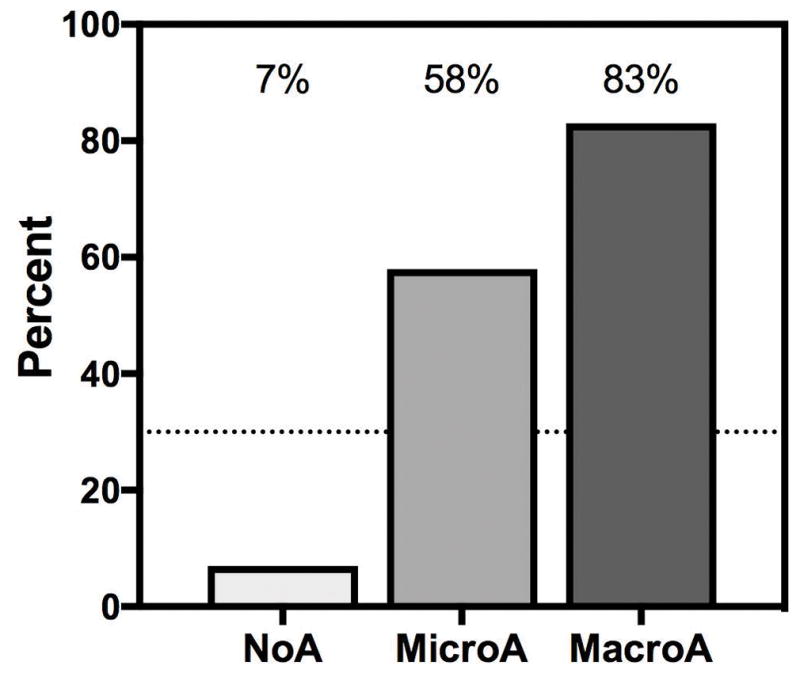
The primary endpoint of a ≥25% reduction in urinary albumin-to-creatinine ratio (UACR) from baseline was met in 67% (12/18) of participants with albuminuria and 41% (13/32) of all participants. Most participants in the MicroA and MacroA groups met the primary endpoint. The dotted line indicates 30% of participants having a ≥25%reduction in UACR, which was the minimum anticipated proportion of participants with ≥25% reduction in UACR in the MicroA group used for calculation of sample size. P-values are from a two-sided binominal test, assuming that the proportion of significant reduction (≥25%) in UACR without losartan was ≤5%.
Figure 2. Fold-change in albuminuria, glomerular filtration rate (eGFR), and urinary osmolality after losartan therapy by baseline class of albuminuria.

For all panels, a negative fold-change indicates a reduction in the value of the outcome at the final study visit compared to the baseline value. The boxes are Tukey’s hinges, the line is the median value, the whiskers are 5th and 95th percentiles, and the dots are outliers. Panel A: Urinary albumin-to-creatinine ratio (UACR) on first morning void. The light, dotted line indicates a 25% reduction in UACR, the primary outcome measure. For the MicroA and MacroA groups, the median fold-change is below this line (indicating a ≥25% reduction for most participants in those groups). Panel B: eGFR by creatinine clearance on 24-hour urine collection. Panel C: eGFR by measurement of cystatin C. Panel D: Urinary osmolality on first morning void. For each panel, the P-value is from an independent samples Kruskal-Wallis test of the distribution of the outcome across the three classes of baseline albuminuria.
Figure 3. Longitudinal analysis of UACR by class of baseline albuminuria.

The natural log of the fold-change in the UACR by number of weeks following initiation of losartan is shown (the natural log of a fold-change of 1 equals 0). Statistically significant differences are indicated by asterisks. In the MiA group, 6 participants had missing week 26 values.
eGFR
We used two methods to determine eGFR (eGFR-CrCl and eGFR-CysC). At baseline, by both methods, the MicroA group had the highest eGFR, and the MacroA group had the lowest eGFR (Table 1). There were no statistically significant changes in eGFR-CrCl after treatment with losartan (Supplemental Table 5; Figure 2, Panel B). The largest percent change in eGFR-CrCl was in the MicroA group (-24%; 176±17 to 142±19 mL/min/1.73m2; P=0.11; Supplemental Table 5). Similarly, the MicroA group had the highest eGFR-CysC at baseline (Table 1). There was a statistically significant increase in eGFR-CysC after treatment with losartan (P=0.02), driven by an 18% increase in the NoA group (Supplemental Table 5; Figure 2, Panel C).
Measurements of eGFR-CrCl and eGFR-CysC were performed in parallel at 3 study visits (screening, visit 1, and final). The linear correlation between the two was statistically significant (r=0.36, 0.55 and 0.46 for the screening, visit 1 and final visit, respectively; Supplemental Figure 3). Overall, eGFR-CysC was lower than the eGFR-CrCl (Supplemental Figure 3). Bland-Altman plots showed a positive bias for eGFR-CrCl for higher values of eGFR at the screening visit and visit 1 (Supplemental Figure 4). The 95% confidence limits of the difference (eGFR-CrCl – eGFR-CysC) were approximately −100 and 100 mL/minute/1.73 m2, indicating that these eGFR measurements are poor surrogates for each other in individuals with SCA.
Osmolality
At baseline, urine osmolality was lower with increasing class albuminuria (P=0.005; Table 1). There were no statistically significant changes in osmolality after treatment with losartan (P=0.06; Supplemental Table 5; Figure 2, Panel D). The largest percent change (decline) in osmolality was in the MacroA group (−21%, P=0.17; Supplemental Table 5).
Adverse effects
Toxicity requiring discontinuation of losartan occurred in 3 participants: (1) leg cramps; (2) decline in eGFR ≥25% (142→104 mL/minute/1.73 m2); and (3) rise in serum creatinine ≥50% (0.2→0.3 mg/dL). There were no significant changes in systolic blood pressure (−1.2 mmHg, P=0.44) or diastolic blood pressure (−1.5 mmHg, P=0.24) after treatment with losartan (change from baseline value at final visit). No participant had hyperkalemia or angioedema. No other toxicities were identified.
Biomarkers
There were no significant pre-treatment differences in the renal structural injury biomarkers, NAG and KIM-1, by class of baseline albuminuria, and there were no detectable changes in NAG and KIM-1 with losartan therapy (data not shown).
Plasma arginine, asymmetric dimethylarginine (ADMA), and monomethylarginine (MMA) were similar across all groups at baseline (Supplemental Figure 5). Symmetric dimethylarginine (SDMA), which is filtered by the kidney, was elevated in the MicroA and MacroA groups compared to the NoA group. Plasma SDMA, which has been proposed as a measure of renal clearance,32,33 correlated with eGFR-CysC (r=0.45, P=0.01) but not eGFR-CrCl (r=−0.23, P=0.22). SDMA also correlated with UACR (r=0.46, P=0.01), a relationship that was independent of eGFR-CysC. Although baseline arginine, ADMA, MMA, or SDMA did not predict the primary endpoint (reduction in UACR), losartan therapy lowered ADMA in the MicroA and MacroA groups. This effect was especially pronounced when analyzed as the ADMA/SDMA ratio (Supplemental Figure 6). This may reflect decreased production of ADMA, or improved metabolism of ADMA by dimethylarginine dimethylaminohydrolase (DDAH), an effect of ARBs previously observed in vitro.34
Cardiopulmonary Correlates
There were no changes in cardiopulmonary outcomes after losartan therapy. However, there were baseline differences in cardiac phenotype based on the presence or absence of albuminuria. Compared to the NoA group, participants with albuminuria (MicroA and MacroA combined; mean UACR = 373 ± 87 mg/g) had worse diastolic function indicated by lower early-to-late ratios of mitral inflow velocities (E/A; 1.8±0.1 vs. 2.3±0.1, P=0.018) and septal annular velocities (e′/a′; 5±0.2 vs. 2.4±0.2, P<0.001) and significantly greater left atrial volume indices (34.4±2.1 vs. 27±2.8 ml/m2, P=0.039). In addition, the early mitral inflow to annular velocity ratio, lateral E/e′, had a significant correlation with the ADMA/SDMA ratio (r=−0.37, P=0.03) suggesting that the severity of diastolic dysfunction and sickle cell nephropathy are correlated. Systolic function was normal and similar in both groups (shortening fraction 36.9±2% vs. 36.7±1.5%, P=0.94). There were no differences in tricuspid regurgitant jet velocity, left ventricular mass, or left ventricular diameter. Participants with albuminuria had significantly shorter 6-minute walk distance (6MWD): 1182±78 vs. 1472±86 ft, P=0.018. 6MWD also correlated significantly with UACR (r= −0.46, P=0.005) and the diastolic measures, E/A (r=−0.42, P=0.013) and septal e′/a′ (r=−0.45, P=0.015).
Discussion
In this phase-2 study, six months of losartan therapy appeared to decrease urinary albumin excretion in most participants with albuminuria (>30 mg/g; MicroA and MacroA groups). Participants with macroalbuminuria (>300 mg/g; MacroA group) had the greatest response. There were no consistent changes in eGFR or urine osmolality with losartan therapy across all participants, but there was some evidence for increased eGFR in the NoA group and lowered osmolality in the MacroA group. Only 1 participant had clinically significant toxicity (leg cramps). The decline in eGFR or rise in serum creatinine in two participants was not clinically worrisome; in retrospect, the toxicity criteria in the protocol were too strict. We did not observe hyperkalemia or detect changes in blood pressure. We also found that SDMA may be a potential biomarkers of sickle cell nephropathy.
There are no published clinical trials of ARB therapy, such as losartan, for sickle cell nephropathy to compare to our findings. However, ACE inhibition, using captopril or enalapril, for reduction of albuminuria or proteinuria in SCA has been reported in one small randomized trial (N=22)20 and 4 observational studies.12,19,21,22 The randomized trial by Foucan et al.20 demonstrated that mean urinary albumin excretion was lower after 6 months of captopril therapy. Serum creatinine and potassium did not change during the study, but diastolic and systolic blood pressure were lower by 5 to 8 mmHg, respectively, after treatment. The observational studies with different durations of treatment (2 weeks to nearly 9 years), provided supportive evidence that ACE inhibition can reduce urinary albumin excretion in many patients.12,19,21,22 The study by Aoki et al.19 also found that mean arterial blood pressure fell by approximately 8 mmHg. In contrast, we found no change in blood pressure with losartan therapy. An evidenced-based review concluded that patients with SCA and proteinuria “should be treated” with an ACE inhibitor.35 In contrast, a Cochrane review concluded that ACE inhibition to reduce proteinuria and microalbuminuria “may not be indicated until further evidence is obtained”.23 As such, there appears to be equipoise for the use of ACE inhibition and a lack of evidence, until now, for the use of ARBs for sickle cell nephropathy.
This small, phase-2 study has inherent limitations. Mainly, we cannot conclude that losartan is efficacious for sickle cell nephropathy. Rather, the encouraging data generated from this study will inform the design of a phase-3 randomized trial to determine its efficacy. Additionally, these findings may not be generalizable to all individuals with SCA, because we selected participants without many serious complications of SCA and excluded those receiving chronic transfusion therapy. Chronic transfusions were an exclusion criterion because of the potentially confounding effect of deferasirox-related nephropathy. Consequently, individuals with severe cerebral vasculopathy were also not studied. The effects of losartan in these sub-populations are not known. Although we did not observe hypotension, and other adverse effects were infrequent, studies with more participants of longer duration are needed to assess toxicity. Finally, it is not known whether reduction of albuminuria slows the progression of sickle cell nephropathy. Long-term studies will be needed to determine this. Indeed, given that losartan is being studied as a potential, long-term, disease-modifying therapy, phase 3 trials should examine a longer duration of treatment than the 6 months we studied here.
The reported longitudinal variation of UACR in SCA36 poses challenges to study design. Indeed, we observed that some participants changed class of albuminuria in the 2-week interval between the initial screening visit and visit 1, the immediate pre-losartan study visit (Supplemental Figure 1). To help account for this in analyses, we used the mean of the screening and visit 1 measurements as the value to define the baseline class of albuminuria and to calculate change from baseline for UACR and the secondary outcomes, eGFR and osmolality. We also observed that a few participants had large (2.5 to 3.5-fold) increases in UACR while on losartan (Supplemental Figure 2). We suspect this was due, at least partly, to decreased adherence to losartan near the end of the study period (a “rebound” effect). A definitive trial will need to account for the longitudinal variability in UACR and optimize adherence to study medication to be able to demonstrate a therapeutic benefit of losartan.
Estimation of GFR in SCA using common laboratory methods is challenging. Creatinine secretion by the proximal tubule is increased in SCA,37 so estimates that use serum creatinine or urinary creatinine clearance can over-estimate GFR.38 In contrast to creatinine-based methods, a cystatin C-based method is more accurate in the setting hyperfiltration,39 and may be more sensitive to early decline in GFR in SCA.40,41 However, inflammation in SCA could increase cystatin C production and affect the estimation of GFR.42 Because eGFR was not the primary outcome measure in this study, we used both eGFR-CysC and eGFR-CrCl to monitor for toxicity of losartan, rather than a more definitive method, to decrease the burden of the study to participants. Inulin is not available in the United States to perform the gold standard GFR method. In a phase 3 study of losartan for SCA, we plan to use eGFR-CysC and a method based on clearance of iohexol or a radioisotope, and, possibly, novel approaches based on SDMA.32,33
Both ARBs and ACE inhibitors may cause acute kidney injury in the event of dehydration. Therefore, the use of either class therapy needs to be carefully considered and monitored in individuals with SCA, who may be prone to dehydration. During treatment with these medications, serial laboratory monitoring, ongoing education of patients about the need to maintain adequate fluid intake, and heightened clinical vigilance about fluid status during acute illnesses are all necessary. The lack of changes in the structural injury biomarkers, NAG and KIM-1, suggest that losartan therapy was tolerated well by the kidney in this study.
Further study of arginine-related biomarkers is also warranted, as we found that elevated SDMA was associated with micro- and macroalbuminuria and lower eGFR. Studies in other forms of renal disease similarly showed that higher SDMA levels were associated with reduced renal function.43,44 Furthermore, ADMA, a potent inhibitor of endothelial nitric oxide synthase, appears to have been decreased by losartan therapy. This effect is similar to in vitro models showing that ARBs decreased intracellular oxidative stress and improved metabolism of ADMA by DDAH.34
Individuals with SCA can develop a cardiomyopathy with restrictive physiology.45,46 We found that baseline albuminuria was associated with echocardiographic evidence of restrictive physiology. Although participants with albuminuria (MicroA and MacroA) had a lower Hb concentration than those without (NoA) (Table 1), these echocardiographic features are not readily explained by anemia alone, because there were no differences in left ventricular mass or diameter, which are direct consequences of an anemia-related hyperdynamic state. While the diastolic dysfunction observed in these participants was not severe, the measures of diastolic function that have been associated with early mortality (E/A, e′/a′, and E/e′),47 were also associated with markers of nephropathy and impaired functional tolerance. Treatment with losartan for 6 months had no measurable effects on echocardiographic parameters or 6MWD. In contrast, enalapril given for 36 months to 9 adults appeared to prevent the cardiac remodeling associated with SCA (e.g., increase in left ventricular mass and wall thickness over time).48 If it is effective, perhaps only 6 months of losartan therapy is insufficient to measurably affect cardiac structure and function, or the age at initiation of therapy is important.
In summary, losartan appeared to decrease urinary albumin excretion in most participants with albuminuria (>30 mg/g). Those with baseline macroalbuminuria (>300 mg/g) appeared to have the greatest benefit, nearly all of whom showed a response. Blood pressure did not change with losartan, in contrast to reports of ACE inhibition. Toxicity of losartan was mild and infrequent; hypotension and hyperkalemia were not encountered. Using these data, a phase-3, randomized, placebo-controlled trial is being designed to determine the efficacy of losartan for individuals with SCA and micro- or macroalbuminuria.
Supplementary Material
Acknowledgments
This trial is supported by NIH-NHLBI grant R34HL108752 (P. Malik) and registered at clinicaltrials.gov (NCT01479439).
We thank the following additional members of the clinical research team: Akron Children′s Hospital: Lisa Sidebotham and Jill Bradisse; Cincinnati Children’s Hospital Medical Center and University of Cincinnati College of Medicine: Rubina Dosani and Kelly Thueneman; Nationwide Children’s Hospital: Heidi Ziegler and Dianna Glynn; Sickle Cell Branch, National Heart, Lung, and Blood Institute, National Institutes of Health: Katherine Roskom and Anna Conrey; Texas Children’s Hospital: Bogdan Dinu; University of Illinois at Chicago: and Michel Gowari and Lani Krauz; University of Louisville: Jennifer Comings and Tressa Bratton; and U.T. Southwestern Medical Center: Pamela Kurian. We also thank the members of the DSMB: Patrick Brophy, University of Iowa Children’s Hospital; Stuart Goldstein, Cincinnati Children’s Hospital Medical Center; Joseph Palumbo, Cincinnati Children’s Hospital Medical Center; and Rakesh Shukla, University of Cincinnati College of Medicine.
Footnotes
AUTHORSHIP
Study design: PM, PD, CTQ, MT, ABR
Recruitment of participants: CTQ, SS, VG, CF, SC, PB, AG, AR, AN, ON
Study coordination: LM
Laboratory measurements: CT, PD, HA, YY
Analysis of data: CTQ, ON, AL, PM
Writing and editing the manuscript: CTQ, ON, PM, SS, VG, CF, SC, PB, AG, AN, CT, LM, MT, PD, HA, YY
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
References
- 1.Saborio P, Scheinman JI. Sickle cell nephropathy. J Am Soc Nephrol. 1999;10(1):187–192. doi: 10.1681/ASN.V101187. [DOI] [PubMed] [Google Scholar]
- 2.Becker AM. Sickle cell nephropathy: challenging the conventional wisdom. Pediatr Nephrol. 2011;26(12):2099–2109. doi: 10.1007/s00467-010-1736-2. [DOI] [PubMed] [Google Scholar]
- 3.Asnani M, Serjeant G, Royal-Thomas T, Reid M. Predictors of renal function progression in adults with homozygous sickle cell disease. Br J Haematol. 2016;173(3):461–468. doi: 10.1111/bjh.13967. [DOI] [PubMed] [Google Scholar]
- 4.Guasch A, Cua M, Mitch WE. Early detection and the course of glomerular injury in patients with sickle cell anemia. Kidney Int. 1996;49(3):786–791. doi: 10.1038/ki.1996.109. [DOI] [PubMed] [Google Scholar]
- 5.McPherson Yee M, Jabbar SF, Osunkwo I, et al. Chronic kidney disease and albuminuria in children with sickle cell disease. Clin J Am Soc Nephrol. 2011;6(11):2628–2633. doi: 10.2215/CJN.01600211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dharnidharka VR, Dabbagh S, Atiyeh B, Simpson P, Sarnaik S. Prevalence of microalbuminuria in children with sickle cell disease. Pediatr Nephrol. 1998;12(6):475–478. doi: 10.1007/s004670050491. [DOI] [PubMed] [Google Scholar]
- 7.Pham PT, Pham PC, Wilkinson AH, Lew SQ. Renal abnormalities in sickle cell disease. Kidney Int. 2000;57(1):1–8. doi: 10.1046/j.1523-1755.2000.00806.x. [DOI] [PubMed] [Google Scholar]
- 8.Alvarez O, Montane B, Lopez G, Wilkinson J, Miller T. Early blood transfusions protect against microalbuminuria in children with sickle cell disease. Pediatr Blood Cancer. 2006;47(1):71–76. doi: 10.1002/pbc.20645. [DOI] [PubMed] [Google Scholar]
- 9.Guasch A, Navarrete J, Nass K, Zayas CF. Glomerular involvement in adults with sickle cell hemoglobinopathies: prevalence and clinical correlates of progressive renal failure. J Am Soc Nephrol. 2006;17(8):2228–2235. doi: 10.1681/ASN.2002010084. [DOI] [PubMed] [Google Scholar]
- 10.Wesson DE. The initiation and progression of sickle cell nephropathy. Kidney Int. 2002;61(6):2277–2286. doi: 10.1046/j.1523-1755.2002.00363.x. [DOI] [PubMed] [Google Scholar]
- 11.Guasch A, Cua M, You W, Mitch WE. Sickle cell anemia causes a distinct pattern of glomerular dysfunction. Kidney Int. 1997;51(3):826–833. doi: 10.1038/ki.1997.116. [DOI] [PubMed] [Google Scholar]
- 12.Falk RJ, Scheinman J, Phillips G, Orringer E, Johnson A, Jennette JC. Prevalence and pathologic features of sickle cell nephropathy and response to inhibition of angiotensin-converting enzyme. N Engl J Med. 1992;326(14):910–915. doi: 10.1056/NEJM199204023261402. [DOI] [PubMed] [Google Scholar]
- 13.Gosmanova EO, Zaidi S, Wan JY, Adams-Graves PE. Prevalence and progression of chronic kidney disease in adult patients with sickle cell disease. J Investig Med. 2014;62(5):804–807. doi: 10.1097/01.JIM.0000446836.75352.72. [DOI] [PubMed] [Google Scholar]
- 14.Powars DR, Elliott-Mills DD, Chan L, et al. Chronic renal failure in sickle cell disease: risk factors, clinical course, and mortality. Ann Intern Med. 1991;115(8):614–620. doi: 10.7326/0003-4819-115-8-614. [DOI] [PubMed] [Google Scholar]
- 15.Abbott KC, Hypolite IO, Agodoa LY. Sickle cell nephropathy at end-stage renal disease in the United States: patient characteristics and survival. Clin Nephrol. 2002;58(1):9–15. doi: 10.5414/cnp58009. [DOI] [PubMed] [Google Scholar]
- 16.McClellan AC, Luthi J-C, Lynch JR, et al. High one year mortality in adults with sickle cell disease and end-stage renal disease. Br J Haematol. 2012;159(3):360–367. doi: 10.1111/bjh.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang KH, Nayak RC, Roy S, et al. Vasculopathy-associated hyperangiotensinemia mobilizes haematopoietic stem cells/progenitors through endothelial AT2R and cytoskeletal dysregulation. Nat Commun. 2015;6:5914. doi: 10.1038/ncomms6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roy S, Konstantinidis DG, Rizvi T, et al. Increased oxidative stress in sickle cell disease activates the renin-angiotensin-TGF-β pathway to mediate sickle nephropathy. Blood. 2013;122(21):2211–2211. [Google Scholar]
- 19.Aoki RY, Saad ST. Enalapril reduces the albuminuria of patients with sickle cell disease. Am J Med. 1995;98(5):432–435. doi: 10.1016/S0002-9343(99)80341-6. [DOI] [PubMed] [Google Scholar]
- 20.Foucan L, Bourhis V, Bangou J, Mérault L, Etienne-Julan M, Salmi RL. A randomized trial of captopril for microalbuminuria in normotensive adults with sickle cell anemia. Am J Med. 1998;104(4):339–342. doi: 10.1016/s0002-9343(98)00056-4. [DOI] [PubMed] [Google Scholar]
- 21.Fitzhugh CD, Wigfall DR, Ware RE. Enalapril and hydroxyurea therapy for children with sickle nephropathy. Pediatr Blood Cancer. 2005;45(7):982–985. doi: 10.1002/pbc.20296. [DOI] [PubMed] [Google Scholar]
- 22.McKie KT, Hanevold CD, Hernandez C, Waller JL, Ortiz L, McKie KM. Prevalence, prevention, and treatment of microalbuminuria and proteinuria in children with sickle cell disease. J Pediatr Hematol Oncol. 2007;29(3):140–144. doi: 10.1097/MPH.0b013e3180335081. [DOI] [PubMed] [Google Scholar]
- 23.Sasongko TH, Nagalla S, Ballas SK. Angiotensin-converting enzyme (ACE) inhibitors for proteinuria and microalbuminuria in people with sickle cell disease. Cochrane Database Syst Rev. 2015;(6):CD009191. doi: 10.1002/14651858.CD009191.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siragy HM. The angiotensin II type 2 receptor and the kidney. J Renin Angiotensin Aldosterone Syst. 2010;11(1):33–36. doi: 10.1177/1470320309347786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Padia SH, Carey RM. AT2 receptors: beneficial counter-regulatory role in cardiovascular and renal function. Pflugers Arch. 2013;465(1):99–110. doi: 10.1007/s00424-012-1146-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wenzel UO, Krebs C, Benndorf R. The angiotensin II type 2 receptor in renal disease. J Renin Angiotensin Aldosterone Syst. 2010;11(1):37–41. doi: 10.1177/1470320309347787. [DOI] [PubMed] [Google Scholar]
- 27.Pylypchuk GB. ACE inhibitor- versus angiotensin II blocker-induced cough and angioedema. Annals of Pharmacotherapy. 1998;32(10):1060–1066. doi: 10.1345/aph.17388. [DOI] [PubMed] [Google Scholar]
- 28.DeBaun MR, Strunk RC. The intersection between asthma and acute chest syndrome in children with sickle-cell anaemia. Lancet. 2016;387(10037):2545–2553. doi: 10.1016/S0140-6736(16)00145-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Datta V, Ayengar JR, Karpate S, Chaturvedi P. Microalbuminuria as a predictor of early glomerular injury in children with sickle cell disease. Indian J Pediatr. 2003;70(4):307–309. doi: 10.1007/BF02723586. [DOI] [PubMed] [Google Scholar]
- 30.Zandbergen AAM, Baggen MGA, Lamberts SWJ, Bootsma AH, de Zeeuw D, Ouwendijk RJT. Effect of losartan on microalbuminuria in normotensive patients with type 2 diabetes mellitus. A randomized clinical trial. Ann Intern Med. 2003;139(2):90–96. doi: 10.7326/0003-4819-139-2-200307150-00008. [DOI] [PubMed] [Google Scholar]
- 31.Alkaitis MS, Nardone G, Chertow JH, Ackerman HC. Resolution and quantification of arginine, monomethylarginine, asymmetric dimethylarginine, and symmetric dimethylarginine in plasma using HPLC with internal calibration. Biomed Chromatogr. 2016;30(3):294–300. doi: 10.1002/bmc.3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tutarel O, Denecke A, Bode-Böger SM, et al. Symmetrical dimethylarginine outperforms CKD-EPI and MDRD-derived eGFR for the assessment of renal function in patients with adult congenital heart disease. Kidney Blood Press Res. 2011;34(1):41–45. doi: 10.1159/000322614. [DOI] [PubMed] [Google Scholar]
- 33.El-Khoury JM, Bunch DR, Hu B, Payto D, Reineks EZ, Wang S. Comparison of symmetric dimethylarginine with creatinine, cystatin C and their eGFR equations as markers of kidney function. Clin Biochem. 2016;49(15):1140–1143. doi: 10.1016/j.clinbiochem.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 34.Luo Z, Teerlink T, Griendling K, Aslam S, Welch WJ, Wilcox CS. Angiotensin II and NADPH oxidase increase ADMA in vascular smooth muscle cells. Hypertension. 2010;56(3):498–504. doi: 10.1161/HYPERTENSIONAHA.110.152959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lottenberg R, Hassell KL. An evidence-based approach to the treatment of adults with sickle cell disease. Hematology Am Soc Hematol Educ Program. 2005;2005(1):58–65. doi: 10.1182/asheducation-2005.1.58. [DOI] [PubMed] [Google Scholar]
- 36.Shatat IF, Qanungo S, Hudson S, Laken MA, Hailpern SM. Changes in urine microalbumin-to-creatinine ratio in children with sickle cell disease over time. Front Pediatr. 2016;4:106. doi: 10.3389/fped.2016.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allon M. Renal abnormalities in sickle cell disease. Arch Intern Med. 1990;150(3):501–504. [PubMed] [Google Scholar]
- 38.Sharpe CC, Thein SL. Sickle cell nephropathy - a practical approach. Br J Haematol. 2011;155(3):287–297. doi: 10.1111/j.1365-2141.2011.08853.x. [DOI] [PubMed] [Google Scholar]
- 39.Huang S-HS, Sharma AP, Yasin A, Lindsay RM, Clark WF, Filler G. Hyperfiltration affects accuracy of creatinine eGFR measurement. Clin J Am Soc Nephrol. 2011;6(2):274–280. doi: 10.2215/CJN.02760310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Voskaridou E, Terpos E, Michail S, et al. Early markers of renal dysfunction in patients with sickle cell/beta-thalassemia. Kidney Int. 2006;69(11):2037–2042. doi: 10.1038/sj.ki.5000248. [DOI] [PubMed] [Google Scholar]
- 41.Alvarez O, Zilleruelo G, Wright D, Montane B, Lopez-Mitnik G. Serum cystatin C levels in children with sickle cell disease. Pediatr Nephrol. 2006;21(4):533–537. doi: 10.1007/s00467-006-0033-6. [DOI] [PubMed] [Google Scholar]
- 42.Knight EL, Verhave JC, Spiegelman D, et al. Factors influencing serum cystatin C levels other than renal function and the impact on renal function measurement. Kidney Int. 2004;65(4):1416–1421. doi: 10.1111/j.1523-1755.2004.00517.x. [DOI] [PubMed] [Google Scholar]
- 43.Schepers E, Barreto DV, Liabeuf S, et al. Symmetric dimethylarginine as a proinflammatory agent in chronic kidney disease. Clin J Am Soc Nephrol. 2011;6(10):2374–2383. doi: 10.2215/CJN.01720211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kielstein JT, Böger RH, Bode-Böger SM, et al. Marked increase of asymmetric dimethylarginine in patients with incipient primary chronic renal disease. J Am Soc Nephrol. 2002;13(1):170–176. doi: 10.1681/ASN.V131170. [DOI] [PubMed] [Google Scholar]
- 45.Niss O, Quinn CT, Lane A, et al. Cardiomyopathy with Restrictive Physiology in Sickle Cell Disease. JACC Cardiovasc Imaging. 2016;9(3):243–252. doi: 10.1016/j.jcmg.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bakeer N, James J, Roy S, et al. Sickle cell anemia mice develop a unique cardiomyopathy with restrictive physiology. Proc Natl Acad Sci U S A. 2016;113(35):E5182–E5191. doi: 10.1073/pnas.1600311113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sachdev V, Machado RF, Shizukuda Y, et al. Diastolic dysfunction is an independent risk factor for death in patients with sickle cell disease. J Am Coll Cardiol. 2007;49(4):472–479. doi: 10.1016/j.jacc.2006.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lima CSP, Ueti OM, Ueti AA, Franchini KG, Costa FF, Saad STO. Enalapril therapy and cardiac remodelling in sickle cell disease patients. Acta Cardiol. 2008;63(5):599–602. doi: 10.2143/AC.63.5.2033227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.