

Abstract
Diuretics are commonly prescribed for treatment in patients with hypertension, edema or heart failure. Studies on hypertensive and salt-losing disorders and on urea transporters have contributed to better understanding of mechanisms of renal salt and water reabsorption and their regulation. Proteins involved in the regulatory pathways are emerging targets for diuretic and aquaretic therapy. Integrative high-throughput screening, protein structure analysis, and chemical modification have identified promising agents for pre-clinical testing in animals. These include WNK-SPAK inhibitors, ClC-K channel antagonists, ROMK channel antagonists, and pendrin and urea transporter inhibitors. We discuss the potential advantages and side effects of these potential diuretics.
Keywords: Aquaretics, Diuretics, Salt reabsorption, Urine concentration, Ste20-related proline-alanine rich kinase (SPAK), With-no-lysine kinase (WNK)
Introduction
Diuretics have been prescribed for the treatment of edema, hypertension, and congestive heart failure since the 16th century. Historically, the discovery of diuretics has frequently been serendipitous. The ancient diuretics mercurous chloride and organomercurials were discovered from the observation that patients treated with these anti-syphilis medications had increased urine output. Sulfanilamide, the first widely-used diuretics introduced in the early 20th century, was initially used as an anti-bacterial agent and found to inhibit carbonic anhydrase and increase urinary salt and potassium (K+) excretion.1 Despite the successful use of carbonic anhydrase inhibitors in heart failure and hypertension, these diuretics caused the untoward side effect of metabolic acidosis. By modifying the sulfamoyl and amino groups on the sulfonamide, researchers at Merck chemically synthesized chlorothiazide (Diuril).2 The family of thiazide diuretics exhibits strong saluretic effects with minimal or no increase in urinary bicarbonate excretion. Furosemide, also chemically modified from sulfonamide, was introduced in 1962, and became the most potent diuretic in clinical practice to date. The thiazides and furosemide diuretics were later found to inhibit the sodium (Na+)-chloride (Cl-) cotransporter (NCC, SLC12A3) in distal convoluted tubule and the Na+, K+, 2Cl- cotransporter (NKCC2, SLC12A1) in the loop of Henle, respectively.3 These two classes of diuretics have remained among the most widely used diuretics.
In the past three decades, the power of genetics and molecular physiology have revolutionized our understanding of the molecular mechanisms of salt and water handling in renal tubules. The molecular identity of previously known transporters, their regulators, and new transporters and regulatory pathways have been discovered. These ion channels, transporters, and regulatory molecules have attracted attention from pharmaceutical companies or research centers as targets for potential diuretic therapeutics. Current strategies of diuretic development have evolved from serendipity to molecular specificity. Future diuretics are expected to be able to specifically inhibit a critical target involved in a pathway of salt or water regulation in the kidney. The goals are not only to minimize off-target effects but also to make clinical responses more predictable. Also, novel diuretics should surpass current diuretics in potency and safety or could be used in conjunction with current diuretics to reduce diuretic resistance due to compensatory responses. In this review, we discuss the new understanding of mechanisms of renal salt handling and progress in the development of new diuretics.
Mechanisms of Salt Diuresis
Urine is composed of osmoles and water, both of which can be independently regulated by the kidney. Besides glomerular filtration rate, the urine flow rate is determined by the total urinary osmole excretion rate and free water clearance rate. Similar to the composition of serum, Na+ and Cl- are the major cationic and anionic osmoles in glomerular filtrate. Filtered NaCl is sequentially reabsorbed by proximal tubule (∼60%), loop of Henle (∼25%), distal convoluted tubule (∼10%), connecting tubule, and collecting duct (∼5%). Most traditional diuretics, except spironolactone, inhibit NaCl reabsorption by inhibiting apical Na+ transporters or channels in different segments of the renal tubule after they are secreted by organic anion or cation transporters in the proximal tubule. The increased salt delivery to the downstream tubule not only creates negative salt balance but also offsets the osmotic gradient between lumen and interstitium and reduces water absorption in the collecting duct. Since the 1990s, studies on familial hypertensive or hypotensive disorders have identified the primary Na+ transporters responsible for urinary salt reabsorption, such as NCC in the distal convoluted tubule in Gitelman's syndrome,4 NKCC2 in the loop of Henle in Bartter's syndrome,5 and the epithelial Na+ channel (ENaC, SCNN1) in the connecting tubule and cortical collecting duct in Liddle's syndrome and autosomal recessive pseudohypoaldosteronism type I (PHAI).6, 7 These findings highlight the importance of salt reabsorption in blood pressure. In the 21st century, studies on other Mendelian disorders with abnormal blood pressure further deciphered the mechanisms of salt handling in the renal tubule, most notably with-no-lysine (WNK) kinases in pseudohypoaldosteronism type II (PHAII, also known as familial hyperkalemic hypertension or Gordon's syndrome).8 WNK kinases do not directly transport Na+ but regulate the activities of NKCC2, NCC, and ENaC.9 Na+ reabsorption is virtually always coupled with transport of other ions. For example, apical NaCl reabsorption via NKCC2 in the loop of Henle requires apical K+ recycling via the renal outer medullary K+ channel (ROMK, KCNJ1) and basolateral Cl- exit via voltage-gated Cl- channel ClC-Kb channels (CLCNKB), and mutations of each can cause Bartter's syndrome.5 Similarly, the inwardly-rectifying K+ Kir4.1 (KCNJ10) channel is essential for Na+ reabsorption in the distal convoluted tubule and the Cl--HCO3- exchanger pendrin (SLC26A4) for Na+ reabsorption in the cortical collecting duct. Mutations of Kir4.1 and pendrin cause EAST/SeSAME syndrome with Gitelman's-like tubulopathy and Pendred syndrome, respectively.10-12 These proteins are potential targets for diuretics development (Figure 1).
Figure 1. The mechanisms of salt reabsorption in the distal nephron and the related new classes of diuretics.
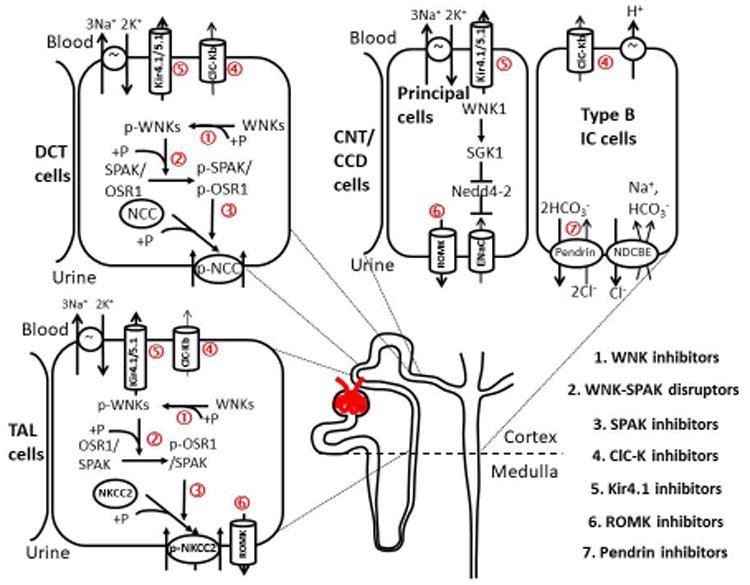
The WNK-SPAK/OSR1-N(K)CC pathway regulates the activity of NCC in the distal convoluted tubule (DCT) and NKCC2 in the loop of Henle through a kinase-dependent phosphorylation cascade. SPAK dominates NCC phosphorylation in DCT while both OSR1 and SPAK activate NKCC2. WNK1 was reported to enhance ENaC in the connecting tubule and cortical collecting duct through SKG1-mediated Nedd4-2 phosphorylation and inactivation, which inhibits the ubiquitination and endocytosis of ENaC. This stimulatory effect of WNK1 on SGK1 is kinase activity independent and may need to cooperate with 3-Phosphoinositide-dependent protein kinase (PDK). The ClC-Kb chloride channel is expressed in the basolateral membrane of TAL, DCT, and non-principal cells of connecting tubule and cortical collecting duct and functionally couples with Na+ transporters in the apical membrane. The potassium Kir4.1/5.1 channel is also localized in the basolateral membrane of the distal nephron and is essential to keep the normal resting membrane potential (MP) and Na+, K+ ATPase activity. ROMK channel provides K+ efflux and recycling to maintain NKCC2 activity in TAL and K+ secretion in the connecting tubule (CNT) and cortical collecting duct (CCD). The Cl--HCO3- exchanger pendrin modulates NaCl reabsorption via ENaC in principal cells and NDCBE in type B intercalated cells. Potentially novel diuretics are listed in the right lower corner, and the molecular targets of these agents are marked in the figure (+P: phosphorylation, ↑: stimulation, ┬: inhibition).
The WNK Pathway for Sodium Handling in the Distal Nephron
WNKs are cytoplasmic serine-threonine protein kinases, which phosphorylate and regulate downstream substrates. WNKs were named because of the lack of the catalytic lysine involved in ATP binding in the canonical β-strand 3, which is instead found in the glycine-rich loop in β-strand 2 in the kinase domain.13 This position of the catalytic lysine, which is unique amongst the serine-threonine kinases, has been exploited in the development of specific WNK inhibitors, as we will describe later. Four members of the mammalian WNK kinase family (WNK1-4) have been identified since the initial cloning of WNK1.14 WNK kinases share a conserved kinase domain and the subsequent autoinhibitory domain, and several coiled-coil motifs, but otherwise are heterogeneous, particularly in the C-terminus. WNKs are widely expressed, including in heart, skeletal muscle, lung, and kidney.15 In kidney, WNK1/3/4 and kidney-specific WNK1 (KS-WNK1), a short isoform of WNK1 transcribed from a distinct exon 4A, along with other splice isoforms are present in renal tubules with a differential distribution. Full-length WNK1 and WNK3 are expressed throughout the nephron while WNK4 and KS-WNK1 are highly localized to the distal convoluted tubule.16 WNK3 knockout mice have trivial alterations in kidney function, perhaps due in part to the relatively small amount of WNK3 and compensation by WNK1 and WNK4.17
In humans, both deletion of intron 1 of the WNK1 gene and missense mutations of WNK4 result in a hereditary hypertensive disorder, PHAII, characterized by hyperkalemia, metabolic acidosis, and hypertension with good therapeutic response to thiazides, indicating hyperactivity of NCC.8 This WNK1/4-NCC pathway has been under active investigation for more than a decade. The consequence of intron 1 deletion is the increased expression of full-length WNK1 and NCC in the distal convoluted tubule, and PHAII mutations in WNK4 also result in increased expression, as described below. Early studies suggested that WNK4 inhibited NCC by antagonizing WNK1 through a kinase-independent interaction.18 Studies using animal models, in vitro biochemistry and heterologous expression discovered that both WNK1 and WNK4 activate NCC through phosphorylating and activating Ste20-related proline/alanine-rich kinase (SPAK, STK39) and the closely related oxidative stress-responsive 1 (OSR1).19 WNKs bind the CCT (conserved carboxyl-terminal) domain, also known as the PF2 (PASK/Fray homology 2) domain, of SPAK/OSR1 via RFxV motifs (Figure 2). The binding facilitates the phosphorylation of the T-loop threonine in the SPAK/OSR1 kinase domain and a serine in the S-motif of SPAK/OSR1. The active SPAK/OSR1 then contacts the N-terminal RFxV/I motifs of SLC12 cation-chloride cotransporters including NCC, NKCC1 (SLC12A2), and NKCC2 and phosphorylates a cluster of conserved threonine and serine residues in the N-terminus of these cotransporters to activate them.19 Chronic stimulation of NKCC2 and NCC in the kidney enhances urinary NaCl reabsorption and causes positive salt balance and hypertension. WNKs also stimulate serum- and glucocorticoid-induced protein kinase (SGK) 1, and the epithelial Na+ channel (ENaC) in the cortical collecting duct, through kinase-independent mechanisms.20
Figure 2. The activation cascade of the WNK-SPAK/OSR1-N(K)CC pathway and the related novel diuretics.
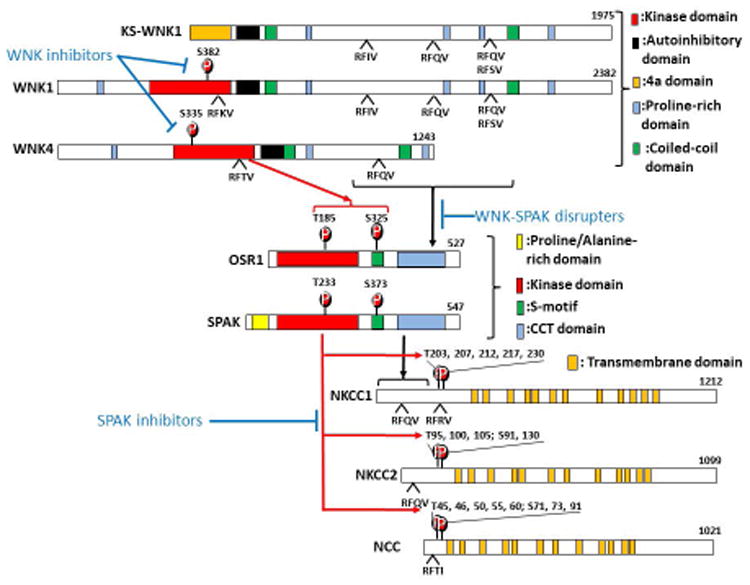
(A) Domain structures of WNKs, SPAK/OSR1, and NKCC1/NKCC2/NCC are shown. Autophosphorylation of WNK kinase (S382 and S335 in WNK1 and WNK4 respectively) is required for WNK activation and subsequent phosphorylation of SPAK and OSR1 (T233 and T185 in the activation loop and S373 and S325 in the S-motif of SPAK and OSR1 respectively). This process requires the interaction between RFxV motifs of WNKs and the CCT domain of SPAK/OSR1. The activated SPAK/OSR1 binds to the N-terminal RFxV/I motifs on their substrates via the CCT domain and phosphorylates a cluster of conserved threonine and serine residues. WNK inhibitors prevent the autophosphorylation of WNKs. WNK-SPAK disrupters interfere with the interaction between WNK and SPAK/OSR1. SPAK inhibitors inhibit SPAK kinase activity and N(K)CC phosphorylation and activation. These novel diuretic agents are highlighted in blue font. The red arrow denotes kinase-dependent phosphorylation. Black arrow represents protein-protein interactions. The blue line indicates pharmacological inhibition.
The WNK1/4-NCC pathway is actively regulated under physiological conditions. Several hormones, including insulin, angiotensin II, and aldosterone, activate WNKs through their receptors in the distal nephron. However, the signaling cascades between these receptors and WNKs are mostly unknown, except the insulin-stimulated phosphatidylinositol 3-kinase-Akt/SGK-WNK pathway.21 Recently, exome sequencing of PHAII patients without WNK1 or WNK4 mutations identified two new pathogenic genes resulting in PHAII when mutated, KLHL3 and CUL3.22, 23 KLHL3 and CUL3 encode a substrate adaptor Kelch-like protein 3 (KLHL3) and a scaffold protein cullin3 (CUL3), respectively, for a cullin3-based E3 ubiquitin ligase, which ubiquitinates WNK kinases for proteasome-mediated degradation. Angiotensin II was shown to activate WNK4 by blocking the binding of KLHL3 to WNK4 via a protein kinase C-dependent pathway.24 PHAII mutations in KLHL3, cullin3, and an acidic region of WNK4 also impaired binding between the cullin3 ubiquitin ligase and WNK4.22, 23 Compared to PHAII patients with WNK1 or WNK4 mutations, PHAII patients with CUL3 or KLHL3 mutations had more severe hyperkalemia, metabolic acidosis and earlier onset of hypertension, likely due to the synchronous increase of all WNK kinases.22, 23 Either way, the abundance of WNK1 and WNK4 are elevated in PHAII, consistent with gain-of-function in WNK signaling resulting in PHAII.
Other than protein degradation, the autophosphorylation and kinase activity of WNKs is highly sensitive to intracellular Cl- concentration (Figure 3). WNK kinases have been proposed to be Cl- sensing kinases for more than a decade, since low intracellular Cl- concentration stimulated WNKs. Recently, a Cl--binding pocket consisting of Leu369, Leu371, Phe283, and Leu299 in WNK1 was discovered by X-ray crystallographic studies (Figure 4A).25 Near the catalytic lysine, a Cl- ion bound in this pocket prevents autophosphorylation and activation of WNK1. In contrast, Cl--binding site mutants and low intracellular Cl- level reduce Cl- binding and increase the activity of WNK1. Subsequent studies have shown that Cl- also inhibits WNK3 and WNK4 activity, with WNK4 the most sensitive to Cl- inhibition in vitro.26, 27 In kidney, this Cl--sensing character of WNKs has been proposed to regulate NCC activity in response to the physiological changes of plasma K+ level.26 Hypokalemia hyperpolarizes the membrane potential of the basolateral membrane, enhancing Cl- efflux and reducing intracellular Cl- concentration. The decrease of intracellular Cl- then allows WNK kinase to phosphorylate itself and the downstream SPAK and NCC.28 Hyperkalemia is expected to exert the opposite effect, but a recent in vitro study suggests that hyperkalemia induces rapid NCC dephosphorylation through a SPAK/OSR1-independent pathway.29 Other conditions that could increase intracellular Cl- concentration, such as inhibition of the basolateral Kir4.1 or ClC-Kb channel, may also inhibit the WNK-SPAK/OSR-NCC pathway in a manner similar to hyperkalemia (see below in Kir4.1 and ClC-Kb section) (Figure 3).
Figure 3. Regulation of the WNK-SPAK/OSR1-NCC pathway in the distal convoluted tubule.
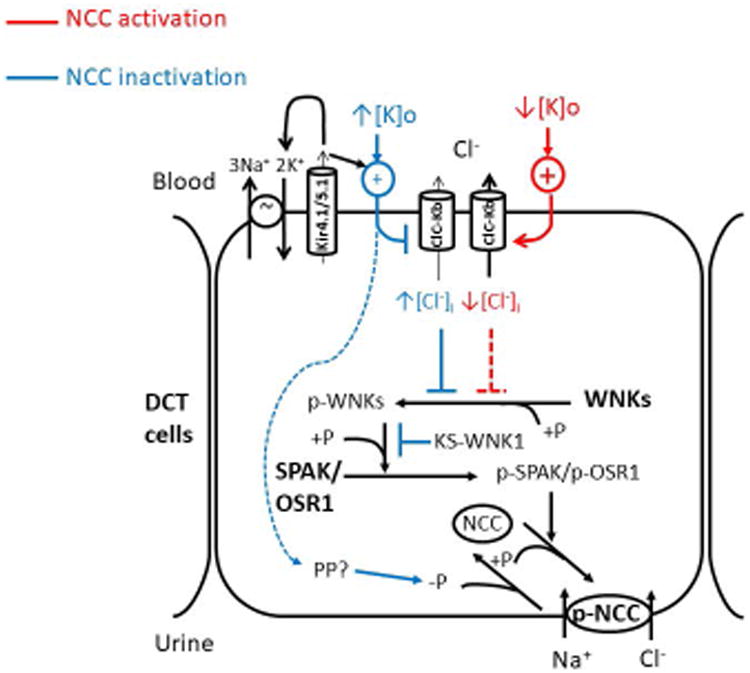
Autophosphorylation on the T-loop serine (S382 in WNK1, S335 in WNK4) activates WNK kinases. This process is inhibited by the Cl- ion binding to the Cl- sensing pocket. The plasma K+ level affects membrane potential (MP) and Cl- efflux via the ClC-Kb channel. Hypokalemia (↓[K]o) releases the Cl--sensing inhibition on WNK by hyperpolarizing MP, increasing Cl- efflux and reducing intracellular Cl- level ([Cl-]i). Hyperkalemia (↑[K]o) is supposed to do the opposite or inactivates NCC through an unknown SPAK/OSR1-independent protein phosphatase (PP) pathway (blue dashed arrow). Kir4.1/5.1 functions to maintain normal MP of distal convoluted tubule (DCT), together with the Na+, K+ ATPase and ClC-Kb channel. Inhibition of Kir4.1 results in a depolarized MP and increased intracellular Cl- concentration. Activated WNKs switch on SPAK/OSR1-NCC signaling through a phosphorylation cascade. Other kinases may phosphorylate NCC since Spak and Osr1 double knockout mice still preserved some phosphorylated NCC. KS-WNK1 may exert competitive inhibition on WNK1 through the interaction with WNK1 downstream substrates. Red lines and blue lines denote stimulatory and inhibitory regulations on NCC, respectively. +P: phosphorylation; -P: dephosphorylation.
Figure 4. New allosteric WNK and SPAK/OSR1 inhibitors.
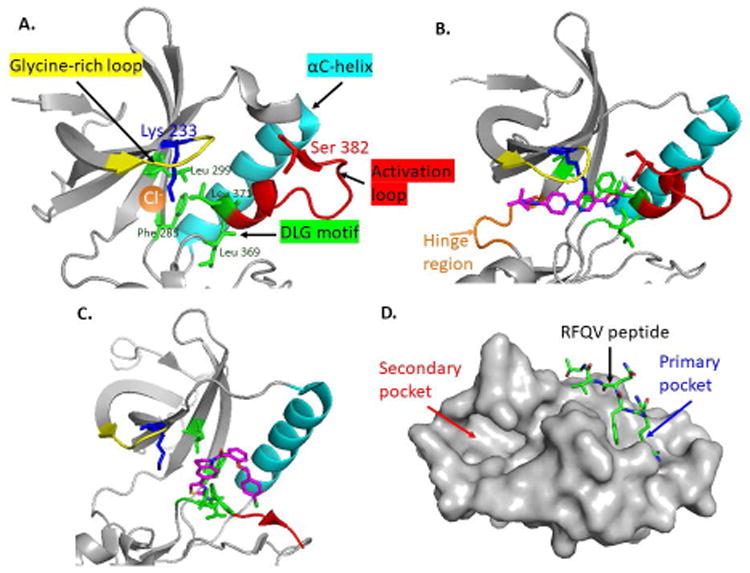
(A) The Cl- binding pocket of WNK1 (PDB: 3FPQ) is surrounded by the glycine-rich loop (yellow), αC-helix (cyan), and activation loop (red). The Cl- (orange dot) forms hydrophobic interactions with Leu369 and Leu371 (green sticks) in the DLG motif (green) and with Phe283 and Leu299 in β3 & β4 helix, respectively. The catalytic lysine (Lys233) and T-loop serine (Ser382) are shown in blue and red sticks. (B) The crystal structure of WNK1 complexed with WNK463 (PDB: 5DRB). WNK463 (magenta) contacts the hinge region (orange loop) of the ATP-binding site and fits perfectly into the narrow tunnel of the catalytic site and interferes with ATP binding of WNKs. (C) ATP non-competitive WNK473 (magenta) binds only to the back pocket (surrounded by DLG sequence, activation loop, and αC-helix) of WNKs but not the ATP-binding site (PDB: 5TF9). (D) The OSR1 CCT domain (PDB: 2V3S) shows the primary pocket (blue arrow) for RFQV peptide docking and secondary pocket where novel SPAK/OSR1 inhibitors bind (red arrow). We drew these figures using Pymol and the open PDB files.
Among all WNK isoforms, KS-WNK1 is the only isoform that is known to inhibit salt reabsorption in the kidney. KS-WNK1 lacks a complete kinase domain but has an otherwise intact C-terminus. In vitro, KS-WNK1 antagonizes the effects of full-length WNK1 through competitively binding WNK1's substrates.30 The functions of KS-WNK1 are best demonstrated by the phenotypes of KS-WNK1 mouse models. Two KS-WNK1 knockout mice displayed increased activity of NCC and mild hypertension.31,32 In contrast, transgenic mice overexpressing KS-WNK1 became relatively hypotensive and excreted more urinary K+.32 Dietary K+ regulates WNK1 isoform expression. Restriction of K+ intake increased full length-WNK1 but decreased KS-WNK1 while high dietary K+ enhanced KS-WNK1 and the ratio of KS-WNK1 to full-length WNK1.33 This modification of WNK1 isoforms adjusts the amounts of distal salt delivery to ensure a proper urinary K+ excretion in response to dietary K+. Other intracellular regulatory mechanisms of WNKs include the oligomerization between different WNKs and changing subcellular localization.9 In summary, WNK kinases are important regulators of NaCl reabsorption in the distal nephron. Agents targeting the KLHL3/CUL3-WNK1/4-SPAK/OSR1-NCC/NKCC2 pathway could be promising to induce solute diuresis and reduce blood pressure. Here, we introduce novel agents that exert inhibitory effects on this pathway.
WNK kinase inhibitor
WNK kinase inhibitors have been pursued by pharmaceutical companies for more than a decade. Researchers from Novartis recently reported the first orally bioavailable pan-WNK-kinase inhibitor, WNK463.34 The prototype of WNK463 came from a hit in a high-throughput screen for small molecule inhibitors of WNK1 catalytic activity. WNK463 exhibited strong affinity and potent inhibition of all WNK kinases in vitro (KD ∼4 nM for WNK1 and WNK4) and reduced the phosphorylation of OSR1 in HEK293 cells. In the crystal structure of WNK463 in complex with WNK1 S382A (kinase-dead) kinase domain (PDB: 5DRB) (Figure 4B), WNK463 contacts the hinge region of the ATP binding site and fits in the narrow tunnel of the catalytic site, a unique structure created by the atypical placement of Lys233 in subdomain 1 of WNK1. Accordingly, WNK463 displayed a high specificity to WNK kinases and inhibited only 2 out of the other 422 human kinases tested at an extremely high inhibitor concentration. In both spontaneously hypertensive rats and WNK1 transgenic mice, WNK463 showed a remarkable saluretic and kaliuretic effect (urine output and Na+ excretion up to ∼4-fold of basal state at a dose of 10 mg/kg) and reduced blood pressure in a dose-dependent manner (∼40 mmHg reduction at 10 mg/kg). The amounts of phosphorylated SPAK and OSR1 in the kidney lysate of WNK463-treated rodents were also decreased. Despite the successes of in vitro and animal studies, the development of WNK463 as a therapeutic drug has been halted due to unspecified serious safety events, likely due to the ubiquitous expression of WNKs and the inhibition of all WNK paralogs by WNK463. The same group reported another ATP non-competitive allosteric WNK inhibitor, WNK476, by conducting another high-throughput screen against 1.2 million compounds at high ATP concentrations.35 Distinct from WNK463, WNK476 binds to the back-pocket formed by the outwardly moved αC-helix, activation loop, and glycine-rich loop, adjacent to the ATP binding site of WNKs (PDB: 5TF9) (Figure 4C). WNK476 binds only phosphorylated WNK1 and could be more efficient in high physiological ATP conditions. Further chemical modification and trials in animals are ongoing.
WNK-SPAK binding disrupter
Knockout of WNK1 or OSR1 results in embryonic lethality.36 In contrast, SPAK knockout mice were viable and did not show obvious extra-renal phenotypes except vasodilation due to decreased vascular NKCC1 activation.37 This result suggests that agents that inhibit SPAK may cause less unwanted side effects. The unique CCT (or PF2) domain of SPAK is functionally essential by providing a docking site for its activators (WNKs) and substrates (SLC12 cation-chloride cotransporters) (Figure 4D). Since the CCT domain of SPAK/OSR1 is structurally unique, agents that interrupt protein interactions with the CCT domain could be potentially specific to SPAK/OSR1. Two compounds, STOCK1S-50699 and STOCK2S-26016, were found to disrupt the binding between a WNK4-RFQV peptide and SPAK-CCT in a high-throughput screen testing 17,000 compounds using fluorescent correlation spectroscopy.38 These compounds dose-dependently (at 50∼200 μM range) reduced the phosphorylation of endogenous SPAK and NCC in mammalian kidney cells, but have not been tested in animals. However, knockin mice have been generated in which Leu502 in the CCT domain of SPAK was mutated to Ala, in order to abolish binding to RFXV/I in SPAK binding partners, genetically mimicking the effect of STOCK1S-50699 and STOCK2S-26016. These mice had decreased NCC, NKCC1 and NKCC2 phosphorylation and decreased blood pressure.39 Further testing will be required to determine pharmacokinetics, pharmacodynamics, and toxicity in animal models, with chemical modification as needed to improve bioavailability and potency. While the genetic data suggest that inhibition of SPAK binding to its substrates will not be overly toxic, it is unknown whether interfering with OSR1 binding to substrates will have additional toxicity.
SPAK kinase inhibitor
Another study conducted a high-throughput screen of a ∼20,000 compound small molecule library and identified compound STOCK1S-14279, which inhibited the phosphorylation of NKCC2 by constitutively active SPAK T233E with an IC50 of 0.26 μM in a non-ATP-competitive manner.40 However, repeated injection of STOCK1S-14279 in mice caused lethality. In a library of 840 existing drugs, Closantel, an antiparasitic agent in livestock, showed similar structure and effect of STOCK1S-14279 in vitro. Moreover, Closantel induced a rapid-onset (< 30 mins) and short-lasting hypotensive effect after a single dose (20 mg/kg) intraperitoneal injection in mice, likely due to the vasorelaxing effect through NKCC1 inhibition in vascular smooth muscle. In mouse kidney, Closantel inhibited NCC phosphorylation both acutely and chronically, but only chronic treatment (300 mg/kg oral) of Closantel reduced NKCC2 phosphorylation. The cause of this discrepancy remains unclear but may be due to the cross-reaction and concurrent inhibition of OSR1 by Closantel in chronic Closantel treatment. In addition, the level of phosphorylated NKCC2 does not always reflect the activity of NKCC2. For example, although NKCC2 phosphorylation is increased in SPAK knockout mice,37 ex vivo micro-perfusion experiments showed that 70∼80% of Na+ flux in the thick ascending limb of Henle's loop, which is mostly due to NKCC2 activity, is SPAK-dependent.41 The remainder could be mediated by OSR142 and other Ste20 kinase-independent NKCC2 stimulation pathways, such as AMP-activated protein kinase (AMPK, PRKA), calcium-binding protein (CAB39), cAMP, or other kinases.43-45 A recent study confirmed that Closantel and another structurally similar anti-parasitic agent, Rafoxanide, inhibited the kinase activity of OSR1 T185E and SPAK T233E (constitutively active mutants) in vitro through binding to the secondary pocket of the CCT domain (Figure 4D).46 Surprisingly, STOCK1S-50699, a WNK-SPAK disrupter, also bound to the secondary pocket and inhibited OSR1 kinase activity. Therefore, STOCK1S-50699 may have dual actions and high potency against SPAK/OSR1. Overall, the role of SPAK in NKCC2 regulation is not as clear as its dominant role in NCC activation. Of note, chronic treatment with Closantel neither reduced blood pressure nor increased urinary salt excretion, possibly due to downstream adaptation in the collecting duct. Further modification of STOCK1S-14279 or Closantel and in vivo testing in animals are warranted.
Taken together, WNK463, a specific pan-WNK kinase inhibitor, has become a proof-of-concept agent that elegantly demonstrates the powerful role of WNK kinases in controlling renal salt and K+ handling. However, the clinical application of WNK463 seems limited by the ubiquitous expression of WNKs in other critical organs (Figure 5). Further modification of WNK463 to enhance its specificity to WNK4 kinase might be a solution to avoid serious side effects since WNK4 is highly concentrated in the distal convoluted tubule and WNK4 knockout mice did not show severe extra-renal phenotypes. Like WNK4, SPAK inhibition is a theoretically practical choice to inhibit the WNK-N(K)CC pathway due to the relatively milder phenotype of SPAK knockout mice compared to the embryonic lethality of WNK1 or OSR1 knockout mice. However, the goal of producing a SPAK-specific agent is challenging due to the similarity between SPAK and OSR1. Closantel, an existing drug with anti-SPAK effects, displayed some promising results of reducing phosphorylation of NCC and NKCC2 in kidneys, and NKCC1 in aorta, but had a short-acting antihypertensive effect. More experiments are required to realize a potent and safe anti-WNK-SPAK-NCC diuretic.
Figure 5. The tissue expression pattern of the novel diuretic target genes.
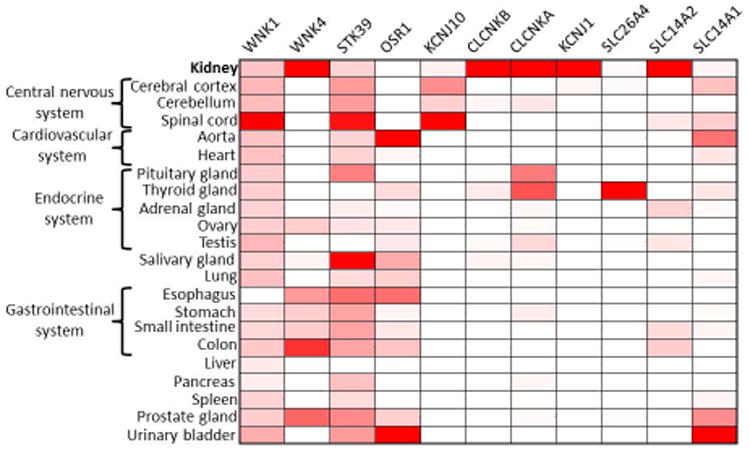
Heat map represents the abundance (red: presence, white: absence) of 11 potential targets of novel diuretics or aquretics (columns) across 22 human tissues (rows) based on public RNA-seq data (the Genotype-Tissue Expression (GTEx) Project, etc.). Color intensity represents fractional density expression of each mRNA across all tissues.
Alternative Routes to Manipulate Na+ Transport in the Distal Nephron
Inwardly rectifying Kir4.1 in the distal convoluted tubule
In 2009, two groups reported that human KCNJ10 mutations lead to EAST syndrome characterized by epilepsy, ataxia, sensorineural deafness, and Gitelman's-like tubulopathy.10, 11 The KCNJ10 gene encodes an inwardly rectifying K+ Kir4.1 channel that is expressed in brain, inner ear, and kidney. Kir4.1 forms a heterotetramer with Kir5.1 in the basolateral membrane of the distal nephron ranging from thick ascending limb to collecting duct. Kir4.1/5.1 maintains the normal resting membrane potential of distal nephron epithelial cells close to the equilibrium potential of K+. In addition, by recycling K+, Kir4.1/5.1 is critical for the activity of the basolateral Na+, K+ ATPase for extrusion of Na+ into the circulation. Since EAST syndrome patients manifest a renal phenotype similar to Gitelman's syndrome, the major renal defect of KCNJ10 mutation likely occurs in the distal convoluted tubule. This notion is supported by the finding that genetic deletion of Kir4.1 abolished the basolateral K+ conductance and depolarized the membrane potential of epithelial cells of distal convoluted tubule.47 Also, expression of Kir4.1 disease mutants in distal convoluted tubule cells increased intracellular Cl- concentration because the depolarized resting membrane potential suppressed the Cl- efflux via the ClC-Kb channel.27 Kidney-specific Kir4.1 knockout mice recapitulated Gitelman's syndrome with decreased phosphorylation of NCC, NKCC2, and SPAK in renal tubules, perhaps through Cl--mediated inhibition of WNK kinase activity.48 The NCC activity of Kir4.1 knockout mice did not respond to changes in dietary and plasma K+, likely due to the persistently depolarized membrane potential of distal convoluted tubule.
So far, there is no known specific Kir4.1 inhibitor. Pharmacological inhibition of Kir4.1 is expected to decrease NCC and possibly NKCC2 activity and cause salt diuresis. Kir4.1 also functions in the connecting tubule and cortical collecting duct and contributes to the polarized basolateral membrane potential. ENaC expression was increased in Kcnj10 knockout mice, but the amiloride-sensitive Na+ flux was not measured.49 The broad distribution of Kir4.1, including important roles in the nervous system (Figure 5) that explain the neurological phenotypes in patients with KCNJ10 mutations, and the fact that global KCNJ10 deletion led to an early fatality 2 weeks after birth, raise concerns about potential side effects.47
Voltage-gated ClC-Kb chloride channel in the loop of Henle and distal convoluted tubule
Similar to the Kir4.1 channel, the ClC-Kb Cl- channel is also localized to the basolateral membrane of thick ascending limb, distal convoluted tubule, connecting tubule, and intercalated cells of collecting duct. Human mutations in the CLCNKB gene, encoding ClC-Kb, lead to classic Bartter's syndrome featuring urinary Na+, K+, and divalent cation wasting, hypokalemia, and metabolic alkalosis, resembling the phenotype of defective NKCC2.5 In addition, some patients with CLCNKB mutations manifest a milder phenotype mimicking Gitelman's syndrome due to defective NCC. These clinical phenotypes highlight the important role of ClC-Kb in the loop of Henle and distal convoluted tubule. The Clcnk2 (mouse ortholog of human CLCNKB) mouse model has not been established until recently. The Clcnk2 knockout mice had antenatal Bartter's-like syndrome with growth retardation, hyperprostaglandin E, and severe salt-losing tubulopathy with hypotension.50, 51 In Clcnk2 knockout mice, the phosphorylated and total NCC levels were remarkably decreased, while levels of NKCC2 phosphorylation were highly variable. In diuretic challenge tests, Clcnk2 knockout mice had blunted responses to furosemide and thiazide, suggesting decreased activity of NCC and NKCC2. The difference between the highly variable phosphorylated NKCC2 level and the blunted response to furosemide test further emphasizes that phosphorylated NKCC2 may not accurately reflect NKCC2 activity. Although the mechanisms of NCC and NKCC2 deactivation in Clcnk2 knockout mice are still unclear, it is proposed that basolateral ClC-Kb channel deletion leads to increased intracellular Cl- concentration, which binds and inhibits the kinase activity of WNK and switches off the WNK-SPAK-N(K)CC pathway in the distal convoluted tubule and loop of Henle. The phenotype of Clcnk2 knockout mice was more like antenatal Bartter's syndrome rather than classic Bartter's syndrome with highly variable phenotypes. The severe phenotype of Clcnk2 knockout mice likely results from the efficient genetic ablation and nearly abolished ClC-K2 current. Also, there was no detectable 40-pS ClC-K1 channel in isolated Clcnk2 knockout distal renal tubules, suggesting that ClC-K1, a homologous chloride channel mostly distributed in renal medulla, did not compensate for ClC-K2 in renal cortex. Whether ClC-Ka (human ortholog of ClC-K1) compensates for ClC-Kb in human is still unknown. The phenotypic heterogeneity in patients with CLCNKB mutations could be due to variable degrees of remaining basolateral Cl- conductance in the thick ascending limb and distal convoluted tubule provided by the mutant ClC-Kb channels52 or other Cl- channels, such as ClC-Ka or pseudo-cystic fibrosis transmembrane conductance regulator (pseudo-CFTR) channel.53 In the collecting duct, ClC-Kb dominates the basolateral Cl- conductance in type A and type B intercalated cells, but not in principal cells.50, 54 The function of ClC-Kb in intercalated cells is still unclear but likely mediates Cl- absorption or Cl- recycling across the basolateral membrane.54, 55 Other Cl- transporters, such as K+, Cl- cotransporter or anion exchangers, are present in these segments and may compensate for the loss of ClC-Kb.
Two benzofuran derivatives, MT-189 and RT-93, were chemically synthesized by Liantonio A, et al with affinity for ClC-Ka/Kb of KD < 10 μM.56 When orally fed to rats, MT-189 and RT-93 increased urine flow by ∼60% at the dosage of 50 mg/kg and significantly reduced blood pressure by ∼12% at the dosage of 200 mg/kg. The total urinary salt excretion, however, was not increased, suggesting that the renal effect of these two agents was mainly water diuresis instead of salt diuresis. It is possible that the current benzofuran derivatives inhibit ClC-K1 more efficiently than ClC-K2. The ClC-K1 channel in mice is mainly localized in both the apical and basolateral membranes of the thin ascending limb of Henle's loop and is partly responsible for the hyperosmolality of the renal medulla. The Clcnk1 knockout mice (mouse ClC-K1 is equivalent to human ClC-Ka) exhibited nephrogenic diabetes insipidus and had a lower salt concentration in the renal medulla.57 MT-189 treated rats do not show decreased inner medulla osmolality but do have reduced aquaporin 2 (AQP2) expression. The link between ClC-K1 and aquaporin 2 needs further clarification. Since their expression is limited to a few organs, including kidney, brain, inner ear, salivary gland, and thyroid gland (Figure 5), ClC-K channels may represent better targets for novel diuretics in comparison with Kir4.1. In addition, a potent ClC-Ka and ClC-Kb inhibitor can theoretically induce both water diuresis and salt diuresis, similar to the phenotype of severe polyuria and renal salt wasting in patients with bigenic CLCNKA and CLCNKB mutations or BSND mutation.58 The BSND gene encodes barttin (BSND), which is an essential accessory protein for both ClC-Ka and ClC-Kb. Barttin mutation disrupts the function of ClC-Ka and ClC-Kb simultaneously and causes Bartter's syndrome type IV with sensorineural deafness.59 The congenital deafness in these affected infants hints at the potential risk of ototoxicity with non-selective ClC-K inhibitors.
Pendrin inhibitors
Pendrin, encoded by the PDS gene, is a monovalent anion (Cl-, HCO3-, I-, formate) exchanger, which is mainly expressed in thyroid, inner ear, adrenal gland, inflamed airways, and distal nephron.12 Mutations in the PDS gene result in Pendred syndrome, an autosomal recessive disorder with congenital goiter and sensorineural deafness. Pendrin is highly expressed in the apical membrane of type B and non-A, non-B intercalated cells in connecting tubule and cortical collecting duct and functions as a Cl--HCO3- exchanger (one luminal Cl- for a cytoplasmic HCO3-).60 Although pendrin does not transport Na+ itself, it regulates the activity and abundance of ENaC in principal cells through luminal HCO3- levels and functionally couples with Na+-dependent Cl--bicarbonate exchanger (NDCBE, SLC4A8) in type B intercalated cells.61, 62 However, neither patients with Pendred syndrome nor pendrin-deficient mice manifest obvious abnormalities in renal salt handling or acid-base imbalance. The importance of pendrin in salt handling and blood pressure was revealed only in certain conditions when pendrin is normally upregulated, such as salt restriction and disorders of upstream Na+ transporters.63 Pendrin is upregulated in NCC-null mice and SPAK-null mice.64, 65 Double knockout of NCC and pendrin predisposes mice to severe salt wasting and volume depletion.66 Of note, neither NCC nor pendrin single knockout displayed significantly increased urinary NaCl loss or blood pressure change, indicating that NCC function compensates for the loss of pendrin-ENaC function in the cortical collecting duct and vice versa. This notion is supported by the report that a child with Pendred syndrome had an exaggerated response to thiazide,67 and suggests that dual inhibition of NCC and pendrin may be a potent diuretic therapy.
A new pendrin inhibitor, PDSinh-C01, is a pyrazole-thiophenesulfonamide derivative.68 This compound was selected from a high throughput screen of 36,000 synthetic small molecules against human pendrin and modified from the original hit. PDSinh-C01 inhibited ∼85% of pendrin activity with an IC50 1.2 μM in vitro and did not affect the activities of other anion exchangers. In an animal trial, PDSinh-C01 treatment alone did not significantly cause increased urinary salt excretion, acid-base disturbance, or decreased blood pressure, but potentiated the saluretic and kaliuretic effects of furosemide, especially in chronic furosemide use when pendrin is expected to be activated to compensate for the loss of NKCC2 activity. Surprisingly, PDSinh-C01 decreased the effects of thiazide, which seems to be in conflict with the notion of upregulation of NCC. However, it should be noted that thiazide may also inhibit the activity of the pendrin/NDCBE functional complex.62 Overall, since it is likely the last gatekeeper for salt reabsorption, pendrin may be an attractive target for novel diuretics. The effects of PDSinh-C01 on pendrin in other tissues, such as inner ear and thyroid (Figure 5), demands careful evaluation before human trials.
Another compound, PDSinh-A01, which has a tetrahydropyrazolopyridine structure, showed a more potent inhibitory effect on pendrin than PDSinh-C01, and nearly abolished pendrin activity in vitro at a dose of <10 μM.69 PDSinh-A01 also showed a high specificity towards pendrin as compared to the proteins highly similar to pendrin in sequence, such as Cl--anion exchanger (chloride-losing diarrhea protein, SLC26A3). The in vivo effect of PDSinh-A01 in mice has not been reported. Pendrin increases the airway surface liquid depth in IL-13 treated inflamed human airway epithelial cells. Therefore, PDSinh-A01 could be therapeutic for cystic fibrosis patients with defective Cl- and HCO3- transport in airways. Pendrin activity is not affected by most currently used diuretics, including thiazide and furosemide.
ROMK channel inhibitor
The ROMK (Kir1.1) channel encoded by the KCNJ1 gene is the founding member of the Kir inwardly-rectifying K+ channel family. Hypofunctional mutations in ROMK cause antenatal Bartter's syndrome type II. These patients present with prenatal polyhydramnios, metabolic alkalosis, hypotension, failure to thrive, and renal salt wasting, with hypokalemia after a period of neonatal hyperkalemia.5 The potential antihypertensive effect of inhibiting ROMK is also demonstrated by the lower blood pressure found in individuals in the population with heterozygous KCNJ1 mutations or single nucleotide polymorphisms which reduce ROMK current.70, 71 The ROMK channel functions in the apical membrane of Henle's loop to recycle K+ back into the lumen to maintain NKCC2 activity since the luminal K+ level is much lower than Na+ and Cl- in the loop of Henle and becomes the rate-limiting step for NKCC2 activity. In the aldosterone-sensitive distal nephron ranging from late distal convoluted tubule to medullary collecting duct, the apical ROMK channel secretes K+ into the lumen under the regulation of aldosterone. The K+ efflux via ROMK is responsible for K+ excretion in basal conditions. With higher dietary K+ content, the Maxi-K channel (also called BK, KCNMA1), a flow-stimulated K+ channel, is required to maximize urinary K+ secretion. Based on these physiological functions in the kidney, a ROMK inhibitor could be a potent furosemide-like diuretic without excessive K+ wasting and severe hypokalemia, leading to a search for specific ROMK inhibitors. The major obstacle is the specificity towards ROMK since all K+ channels share a similar structure. For example, early compounds, like glyburide, were not clinically useful as diuretics due to their low specificity towards ROMK, and cardiovascular and metabolic effects due to the inhibition of hERG (KCNH2) and ATP-sensitive K+ channels, respectively.72
Scientists at Merck discovered a ROMK inhibitor, Compound A, by chemically modifying an original hit from a high throughput screen of 1.5 million compounds.73 Compound A binds ROMK from the cytoplasmic side with high potency (IC50 < 100 nM) and high specificity (IC50 to hERG > 5 μM). Intravenous or oral administration of compound A enhanced urine output and total urinary salt excretion, but did not affect urine K+ excretion.73 The potency of diuresis and natriuresis (∼4-fold vs. vehicle) of compound A was similar to thiazide diuretics, and the average reduction in mean blood pressure by a continuous intravenous infusion at 1.55 mg/kg/hr was ∼10 mmHg. However, compound A induced QT prolongation. To further improve the specificity toward ROMK, the same group developed MK-7145, which showed superior diuretic and natriuretic effects (∼8-fold increase vs. vehicle) compared to thiazide (∼5-fold) and lowered systolic blood pressure by 20 mmHg at a dose of 10 mg/kg/day with no reported cardiac side effects. The kaliuretic effect was relatively mild with 1.5 ∼2-fold increase, and the plasma K+ level was unchanged.74 In addition, MK-7145 enhanced the antihypertensive effect of thiazide and candesartan. Owing to the success in animal studies, the phase 1b trial (MK-7145-009 AM1) of MK-7145 has been conducted in humans, but results have not been released. Although the lack of kaliuretic effect of ROMK inhibitor could be attractive, this finding needs further evaluation, especially in chronic use, since patients with ROMK mutations develop hypokalemia after infancy due to the compensation by the Maxi-K channel.75 In sum, the targets discussed in this section are not Na+ transporters but support Na+ uptake by other transporters or channels. The combinatorial use of traditional diuretics and these potential novel diuretics could induce a more efficient diuresis and avoid diuretic resistance by blocking compensation by other Na+ transporter pathways.
Mineralocorticoid receptor antagonists
Mineralocorticoid receptor (MR, NR3C2) mediates the effects of aldosterone to conserve Na+ and excrete K+ in the aldosterone-sensitive distal nephron, where the enzyme 11β-hydroxysteroid dehydrogenase type 2 (11βHSD2) inactivates cortisol and allows aldosterone to interact with the ligand binding domain of MR. Patients with autosomal dominant mutation in NR3C2 gene manifest severe urinary salt wasting, failure to thrive, hyponatremia, hyperkalemia, and metabolic acidosis, similar to autosomal recessive PHAI but improving by age 1-3.76 To understand the renal effects of MR, two kidney-specific MR knockout mice were created.77, 78 Both models showed decreased total and active forms of ENaC and NCC in kidneys. MR has been long known to directly stimulate the transcription and apical abundance of ENaC under the control of aldosterone.79 However, the effect of aldosterone and MR on NCC has not been clearly demonstrated until recently. The PY-motif containing WNK1 was found in human kidney and subjective to Nedd4-2 mediated ubiquitination and proteasome degradation. Aldosterone phosphorylated and inactivated Nedd4-2 by SGK1 and thus enhanced the WNK1-SPAK-NCC signaling.80 In MR knockout mice, the NCC level, both total and phosphorylated forms, was reduced. This effect on NCC can be reversed by correcting hyperkalemia in MR knockout mice fed with low potassium diet, suggesting that the change of NCC in MR knockout mice was secondary to the plasma K+ levels.77 According to these findings, pharmacological blockade of MR could lead to suppression of ENaC activity, ROMK-mediated K+ secretion, and WNK-SPAK-NCC signaling, either directly through the SGK1-Nedd4-2 pathway or indirectly through the effect of hyperkalemia.
Spironolactone, the first developed MR antagonist (MRA) in 1957, exerts relatively weak K+-sparing diuresis and undesirable anti-androgenic effects (e.g. gynecomastia, impotence in men) and progesteronic effects (e.g. menstrual irregularity in women) through the non-specific bindings to various steroid receptors. Many attempts have been made to improve the efficacy and specificity of MRA by modifying the structure of spironolactone.81 Eplerenone, the second-generation MRA, is more selective for MR despite lower potency. Evidence of the beneficial effects of adding spironolactone or eplerenone to the traditional treatments for heart failure or chronic kidney disease has accumulated, even at doses without anti-hypertensive or diuretic effect. These benefits are likely mediated by extrarenal MR in heart, vessels, nervous system, macrophage, etc.82 However, spironolactone and eplerenone frequently lead to hyperkalemia and renal function impairment. These renal side effects often limit the further dose escalation for the beneficial effects, such as anti-inflammatory and anti-fibrotic effects and sympathetic nervous system blockade. To counter these renal side effects, third-generation nonsteroidal MRAs, such as finerenone, achieved higher potency and selectivity for extrarenal MR by modifying physicochemical properties (e.g. lipophilicity, polarity, plasma protein binding, etc.) and altering tissue distribution compared with spironolactone and eplerenone.83 Finerenone has demonstrated promising anti-proteinuric effects and reduced cardiovascular events in patients with diabetic nephropathy without disturbing serum K+ level and renal function.84, 85 More than 350 clinical trials (ClinicalTrials.gov) of novel MRAs are ongoing. It will be interesting to follow the diuretic effect of these novel MRAs.
Mechanisms of Urine Concentration
Urine concentration relies on the osmotic gradient that increases progressively from the corticomedullary boundary to the tip of the papilla and proper membrane insertion of aquaporin water channels in collecting duct (Figure 6). These two prerequisites of urine concentration are both tightly regulated by vasopressin. In the outer medulla, the osmotic gradient is maintained by the accumulation of NaCl, which depends on the activity of NKCC2 and the interactions between the loop of Henle and vasa recta, the so-called countercurrent multiplication and exchange.86 In the inner medulla, NaCl and urea accumulation likely contribute to the formation of medullary hyper-osmolality. The high water permeability and low solute permeability of the descending limb of Henle's loop result in increased luminal NaCl concentration, which passively diffuses into the medullary interstitium through the highly Cl--permeable hairpin loop and ascending limb of Henle's loop. Urea is one of the major metabolic wastes and urinary osmoles, especially when dietary protein intake is high. Because most urea has to be excreted, the tubular permeability of urea remains low except the iso-osmotic urea absorption in proximal tubule. Consequently, the high luminal urea concentration in collecting duct could counteract the action of vasopressin and renal tubular water reabsorption.87 The urea transporter A1 and A3 (UT-A1, UT-A3) in the inner medullary collecting duct solve this conundrum by delivering urea into the inner medulla interstitium. The interstitial urea in renal medulla could not be maintained if the blood flow in ascending vasa recta removes urea. A delicate structure, the vascular bundle in the inner strip of the outer medulla, prevents the dissipation of urea in inner medulla.88 The ascending vasa recta co-localizes with descending vasa recta in the vascular bundle center, where the UT-B channel on descending vasa recta allows the urea in ascending vasa recta to quickly recycle to the inner medulla by countercurrent exchange. In addition, UT-A2 on the thin descending limb of short loop, which lines the periphery of the vascular bundle, offers an alternative route for urea to return to the inner medulla through the distal nephron. In contrast, some studies suggested that UT-A2 can outwardly transport urea to ascending vasa recta when pars recta secrets urea,89 which is, however, minuscule in the basal state. Further work will be needed to clarify the role of UT-A2 in urinary concentration mechanisms. Polyuria and water diuresis observed in several urea transporter knockout mice further consolidate the importance of urea transporters in urine concentration. Pharmacological targeting of urea transporters could be a promising strategy for water diuresis (Figure 6).
Figure 6. The mechanisms of NaCl and urea accumulation in renal medulla and the related new classes of aquaretics.
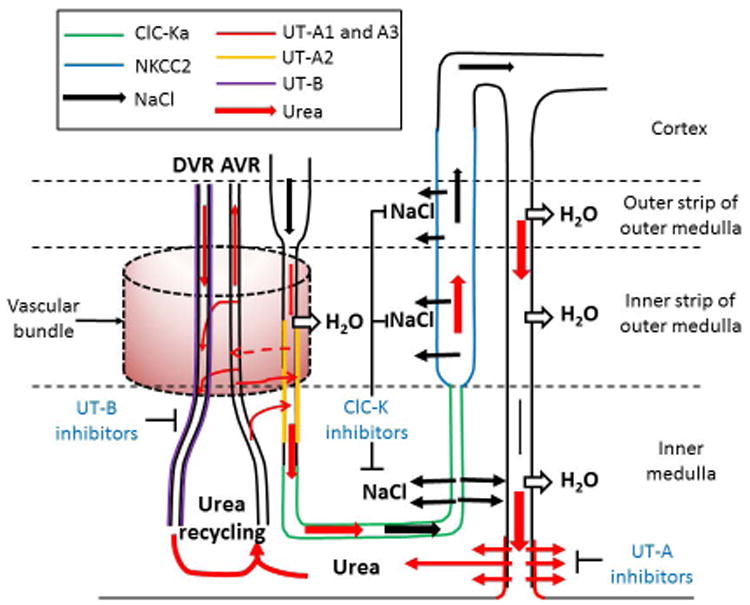
The urinary NaCl concentration (black arrow) increases along the thin descending limb. When urine enters the hairpin loop and thin ascending limb, the highly-concentrated urinary NaCl enters the inner medulla via ClC-Ka on the apical and basolateral membranes. NaCl further accumulates in outer medulla via the active reabsorption by NKCC2 in thick ascending limb.
The urinary urea (red arrow) is concentrated along the nephron. The concentrated urea in inner medullary collecting duct is reabsorbed via UT-A1 and UT-A3 under the stimulation of antidiuretic hormone. Urea leaving the inner medulla via ascending vasa recta (AVR) can be brought back to inner medulla via UT-B on descending vasa recta (DVR) or UT-A2 on thin descending limb. This phenomenon is called urea recycling. UT-A2 may reabsorb urinary urea into adjacent AVR (dashed arrow) and then reenter into DVR or general circulation, although the physiological function of UT-A2 is still unclear. Vascular bundle (dashed, pink cylinder) where close proximity of the DVR and AVR in the center and the AVR and thin descending limb in the periphery prevents removal of medullary urea into the circulation; see text for more details. The dashed line marked the separations of renal cortex, outer medulla, and inner medulla. Each colored line represents the localization of proteins that are involved in NaCl or urea accumulation. For the sake of simplicity, only a long loop of Henle is shown. The classes of potential novel diuretics are highlighted in blue font.
Lessons from human hereditary polyuric disorders and mouse models
Most human hereditary nephrogenic diabetes insipidus results from mutations in the vasopressin receptor V2 (AVPR2 gene) or mutations in the water channel aquaporin 2 (AQP2 gene). The regulation of aquaporin and the use of vasopressin receptor antagonists have been extensively reviewed. Patients with SLC12A1 (NKCC2), CLCNKB (ClC-Kb) or BSDN (barttin) mutation develop Bartter's syndrome and characterized by prenatal polyhydramnios and neonatal polyuria.5 A small group of patients without Kidd blood group antigen (Jka-b-), also known as UT-B, had submaximal urine concentration (∼800 mOsm/Kg.H2O).90 Single nucleotide polymorphisms (V227I, A357T) in UT-A2 have also been reportedly associated with reduced diastolic blood pressure although the underlying mechanism is still unknown.91 These congenital polyuric disorders demonstrate the importance of NaCl reabsorption in the ascending limb of Henle's loop and urea accumulation in inner medulla to the maximal concentrating ability of kidneys. The current model of urine concentration comprises two separate systems for high NaCl transport in the ascending limb of Henle's loop and urea reabsorption in inner medullary collecting duct.
The human SLC14A1 and SLC14A2 genes encode UT-B and UT-A, respectively. In human kidneys, there are at least 3 spliced isoforms of UT-A (UT-A1, A2, A3) and two isoforms of UT-B (UT-B1, B2) due to alternative splicing or transcriptional initiation. These isoforms are differentially expressed in different parts of the kidneys. UT-B is limited to the endothelial cells of descending vasa recta. Genetic ablation of UT-B caused a 50% increase in urine output, mild reduction in maximal urinary osmolality, and increased plasma urea in mice.92 UT-A1 and UT-A3 differentially express in the apical and basolateral membrane of inner medullary collecting duct and mediate urea transport into the innermost portion of renal pyramids, the so-called papilla. UT-A1 and UT-A3 knockout mice urinated 3-fold more urine than wild-type mice on a regular diet and had severely impaired urine concentration.93 The urea concentration in the inner medulla of these mice was two-third lower than in wild-type mice while the medullary NaCl concentration did not change. This finding suggests that the urea accumulation via UT-A1 and A3 may not be the only mechanism to create the axial NaCl gradient in inner medulla.86 Of interest, a low protein diet partly rescued the urinary concentrating defect in UT-A1 and A3 knockout mice while high protein diet aggravated it. This finding suggests that the role of UT-A1 and UT-A3 in urine concentration is more important when dietary protein is high. UT-A2 is present in the descending limb of Henle's loop. Genetic deletion of UT-A2 in mice showed only a mild concentrating defect when the mice were on a low-protein diet.94 In addition, UT-A2 deletion improved the urinary concentration defect in UT-B knockout mice, presumably by reducing the urea loss into circulation via ascending vasa recta.89 The function of UT-A2 is not as clear as other UTs. A recent all-UT-knockout mouse model exhibited a severe urine concentrating defect due to defective urea accumulation in inner medulla, slightly lower blood pressure but no disturbance of renal Na+ transporters activities and internal electrolyte balance.95 The lower blood pressure likely resulted from water diuresis. Also, these mice displayed few extrarenal phenotypes, except early maturation of the male reproductive system, providing substantial evidence supporting the safety of non-selective UT inhibitors.
Comparing the phenotypes between Clcnk1 knockout and all-UT-knockout mice, similar reductions in urinary osmolality (∼40% of wild type) and intramedullary osmolality (20∼40% of wild type) were observed.57, 95 Both mouse models displayed no significant change in urinary salt excretion, plasma electrolyte concentrations, and mild hypotension. In conclusion, NaCl and urea accumulation likely contribute to the hyperosmolality of the inner medulla. Both ClC-Ka inhibitors and non-selective UT inhibitors could be effective aquaretic agents.
Urea transporter inhibitors
High throughput screening assays were developed in the Verkman laboratory using UT-expressing epithelial cells for urea transport assays or erythrocytes with native UT-B for erythrocyte lysis assays. Based on these methods, the Verkman group discovered most of the small-molecule UT inhibitors. Compared to the traditional UT inhibitors, phloretin and dimethylthiourea, these novel selective UT inhibitors showed potent efficacies with IC50s in the low nanomolar range. The discovery of these agents has been elegantly reviewed.96 Here, we focus on four UT inhibitors that have been tested in animals. UTBinh-14, a selective UT-B inhibitor, was a modified derivative of triazolothienopyrimidine selected from a high throughput screen of 100,000 compounds against human UT-B.97 UTBinh-14 binds to the cytoplasmic pore region of UT-B with IC50 ∼10 nM. The water diuretic effect of acute UTBinh-14 intraperitoneal injection was mild (∼80% increase in urine output, 20% reduction in maximal urine osmolality after dDAVP injection). PU-14, another small-molecule thienoquinolin, showed a similar aquaretic effect by inhibiting more UT-B than UT-A.98 This result is not surprising since the relative contribution of UT-B to urine concentration is much less than that of UT-A1. An aryl-thiazole UT-A1 inhibitor, UTAinh-E02, was identified from another high throughput screen of 100,000 small-molecule compounds against rat UT-A1.99 The IC50 of UTAinh-E02 toward UT-A1 and UT-B were 1 μM and 50 μM, respectively. Intravenous injection of 20 mg/kg UTAinh-E02 induced diuretic effects (∼3-fold increase in urine output) and reduced the urine osmolality by 50% within 3 hours in rats. After 3 hours, the urine concentrating ability was recovered, indicating the reversible effect of UTAinh-E02 on UT-A. Compared to the ubiquitous expression of UT-B, the renal-limited expression and dominant function of UT-A1 on renal urea reabsorption make UT-A1 inhibitor a better choice than UT-B inhibitors (Figure 5). However, a non-selective UT inhibitor could still be therapeutic since all-UT-knockout mice displayed severe urine concentrating defect and rare extra-renal phenotypes. Dimethylthiourea, an existing drug selected from urea analog screening, inhibited both UT-A1 and UT-B by noncompetitive binding on the intracellular side with IC50 of 2-3 mM.100 A single intraperitoneal bolus of 500 mg/kg dimethylthiourea increased the 24-hour urine output by 3-fold and reduced urine osmolality by 70%. The urinary urea and K+ clearance rates were also increased. With chronic dimethylthiourea treatment for 7 days, these effects were maintained, and rats developed mild hypokalemia without disturbing urinary salt excretion. The mechanism of hypokalemia is likely through the enhanced activity of flow-stimulated Maxi-K channel.
The differences between aquaretics and diuretics
Aquaretic is defined as an agent that predominantly results in water diuresis without a change in urinary electrolyte excretion. Accordingly, agents that interfere with the action of vasopressin or disrupt the formation of the corticomedullary osmotic gradient could be aquaretics. Currently, vaptans, the vasopressin receptor antagonists, are the only approved aquaretics and have been used in patients with water imbalance disorders, including euvolemic hyponatremia in the syndrome of inappropriate antidiuretic hormone secretion and hypervolemic hyponatremia in heart failure and liver cirrhosis. Of note, aquaretics should not be used in patients with hypovolemic hyponatremia due to the dangers of severe volume depletion and acute kidney injury. Other known agents with aquaretic effect, such as demeclocycline and lithium, have limited clinical use due to their known side effects. In contrast, diuretics enhance urinary salt wasting and cause a negative salt and water balance in extracellular fluid. Therefore, diuretics are the treatment of choice for hypertensive and hypervolemic disorders. Of note, the free water excretion rate is variable among diuretics. Drugs that disrupt the NaCl transport in the ascending limb of Henle's loop, such as furosemide and ClC-K channel inhibitors, could be mixed diuretic and aquaretic since active NaCl reabsorption in this region is critical to the corticomedullary osmotic gradient. Since vasopressin stimulates NCC and ENaC, people have also suggested that vaptan aquaretics may lead to a mild diuretic effect by increasing salt excretion. We compare the characters of diuretics and aquaretics in table 1.
Table 1. The comparisons between aquaretics and diuretics.
| Aquaretics | Diuretics | |
| Free water clearance rate | ++ | (except loop-like diuretics) |
| Saluresis | — | + ∼ ++ |
| Kaliuresis | — ∼ +/- | —∼ ++ |
| Urinary Ca2+excretion | — | ↑(loop-like diuretics) ↓(thiazide-like diuretics) |
| Urine osmolality | ↓↓ | ↓ ∼ ↑ |
| Blood pressure | — ∼ mild↓ | ↓ ∼ ↓↓ |
| GFR | — ∼ mild↓ | ↓ ∼ ↓↓ |
| ECF osmole balance | — ∼ mild↓(urea loss) | ↓↓(NaCl loss) |
| ECF osmolality | ↑ | Occasionally↓ |
| Plasma Na+, Cl- concentrations | ↑ | Occasionally ↓ (especially thiazides) but depend on salt and water balance. |
| Plasma K+ concentration | — ∼ mild ↓ | ↓(most diuretics) ↑(K+-sparing diuretics) |
| Plasma pH | — | ↑(except CAIs and MRAs) |
| Therapeutic resistance | — (not obvious) | Frequent (especially in chronic use) |
| Indications | Water imbalance | Hypertension, ECF volume expansion. |
CAI denotes carbonic anhydrase inhibitor; ECF: extracellular fluid; GFR:glomerular filtration rate; MRA: mineralocorticoid receptor antagonist.
Conclusion
Furosemide and thiazide have been and remain the mainstay treatment for disorders with expanded extracellular volume or edema for the past three decades. One major problem of clinical diuretic use is diuretic resistance. The compensatory upregulation of non-targeted Na+ transporters is an important mechanism underlying diuretic resistance. Given the normal physiological tubular response, this phenomenon is nearly inevitable with chronic diuretic use. Combination diuretic therapy is important. The phenotypes of several double knockout mice suggest the potential outcomes of combination diuretic therapy. Recent advances in the understandings of renal salt handling have laid strong foundations for future promising novel diuretics to achieve this goal (Table 2). Finally, agents that promote water diuresis with minimal saluretic effect, such as UT-A inhibitors, would be particularly beneficial for conditions associated with hyponatremia and refractory edema.
Table 2. Novel classes of diuretics and aquaretics.
| Class | Name | Screen method |
KD in vitro |
IC50 | Mechanism | Salt diuresis |
Kaliuresis | Water diuresis |
Blood pressure |
Side effects | Ref |
|---|---|---|---|---|---|---|---|---|---|---|---|
| WNKs inhibitors | WNK463 | HTS, WNK1 catalytic activity | 4 nM for WNK1/4 | <10 nM | ATP-competitive kinase inhibition | +++ | +++ | — | ↓↓ | Serious safety issues | 34 |
| WNK476 | HTS, WNK1 catalytic activity | 0.19 μM for WNK1 | 0.57 μM | ATP-Non-competitive kinase inhibition | ND | ND | ND | ND | ND | 35 | |
| WNK- SPAK disrupters | STOCK 1S-50699 | HTS, FCS | 32 μM for SPAK | 37μM | Binding to CCT domain of SPAK | ND | ND | ND | ND | ND | 38 |
| STOCK 2S-26016 | HTS, FCS | 20 μM for SPAK | 16μM | Binding to CCT domain of SPAK | ND | ND | ND | ND | ND | 38 | |
| SPAK inhibitors | STOCK 1S-14279 | HTS, SPAK catalytic activity | 7 μM for SPAK | 0.26 μM | ATP- Non-competitive kinase inhibition | ND | ND | ND | ND | Mice died after repeated injection | 40 |
| Closantel | Drug reposition | NS | 8.65μM for OSR | ATP- Non-competitive kinase inhibition | — | — | — | ↓ Short lasting | Possible vasodilation, ↓Heart rate | 40 | |
| Rafoxanide | In silico screen | NS | 8/13μM for OSR /SPAK | ATP- Non-competitive kinase inhibition | ND | ND | ND | ND | ND | 46 | |
| ClC-K inhibitors | MT-189 | Rational modification | 6 μM for ClC-Ka | NS | Inhibitory binding | — | — | + | ↓ | NS | 56 |
| RT-93 | Rational modification | 6 μM for ClC-Kb/a | NS | Inhibitory binding | — | — | + | ↓ | NS | 56 | |
| ROMK inhibitors | Compound A | HTS, chemical modification | NS | <100 nM | Inhibitory binding (cytoplasmic side) | + | — | — | NS | QT prolongation | 73 |
| MK-7145 | modification | NS | 6.6nM | Inhibitory binding | + | —/+ | — | ↓ | NS | 74 | |
| Pendrin inhibitors | PDSinh-C01 | HTS, | NS | 1.2μM | Inhibitory binding (intracellularly) | Not alone* | Not alone* | — | Not alone* | Metabolic alkalosis*, increase airway surface liquid volume | 68 |
| Urea transporter inhibitors | UTBinh-14 | HTS | NS | 10nM hUTB | Inhibitory binding (intracellularly) | — | — | +/- | — | NS | 97 |
| PU-14 | HTS | NS | 1.7μMhUTB | Inhibitory binding (intracellularly) | — | — | +/- | — | NS | 98 | |
| UTAinh-E02 | HTS | NS | 1μM rUTA1 | Inhibitory binding (intracellularly) | — | — | + | — | NS | 99 | |
| DMTU | Urea analog screening | NS | 2-3mM rUTA/B | Inhibitory binding (intracellularly) | — | + | ++ | — | NS | 100 |
HTS denotes high throughput screen; FCS: fluorescent correlation spectroscopy; ND: not done; NS: not shown;
+ when it combined furosemide
Acknowledgments
Grant: Supported by grants from MOST of Taiwan (MOST 103-2628-B-016-001-MY3 to CJC), from National Health Research Institutes of Taiwan (NHRI-EX106-10323SC to CJC), from Tri-Service General Hospital of Taiwan (TSGH-C106-94 to CJC), from National Institutes of Health of the United States (DK110358 to ARR, DK109887 and DK100605 to CLH), and from the American Heart Association (16CSA28530002 to ARR)”.NIH, DK110358 and AHA, 16CSA28530002.
References
- 1.Mann T, Keilin D. Sulphanilamide as a specific inhibitor of carbonic anhydrase. Nature. 1940;146:164–165. [Google Scholar]
- 2.Beyer KH, Jr, Baer JE, Russo HF, Noll R. Electrolyte excretion as influenced by chlorothiazide. Science. 1958;127:146–147. doi: 10.1126/science.127.3290.146. [DOI] [PubMed] [Google Scholar]
- 3.Stokes JB. Electroneutral NaCl transport in the distal tubule. Kidney Int. 1989;36:427–433. doi: 10.1038/ki.1989.212. [DOI] [PubMed] [Google Scholar]
- 4.Simon DB, et al. Gitelman's variant of Bartter's syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet. 1996;12:24–30. doi: 10.1038/ng0196-24. [DOI] [PubMed] [Google Scholar]
- 5.Hebert SC. Bartter syndrome. Curr Opin Nephrol Hypertens. 2003;12:527–532. doi: 10.1097/00041552-200309000-00008. [DOI] [PubMed] [Google Scholar]
- 6.Shimkets RA, et al. Liddle's syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell. 1994;79:407–414. doi: 10.1016/0092-8674(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 7.Chang SS, et al. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nat Genet. 1996;12:248–253. doi: 10.1038/ng0396-248. [DOI] [PubMed] [Google Scholar]
- 8.Wilson FH, et al. Human hypertension caused by mutations in WNK kinases. Science. 2001;293:1107–1112. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- 9.Dbouk HA, Huang CL, Cobb MH. Hypertension: the missing WNKs. Am J Physiol Renal Physiol. 2016;311:F16–27. doi: 10.1152/ajprenal.00358.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bockenhauer D, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med. 2009;360:1960–1970. doi: 10.1056/NEJMoa0810276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scholl UI, et al. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci U S A. 2009;106:5842–5847. doi: 10.1073/pnas.0901749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Everett LA, et al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS) Nat Genet. 1997;17:411–422. doi: 10.1038/ng1297-411. [DOI] [PubMed] [Google Scholar]
- 13.Min X, Lee BH, Cobb MH, Goldsmith EJ. Crystal structure of the kinase domain of WNK1, a kinase that causes a hereditary form of hypertension. Structure. 2004;12:1303–1311. doi: 10.1016/j.str.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 14.Xu B, English JM, Wilsbacher JL, Stippec S, Goldsmith EJ, Cobb MH. WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J Biol Chem. 2000;275:16795–16801. doi: 10.1074/jbc.275.22.16795. [DOI] [PubMed] [Google Scholar]
- 15.Veríssimo F, Jordan P. WNK kinases, a novel protein kinase subfamily in multi-cellular organisms. Oncogene. 2001;20:5562–5569. doi: 10.1038/sj.onc.1204726. [DOI] [PubMed] [Google Scholar]
- 16.O'Reilly M, et al. Dietary electrolyte-driven responses in the renal WNK kinase pathway in vivo. J Am Soc Nephrol. 2006;17:2402–2413. doi: 10.1681/ASN.2005111197. [DOI] [PubMed] [Google Scholar]
- 17.Oi K, et al. A minor role of WNK3 in regulating phosphorylation of renal NKCC2 and NCC co-transporters in vivo. Biol Open. 2012;1:120–127. doi: 10.1242/bio.2011048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang CL, Zhu X, Wang Z, Subramanya AR, Ellison DH. Mechanisms of WNK1 and WNK4 interaction in the regulation of thiazide-sensitive NaCl cotransport. J Clin Invest. 2005;115:1379–1387. doi: 10.1172/JCI22452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richardson C, et al. Activation of the thiazide-sensitive Na+-Cl- cotransporter by the WNK-regulated kinases SPAK and OSR1. J Cell Sci. 2008;121:675–684. doi: 10.1242/jcs.025312. [DOI] [PubMed] [Google Scholar]
- 20.Heise CJ, et al. Serum and glucocorticoid-induced kinase (SGK) 1 and the epithelial sodium channel are regulated by multiple with no lysine (WNK) family members. J Biol Chem. 2010;285:25161–25167. doi: 10.1074/jbc.M110.103432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng CJ, Huang CL. Activation of PI3-kinase stimulates endocytosis of ROMK via Akt1/SGK1-dependent phosphorylation of WNK1. J Am Soc Nephrol. 2011;22:460–471. doi: 10.1681/ASN.2010060681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boyden LM, et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. 2012;482:98–102. doi: 10.1038/nature10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Louis-Dit-Picard H, et al. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat Genet. 2012;44:456–460. doi: 10.1038/ng.2218. [DOI] [PubMed] [Google Scholar]
- 24.Shibata S, et al. Angiotensin II signaling via protein kinase C phosphorylates Kelch-like 3, preventing WNK4 degradation. Proc Natl Acad Sci U S A. 2014;111:15556–15561. doi: 10.1073/pnas.1418342111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Piala AT, Moon TM, Akella R, He H, Cobb MH, Goldsmith EJ. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal. 2014;7:ra41. doi: 10.1126/scisignal.2005050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bazúa-Valenti S, et al. The effect of WNK4 on the Na+-Cl- cotransporter is modulated by intracellular chloride. J Am Soc Nephrol. 2015;26:1781–1786. doi: 10.1681/ASN.2014050470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Terker AS, Zhang C, Erspamer KJ, Gamba G, Yang CL, Ellison DH. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int. 2016;89:127–134. doi: 10.1038/ki.2015.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Terker AS, et al. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab. 2015;21:39–50. doi: 10.1016/j.cmet.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Penton D, et al. Extracellular K+ rapidly controls NaCl cotransporter phosphorylation in the native distal convoluted tubule by Cl--dependent and independent mechanisms. J Physiol. 2016;594:6319–6331. doi: 10.1113/JP272504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subramanya AR, Yang CL, Zhu X, Ellison DH. Dominant-negative regulation of WNK1 by its kidney-specific kinase-defective isoform. Am J Physiol Renal Physiol. 2006;290:F619–624. doi: 10.1152/ajprenal.00280.2005. [DOI] [PubMed] [Google Scholar]
- 31.Hadchouel J, et al. Decreased ENaC expression compensates the increased NCC activity following inactivation of the kidney-specific isoform of WNK1 and prevents hypertension. Proc Natl Acad Sci U S A. 2010;107:18109–18114. doi: 10.1073/pnas.1006128107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Z, Xie J, Wu T, Truong T, Auchus RJ, Huang CL. Downregulation of NCC and NKCC2 cotransporters by kidney-specific WNK1 revealed by gene disruption and transgenic mouse models. Hum Mol Genet. 2011;20:855–866. doi: 10.1093/hmg/ddq525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lazrak A, Liu Z, Huang CL. Antagonistic regulation of ROMK by long and kidney-specific WNK1 isoforms. Proc Natl Acad Sci U S A. 2006;103:1615–1620. doi: 10.1073/pnas.0510609103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamada K, et al. Small-molecule WNK inhibition regulates cardiovascular and renal function. Nat Chem Biol. 2016;12:896–898. doi: 10.1038/nchembio.2168. [DOI] [PubMed] [Google Scholar]
- 35.Yamada K, et al. Discovery and characterization of allosteric WNK kinase inhibitors. ACS Chem Biol. 2016;11:3338–3346. doi: 10.1021/acschembio.6b00511. [DOI] [PubMed] [Google Scholar]
- 36.Xie J, Wu T, Xu K, Huang IK, Cleaver O, Huang CL. Endothelial-specific expression of WNK1 kinase is essential for angiogenesis and heart development in mice. Am J Pathol. 2009;175:1315–1327. doi: 10.2353/ajpath.2009.090094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang SS, et al. SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J Am Soc Nephrol. 2010;21:1868–1877. doi: 10.1681/ASN.2009121295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mori T, et al. Chemical library screening for WNK signaling inhibitors using fluorescence correlation spectroscopy. Biochem J. 2013;455:339–345. doi: 10.1042/BJ20130597. [DOI] [PubMed] [Google Scholar]
- 39.Zhang J, Siew K, Macartney T, O'Shaughnessy KM, Alessi DR. Critical role of the SPAK protein kinase CCT domain in controlling blood pressure. Hum Mol Genet. 2015;24:4545–4558. doi: 10.1093/hmg/ddv185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kikuchi E, et al. Discovery of novel SPAK inhibitors that block WNK kinase signaling to cation chloride transporters. J Am Soc Nephrol. 2015;26:1525–1536. doi: 10.1681/ASN.2014060560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng CJ, Yoon J, Baum M, Huang CL. STE20/SPS1-related proline/alanine-rich kinase (SPAK) is critical for sodium reabsorption in isolated, perfused thick ascending limb. Am J Physiol Renal Physiol. 2015;308:F437–443. doi: 10.1152/ajprenal.00493.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin SH, et al. Impaired phosphorylation of Na(+)-K(+)-2Cl(-) cotransporter by oxidative stress-responsive kinase-1 deficiency manifests hypotension and Bartter-like syndrome. Proc Natl Acad Sci U S A. 2011;108:17538–17543. doi: 10.1073/pnas.1107452108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ortiz PA. cAMP increases surface expression of NKCC2 in rat thick ascending limbs: role of VAMP. Am J Physiol Renal Physiol. 2006;290:F608–616. doi: 10.1152/ajprenal.00248.2005. [DOI] [PubMed] [Google Scholar]
- 44.Davies M, et al. Novel mechanisms of Na+ retention in obesity: phosphorylation of NKCC2 and regulation of SPAK/OSR1 by AMPK. Am J Physiol Renal Physiol. 2014;307:F96–F106. doi: 10.1152/ajprenal.00524.2013. [DOI] [PubMed] [Google Scholar]
- 45.Ponce-Coria J, et al. A novel Ste20-related proline/alanine-rich kinase (SPAK)-independent pathway involving calcium-binding protein 39 (Cab39) and serine threonine kinase with no lysine member 4 (WNK4) in the activation of Na-K-Cl cotransporters. J Biol Chem. 2014;289:17680–17688. doi: 10.1074/jbc.M113.540518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.AlAmri MA, Kadri H, Alderwick LJ, Simpkins NS, Mehellou Y. Rafoxanide and Closantel inhibit SPAK and OSR1 kinases by binding to a highly conserved allosteric site on their C-terminal domains. ChemMedChem. 2017 doi: 10.1002/cmdc.201700077. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 47.Zhang C, et al. KCNJ10 determines the expression of the apical Na-Cl cotransporter (NCC) in the early distal convoluted tubule (DCT1) Proc Natl Acad Sci U S A. 2014;111:11864–11869. doi: 10.1073/pnas.1411705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cuevas CA, et al. Potassium sensing by renal distal tubules requires Kir4.1. J Am Soc Nephrol. 2017 doi: 10.1681/ASN.2016090935.. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Su XT, Zhang C, Wang L, Gu R, Lin DH, Wang WH. Disruption of KCNJ10 (Kir4.1) stimulates the expression of ENaC in the collecting duct. Am J Physiol Renal Physiol. 2016;310:F985–993. doi: 10.1152/ajprenal.00584.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hennings JC, et al. The ClC-K2 chloride channel is critical for salt handling in the distal nephron. J Am Soc Nephrol. 2017;28:209–217. doi: 10.1681/ASN.2016010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grill A, Schieβl IM, Gess B, Fremter K, Hammer A, Castrop H. Salt-losing nephropathy in mice with a null mutation of the Clcnk2 gene. Acta Physiol (Oxf) 2016;218:198–211. doi: 10.1111/apha.12755. [DOI] [PubMed] [Google Scholar]
- 52.Cheng CJ, Lo YF, Chen JC, Huang CL, Lin SH. Functional severity of CLCNKB mutations correlates with phenotypes in patients with classic Bartter's syndrome. J Physiol. 2017 doi: 10.1113/JP274344. in press. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marvão P, et al. Cl- absorption across the thick ascending limb is not altered in cystic fibrosis mice. A role for a pseudo-CFTR Cl- channel. J Clin Invest. 1998;102:1986–1993. doi: 10.1172/JCI4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Palmer LG, Frindt G. Cl- channels of the distal nephron. Am J Physiol Renal Physiol. 2006;291:F1157–1168. doi: 10.1152/ajprenal.00496.2005. [DOI] [PubMed] [Google Scholar]
- 55.Wall SM, Weinstein AM. Cortical distal nephron Cl(-) transport in volume homeostasis and blood pressure regulation. Am J Physiol Renal Physiol. 2013;305:F427–438. doi: 10.1152/ajprenal.00022.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liantonio A, et al. In-vivo administration of CLC-K kidney chloride channels inhibitors increases water diuresis in rats: a new drug target for hypertension? J Hypertens. 2012;30:153–167. doi: 10.1097/HJH.0b013e32834d9eb9. [DOI] [PubMed] [Google Scholar]
- 57.Matsumura Y, et al. Overt nephrogenic diabetes insipidus in mice lacking the CLC-K1 chloride channel. Nat Genet. 1999;21:95–98. doi: 10.1038/5036. [DOI] [PubMed] [Google Scholar]
- 58.Schlingmann KP, et al. Salt wasting and deafness resulting from mutations in two chloride channels. N Engl J Med. 2004;350:1314–1319. doi: 10.1056/NEJMoa032843. [DOI] [PubMed] [Google Scholar]
- 59.Birkenhäger R, et al. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat Genet. 2001;29:310–314. doi: 10.1038/ng752. [DOI] [PubMed] [Google Scholar]
- 60.Royaux IE, et al. Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc Natl Acad Sci U S A. 2001;98:4221–4226. doi: 10.1073/pnas.071516798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pech V. Pendrin modulates ENaC function by changing luminal HCO3- J Am Soc Nephrol. 2010;21:1928–1941. doi: 10.1681/ASN.2009121257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Leviel F, et al. The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest. 2010;120:1627–1635. doi: 10.1172/JCI40145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wall SM, et al. NaCl restriction upregulates renal Slc26a4 through subcellular redistribution: role in Cl- conservation. Hypertension. 2004;44:982–987. doi: 10.1161/01.HYP.0000145863.96091.89. [DOI] [PubMed] [Google Scholar]
- 64.Vallet M, et al. Pendrin regulation in mouse kidney primarily is chloride-dependent. J Am Soc Nephrol. 2006;17:2153–2163. doi: 10.1681/ASN.2005101054. [DOI] [PubMed] [Google Scholar]
- 65.Grimm PR, et al. Integrated compensatory network is activated in the absence of NCC phosphorylation. J Clin Invest. 2015;125:2136–2150. doi: 10.1172/JCI78558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Soleimani M, et al. Double knockout of pendrin and Na-Cl cotransporter (NCC) causes severe salt wasting, volume depletion, and renal failure. Proc Natl Acad Sci U S A. 2012;109:13368–13373. doi: 10.1073/pnas.1202671109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pela I, Bigozzi M, Bianchi B. Profound hypokalemia and hypochloremic metabolic alkalosis during thiazide therapy in a child with Pendred syndrome. Clin Nephrol. 2008;69:450–453. doi: 10.5414/cnp69450. [DOI] [PubMed] [Google Scholar]
- 68.Cil O, Haggie PM, Phuan PW, Tan JA, Verkman AS. Small-molecule inhibitors of pendrin potentiate the diuretic action of furosemide. J Am Soc Nephrol. 2016;27:3706–3714. doi: 10.1681/ASN.2015121312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Haggie PM, Phuan PW, Tan JA, Zlock L, Finkbeiner WE, Verkman AS. Inhibitors of pendrin anion exchange identified in a small molecule screen increase airway surface liquid volume in cystic fibrosis. FASEB J. 2016;30:2187–2197. doi: 10.1096/fj.201600223R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ji W, et al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet. 2008;40:592–599. doi: 10.1038/ng.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fang L, Li D, Welling PA. Hypertension resistance polymorphisms in ROMK (Kir1.1) alter channel function by different mechanisms. Am J Physiol Renal Physiol. 2010;299:F1359–1364. doi: 10.1152/ajprenal.00257.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Clark MA, Humphrey SJ, Smith MP, Ludens JH. Unique natriuretic properties of the ATP-sensitive K(+)-channel blocker glyburide in conscious rats. J Pharmacol Exp Ther. 1993;265:933–937. [PubMed] [Google Scholar]
- 73.Garcia ML, et al. Pharmacologic inhibition of the renal outer medullary potassium channel causes diuresis and natriuresis in the absence of kaliuresis. J Pharmacol Exp Ther. 2013;348:153–164. doi: 10.1124/jpet.113.208603. [DOI] [PubMed] [Google Scholar]
- 74.Tang H, et al. Discovery of MK-7145, an oral small molecule ROMK inhibitor for the treatment of hypertension and heart failure. ACS Med Chem Lett. 2016;7:697–701. doi: 10.1021/acsmedchemlett.6b00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bailey MA, et al. Maxi-K channels contribute to urinary potassium excretion in the ROMK-deficient mouse model of Type II Bartter's syndrome and in adaptation to a high-K diet. Kidney Int. 2006;70:51–59. doi: 10.1038/sj.ki.5000388. [DOI] [PubMed] [Google Scholar]
- 76.Geller DS, et al. Mutations in the mineralocorticoid receptor gene cause autosomal dominant pseudohypoaldosteronism type I. Nat Genet. 1998;19:279–281. doi: 10.1038/966. [DOI] [PubMed] [Google Scholar]
- 77.Terker AS, et al. Direct and indirect mineralocorticoid effects determine distal salt transport. J Am Soc Nephrol. 2016;27:2436–2445. doi: 10.1681/ASN.2015070815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Canonica J, et al. Adult nephron-specific MR-deficient mice develop a severe renal PHA-1 phenotype. Pflugers Arch. 2016;468:895–908. doi: 10.1007/s00424-015-1785-2. [DOI] [PubMed] [Google Scholar]
- 79.Rossier BC, Baker ME, Studer RA. Epithelial sodium transport and its control by aldosterone: the story of our internal environment revisited. Physiol Rev. 2015;95:297–340. doi: 10.1152/physrev.00011.2014. [DOI] [PubMed] [Google Scholar]
- 80.Roy A, et al. Alternatively spliced proline-rich cassettes link WNK1 to aldosterone action. J Clin Invest. 2015;125:3433–3448. doi: 10.1172/JCI75245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Larik FA, et al. Synthetic approaches towards the multi-target drug spironolactone and its potent analogues/derivatives. Steroids. 2017 doi: 10.1016/j.steroids.2016.12.010. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 82.Gomez-Sanchez E, Gomez-Sanchez CE. The multifaceted mineralocorticoid receptor. Compr Physiol. 2014;4:965–994. doi: 10.1002/cphy.c130044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kolkhof P, Nowack C, Eitner F. Nonsteroidal antagonists of the mineralocorticoid receptor. Curr Opin Nephrol Hypertens. 2015;24:417–424. doi: 10.1097/MNH.0000000000000147. [DOI] [PubMed] [Google Scholar]
- 84.Bakris GL, et al. Effect of finerenone on albuminuria in patients with diabetic nephropathy: a randomized clinical trial. JAMA. 2015;314:884–894. doi: 10.1001/jama.2015.10081. [DOI] [PubMed] [Google Scholar]
- 85.Filippatos G, et al. A randomized controlled study of finerenone vs. eplerenone in patients with worsening chronic heart failure and diabetes mellitus and/or chronic kidney disease. Eur Heart J. 2016;37:2105–2114. doi: 10.1093/eurheartj/ehw132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sands JM, Layton HE. Advances in understanding the urine-concentrating mechanism. Annu Rev Physiol. 2014;76:387–409. doi: 10.1146/annurev-physiol-021113-170350. [DOI] [PubMed] [Google Scholar]
- 87.Knepper MA, Miranda CA. Urea channel inhibitors: a new functional class of aquaretics. Kidney Int. 2013;83:991–993. doi: 10.1038/ki.2013.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ren H, et al. Spatial organization of the vascular bundle and the interbundle region: three-dimensional reconstruction at the inner stripe of the outer medulla in the mouse kidney. Am J Physiol Renal Physiol. 2014;306:F321–326. doi: 10.1152/ajprenal.00429.2013. [DOI] [PubMed] [Google Scholar]
- 89.Lei T, et al. Role of thin descending limb urea transport in renal urea handling and the urea concentrating mechanism. Am J Physiol Renal Physiol. 2011;301:F1251–1259. doi: 10.1152/ajprenal.00404.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sands JM, Gargus JJ, Frohlich O, Gunn RB, Kokko JP. Urinary concentrating ability in patients with Jk(a-b-) blood type who lack carrier-mediated urea transport. J Am Soc Nephrol. 1992;2:1689–1696. doi: 10.1681/ASN.V2121689. [DOI] [PubMed] [Google Scholar]
- 91.Ranade K, et al. Genetic variation in the human urea transporter-2 is associated with variation in blood pressure. Hum Mol Genet. 2001;10:2157–2164. doi: 10.1093/hmg/10.19.2157. [DOI] [PubMed] [Google Scholar]
- 92.Yang B, Bankir L, Gillespie A, Epstein CJ, Verkman AS. Urea-selective concentrating defect in transgenic mice lacking urea transporter UT-B. J Biol Chem. 2002;277:10633–10637. doi: 10.1074/jbc.M200207200. [DOI] [PubMed] [Google Scholar]
- 93.Fenton RA, Chou CL, Stewart GS, Smith CP, Knepper MA. Urinary concentrating defect in mice with selective deletion of phloretin-sensitive urea transporters in the renal collecting duct. Proc Natl Acad Sci USA. 2004;101:7469–7474. doi: 10.1073/pnas.0401704101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Uchida S, Sohara E, Rai T, Ikawa M, Okabe M, Sasaki S. Impaired urea accumulation in the inner medulla of mice lacking the urea transporter UT-A2. Mol Cell Biol. 2005;25:7357–7363. doi: 10.1128/MCB.25.16.7357-7363.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jiang T, et al. Generation and phenotypic analysis of mice lacking all urea transporters. Kidney Int. 2017;91:338–351. doi: 10.1016/j.kint.2016.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Esteva-Font C, Anderson MO, Verkman AS. Urea transporter proteins as targets for small-molecule diuretics. Nat Rev Nephrol. 2015;11:113–123. doi: 10.1038/nrneph.2014.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yao C, Anderson MO, Zhang J, Yang B, Phuan PW, Verkman AS. Triazolothienopyrimidine inhibitors of urea transporter UT-B reduce urine concentration. J Am Soc Nephrol. 2012;23:1210–1220. doi: 10.1681/ASN.2011070751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li F, et al. A novel small-molecule thienoquinolin urea transporter inhibitor acts as a potential diuretic. Kidney Int. 2013;83:1076–1086. doi: 10.1038/ki.2013.62. [DOI] [PubMed] [Google Scholar]
- 99.Esteva-Font C, et al. Diuresis and reduced urinary osmolality in rats produced by small-molecule UT-A-selective urea transport inhibitors. FASEB J. 2014;28:3878–3890. doi: 10.1096/fj.14-253872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cil O, et al. Salt-sparing diuretic action of a water-soluble urea analog inhibitor of urea transporters UT-A and UT-B in rats. Kidney Int. 2015;88:311–320. doi: 10.1038/ki.2015.138. [DOI] [PMC free article] [PubMed] [Google Scholar]