


Abstract
Filamentous bacteriophages are particularly efficient for the expression and display of combinatorial random peptides. Two phage proteins are often employed for peptide display: the infectivity protein, PIII, and the major coat protein, PVIII. The use of PVIII typically requires the expression of two pVIII genes: the wild-type and the recombinant pVIII gene, to generate mosaic phages. ‘Type 88’ vectors contain two pVIII genes in one phage genome. In this study a novel ‘type 88’ expression vector has been rationally designed and constructed. Two factors were taken into account: the insertion site and the genetic stability of the second pVIII gene. It was found that selective deletion of recombinant genes was encountered when inserts were cloned into either of the two non-coding regions of the phage genome. The deletions were independent of recA yet required a functional F-episome. Transcription was also found to be a positive factor for deletion. Taking the above into account led to the generation of a novel vector, designated fth1, which can be used to express recombinant peptides as pVIII chimeric proteins in mosaic bacteriophages. The fth1 vector is not only genetically stable but also of high copy number and produces high titers of recombinant phages.
INTRODUCTION
Combinatorial phage display peptide libraries provide an effective means to study protein–protein interactions. This technology relies on the production of very large collections of random peptides associated with their corresponding genetic blueprints (1–6). Presentation of the random peptides is accomplished by constructing chimeric proteins expressed on the outer surface of filamentous bacteriophages (M13, fd and f1) making them amenable to specialized screening assays [referred to as biopanning (7)]. Affinity isolation and identification of peptides that bind to receptors (8–11), enzymes (12,13) or antibodies (1,14–18) have been accomplished using such combinatorial libraries.
Filamentous bacteriophages are non-lytic, male-specific bacteriophages which infect Escherichia coli cells carrying an F-episome (for review see 19). Filamentous phage particles contain a circular ssDNA genome (the + strand). The phage binds to the F-pilus of the bacterium followed by entry of the ssDNA genome into the host. The dsDNA replicating form is generated by initiating DNA synthesis at the phage ori(–) structure. Single-strand (+) DNAs are formed by additional rounds of synthesis, starting at the ori(+) structure, and are packaged into phage particles without causing lysis or apparent damage to the host.
Two structural proteins of the phage, PIII and PVIII proteins, have been used for peptide display. The PIII protein, which exists in five copies per phage (20,21), can tolerate extensive modifications without markedly affecting PIII function (22–26).
The PVIII protein is the major coat protein of the phage present in about 2700 copies per phage (27). PVIII is synthesized as a precoat protein containing a 23 amino acid leader peptide, which is cleaved to yield a mature 50 residue transmembrane protein (28). Use of PVIII as a display scaffold is limited to peptides no longer than six residues, as larger inserts interfere with phage assembly (29–30). Introduction of larger peptides, however, is possible in systems where mosaic phages are produced by in vivo mixing of the recombinant, peptide-containing, PVIII proteins with wild-type PVIII (15,29,31). This enables the incorporation of the chimeric PVIII proteins at low density (tens to hundreds of copies per particle) on the phage surface, interspersed with wild-type coat proteins, during the assembly of phage particles. Two systems have been used that enable the generation of mosaic phages; the ‘type 8+8’ and ‘type 88’ systems as designated by Smith (32).
The ‘type 8+8’ system has two pVIII genes existing in two different genetic units (15,29,31). The recombinant pVIII gene is located on a phagemid. The wild-type PVIII protein is supplied by superinfecting phagemid-harboring bacteria with a helper phage. Therefore, two types of particles are secreted by such bacteria, helper and phagemid, both of which incorporate a mixture of recombinant and wild-type PVIII proteins.
The ‘type 88’ system benefits by containing the two pVIII genes in one and the same infectious phage genome. This obviates the need for a helper phage and superinfection. Furthermore, only one type of mosaic phage is produced. The question arises, however, where should one introduce the second pVIII gene within the filamentous phage genome for efficient expression and genetic stability?
The 10 proteins encoded by the phage (PI–PX) are all essential for production of infectious progeny (19). The genes for the proteins are organized in two tightly packed transcriptional units separated by two non-coding regions (33). One non-coding region, the ‘intergenic region’ (situated between the pIV and pII genes) contains the (+) and (–) origins of DNA replication and the packaging signal of the phage. This region can tolerate deletions or the insertion of foreign DNAs at several sites (34–38). The second non-coding region of the phage is located between the pVIII and pIII genes, and has been used to incorporate foreign recombinant genes as was illustrated by Krebber et al. (39).
A major point for concern for type 88 vectors is the genetic stability of the ultimate vector and its derivatives.
In this study, we have critically examined the attributes of the two non-coding regions of the fd filamentous phage as potential sites for insertion of a second recombinant pVIII gene and its genetic stability. In doing so we have designed and constructed an efficient type 88 phage display expression system.
MATERIALS AND METHODS
Bacterial strains, phages, reagents and general techniques
Escherichia coli strains used in this study are given in Table 1. The wild-type fd filamentous phage and fd-tet vector were kindly provided by G.P.Smith (University of Missouri, Columbia, MO). The M13K07 phage was purchased from New England Biolabs Inc. (NEB). DNA was isolated using the alkaline lysis procedure and purified on a cesium chloride gradient as described previously (40). All the restriction enzymes used were purchased from NEB and the digestions were performed following the manufacturer’s instructions. The monoclonal antibody GV4H3 was produced from a Balb/c mouse immunized with the HIV-1 envelope protein gp120 (41). The oligonucleotides used in this study are given in Table 2.
Table 1. Bacterial strains.
| Strain |
Genotype |
| K802 | F– e14–(McrA–) lacY1 or Δ(lac)6 supE44 galK2 galT22 rfbD1 metB1 mcrB1 hsdS3(rK– mK–) |
| K91KAN | A derivative of K91 (Hfr-Cavalli, thi) in which the ‘mini-Kan hopper’ element was inserted in the lacZ gene of K91 rendering this strain kanamycin resistant (7) |
| DH5α | F– endA1 hsdR17(rK– mK+) supE44 thi-1 recA1 gyrA(Nalr) relA1 Δ(lacIZYA-argF)U169 deoR (φ80dlacΔ(lacZ)M15) |
| DH5αF′ | F′ endA1 hsdR17(rK– mK+) supE44 thi-1 recA1 gyrA(Nalr) relA1 Δ(lacIZYA-argF)U169 deoR (φ80dlacΔ(lacZ)M15) |
| JM109 | F′[traD36 lacIq Δ(lacZ)M15 proA+B+] e14–(McrA–) Δ(lac-proAB) thi gyrA96(Nalr) endA1 hsdR17(rK– mK+) relA1 supE44 recA1 |
| XL1-Blue | F′[::Tn10 proA+B+ lacIq Δ(lacZ)M15] recA1 endA1 gyrA96(Nalr) thi hsdR17(rK– mK+) supE44 relA1 lac |
| MC1061 | F– araD139 Δ(ara-leu)7696 galE15 galK16 Δ(lac)X74 rpsL(Strr) hsdR2(rK– mK+) mcrA mcrB1 |
| NM554 | F– araD139 Δ(ara-leu)7696 galE15 galK16 Δ(lac)X74 rpsL(Strr) hsdR2(rK– mK+) mcrA mcrB1 recA13 |
Table 2. Oligonucleotides used.
| Oligonucleotide |
Sequence |
| ON1 | 5′-GCCTTCGTAGTGGCATTACG-3′ |
| ON2 | 5′-AGCCCGCTCATTAGGCGGGCTTCATTACCGGCCACGTCGGCCACCCTCAGCAGCGAAAGAC-3′ |
| ON3 | 5′-CGGTAATGAAGCCCGCCTAATGAGCGGGCTTTTTTTTGGCCTCTGGGGCCGATCCCGCAAAAGCGGCC-3′ |
| ON4 | 5′-GGTCAGACGATTGGCCTTG-3′ |
| ON5 | 5′-GAGCTCGCTAGCGCTCGAGCCAAAAAAAAAGGCTCCAAAAGG-3′ |
| ON6 | 5′-CTAGAGCAGGGTCCAGCTAA-3′ |
| ON7 | 5′-CTGGACCCTGCT-3′ |
| ON8 | 5′-TCGAGCTGTTGACAATTAATCATCGGCTCGTATAATGTGTGGAATTGTGAGCGGATAACAATTGAGCT-3′ |
| ON9 | 5′-CAATTGTTATCCGCTCACAATTCCACACATTATACGAGCCGATGATTAATTGTCAACAGC-3′ |
| ON10 | 5′-CCGTGCATCTGTCCTCGTTC-3′ |
| ON11 | 5′-CAGGCTTAAGCATCGACGTCTTATCAAGACGCCTTGCTTGTAAACTTTTTGAATAACTTGATACCGATAGTTGCGC-3′ |
| ON12 | 5′-GTCTTGATAAGACGTCGATGCTTAAGCCTGGCTAGCCATCAGATCTGAGTCGGCCGCTGTTTAAGAAATTCACCTCG-3′ |
| ON13 | 5′-GCCGTACCGCTAGCATTAGAAAAACTCATCGAGC-3′ |
| ON14 | 5′-GCTTCCTGACAGGAGGCCGTTTTGTTTTGCAGCCCACCTGAGCTCCCAGCTTAAGGTGTCTCAAAATCTCTGATG-3′ |
| ON15 | 5′-GGCTGAGGGACGTCGAGGGCATGCGTACCCGATAAAAGCGGCTTCCTGACAGGAGGCCG-3′ |
| ON16 | 5′-AGCCTTTCAGGTCAGAAGGG-3′ |
| ON17 | 5′-ATTCATGCCGGAGAGGGTAG-3′ |
| ON18 | 5′-CATAACACCCCTTGTATTACTG-3′ |
| ON19 | 5′-GCTTACATAAACAGTAATACAAGGGGTGTTATGAATAGTTCGACAAAGATCGC-3′ |
| ON20 | 5′-GGATCGAGAGCTAGCATAACTAAGCACTTGTCTCCTG-3′ |
| ON21 | 5′-CGGTCAGTGCAGATCTGCACTGCTCGAGCTGTTGACAATTAATC-3′ |
| ON22 | 5′-GCTCGAGCTGTTGACAATTAATCATCGGCTCGTATAATGTGTGGAATTGTGAGCGGATAACAATTTC-3′ |
| ON23 | 5′-GCTCGAGCTGTTGACAATTAATCATCGGCTCGTATAATGTGTGGAATTGTGAGCGGATAACAATTTC-3′ |
| ON24 | 5′-GCTTAACATTGGGACCAATGTTGCGACCGCCACACTCGCCTTCAAAACAAGGCTTTTCTTCAT-3′ |
| ON25 | 5′-CTTCATTACCGGCCACGTTGGCCGCCTTCCGCCGCAAAGCTTAACATTGGGACCAATGTTG-3′ |
| ON26 | 5′-GGAAGGCGGCCAACGTGGCCGGTAATGAAGCCCGCCTAATGAGCGGGCTTTTTTTTGGCCTCTGGGGC-3′ |
| ON27 | 5′-CTTTTTTTTGGCCTCTGGGGCCGACCCTGCGAAGGCAGCATTCGATAGTTTACAGGCAAGCGC-3′ |
| ON28 | 5′-ACGACGACCATTGCCCAAGCGTAGCCTATGTACTCAGTTGCGCTTGCCTGTAAACTATCG-3′ |
| ON29 | 5′-AACAGTTTTATGCCTATGGTCGCTCCTACGATAACGACGACCATTGCCCAAGCG-3′ |
| ON30 | 5′-CGCTATCAACTGGCTTTAGACGTAAACTTCTTGAACAGTTTTATGCCTATGGTCGC-3′ |
| ON31 | 5′-CGGTAGGTGACGGCCGCTATCAACTGGCTTTAGACG-3′ |
| ON32 | 5′-TGCCGGCGCTCCCGCTGGTTTCGCTATCCTGTCAGCACCCGGGTCTG-3′ |
| ON33 | 5′-ACCCGGGTGCTGACAGGATAGCGAAACCAGCGGGAGCGCCGGCACGT-3′ |
Vector construction
In this study, two novel vector systems were constructed. The rationale for their compositions and structures is described in the Results. Detailed diagrams of the vectors and sub-structures are given in the Figures as indicated. Following are the details of the specific steps taken to construct the vectors.
Construction of ftac88
The ftac88 vector (for a detailed map see Fig. 4) was constructed via the following steps. First, the recombinant pVIII gene, designated pVIIISTS (for orientation see Fig. 3A), was generated by introducing a 62 bp insert three codons downstream to the leader peptide using the ‘SOEing’ PCR mutagenesis (42). For this, four oligonucleotides were used. ON1 and ON2 were used to PCR amplify a 169 bp fragment from fd. ON3 and ON4 were used to PCR generate a 899 bp fragment from fd. ON2 and ON3 each contain 5′ extensions corresponding to the 62 bp insert. Thus, the resulting PCR fragments contain an identical 30 bp stretch of the novel sequence. The two fragments were purified from agarose gel, mixed and used as a combined template for PCR amplification using the oligonucleotides ON1 and ON4 to generate the final 1071 bp product containing the modified pVIII gene, part of pIII gene and the flanking BamHI–SnaBI sites. The PCR reaction was performed by using the Taq polymerase, and the resulting 1071 bp fragment was directly ligated with the linearized pGEM-T vector (Promega Corporation, WI), generating the pGEM-T(p8STS) construct (Fig. 3B).
Figure 4.
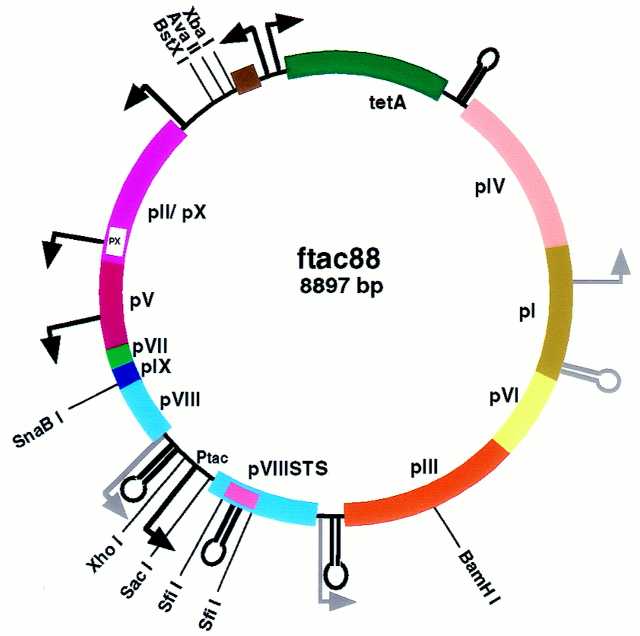
Genome organization in the ftac88 vector. Most of the tetR gene was exchanged for a linker containing the AvaII site, thus eliminating the second SnaBI site previously situated in this region (for comparison see Fig. 1C). The modified pVIIISTS gene is situated between the duplicated overlapping promoter/terminator structures, without disrupting the two phage transcriptional units. Note the pVIIISTS gene is under the control of a tac promoter (Ptac).
Figure 3.
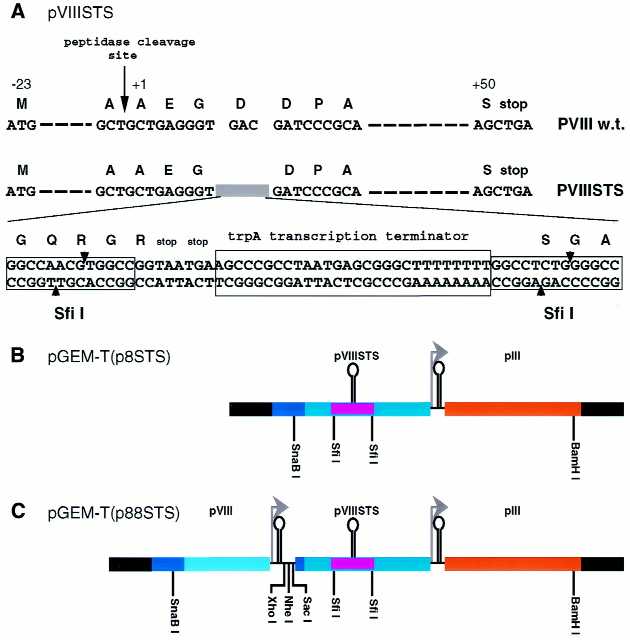
Construction of two tandem pVIII genes. (A) Detailed comparison between the wild-type pVIII gene and the recombinant pVIII gene, designated pVIIISTS. In the pVIIISTS gene, the GAC codon for Asp at position 4 of the wild-type pVIII is deleted and replaced by a sequence of 62 bp containing two stop codons and trpA transcription terminator flanked by two SfiI sites (STS insert). (B) pGEM-T(p8STS) construct. The fragment containing the pVIIISTS gene, preceded by a SnaBI restriction site and followed by downstream pIII gene, was introduced into the pGEM-T vector. Black segments are sequences corresponding to the pGEM-T vector. The 62 bp STS insert is indicated by the violet small segment. (C) pGEM-T(p88STS) construct. This construct contains the wild-type pVIII and the modified pVIIISTS genes arranged as tandem repeats. The fragment containing the wild-type pVIII gene starting from the SnaBI site and ending just beyond the overlapping promoter/terminator was introduced into the SnaBI site of pGEM-T(p8STS), thus destroying the former site while concomitantly introducing a new SnaBI site upstream to the wild-type pVIII gene. Furthermore, additional unique sites were introduced between the two pVIII genes.
Next, the wild-type pVIII gene was PCR amplified from fd using the oligonucleotides ON1 and ON5. ON5 has a 5′ extension that contains the restriction sites XhoI, NheI and SacI. The PCR reaction was performed using the Pwo Polymerase (Boehringer Mannheim), resulting in a blunt-ended 335 bp fragment from the SnaBI site to just beyond the overlapping promoter/terminator followed by the restriction sites introduced by ON5. The resulting fragment was ligated with the blunt-ended SnaBI-linearized pGEM-T(p8STS) construct, generating the pGEM-T(p88STS) construct (Fig. 3C).
To remove the SnaBI site in the tetR gene, the fd-tet vector (for detailed maps see Fig. 1) was digested with XbaI and BstXI restriction enzymes, and the 8458 bp fragment was purified from agarose gel. The purified fragment was ligated with a linker containing the AvaII site produced by annealing the two complementary oligonucleotides ON6 and ON7.
Figure 1.
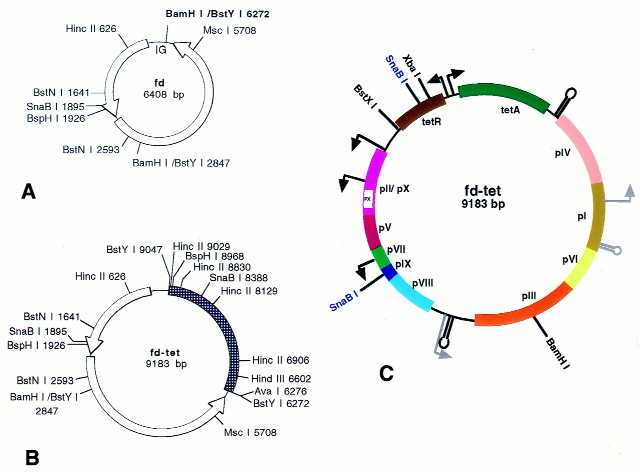
Comparison of fd and fd-tet. Opened arrows indicate the phage-transcriptional units (A and B). The hatched segment indicates the tet fragment introduced into the BamHI site of fd at position 6272 [bold in (A)] in the intergenic region (IG). In (C) the genome organisation is shown: genes pI–pX (colored boxes), promoters (bent arrows), terminators (stem and loop. Strong promoters and terminators are in black. Weak promoters and terminators are in gray. Selected restriction sites are shown (unique sites are in black and non-unique are in blue).Note the bi-directional promoter driving the transcription of genes for tetR (tet repressor protein) and tetA (tetracycline resistance protein).
To exchange the SnaBI–BamHI 952 bp segment of fd-tet for the SnaBI–BamHI 1378 bp segment of the pGEM-T(p88STS) construct containing both the wild-type and recombinant pVIII genes, the AvaII-containing fd-tet derivative was digested with SnaBI/BamHI, and the 7519 bp fragment was purified from agarose gel. In the same manner, the pGEM-T(p88STS) construct was digested with SnaBI/BamHI, and the released 1378 bp fragment was purified from agarose gel. These two fragments were ligated to generate an intermediate vector.
This intermediate vector was digested with SacI and XhoI restriction enzymes, and the 13 bp stuffer was removed by applying the DNA digest on a chroma-spin™ column (Clontech Laboratories, Inc.). The linearized intermediate vector was ligated with a linker containing the tac promoter produced by annealing the two complementary oligonucleotides ON8 and ON9, generating the ftac88 vector.
Construction of fth1
The construction of the fth1 vector (for a detailed map see Fig. 6) was performed through a series of intermediate vectors. The intermediate vector 1 (IV-1; Fig. 7B) was constructed by introducing a DNA segment 10 codons upstream to the stop codon of the wild-type pVIII gene using the ‘SOEing’ PCR mutagenesis described above. Two fragments of 519 and 827 bp were PCR generated from fd using the oligonucleotides ON10/ON11 and ON12/ON4, respectively. The two fragments were mixed and used as a combined template using ON10/ON4 to PCR generate the final 1316 bp fragment. This product was digested with SnaBI and BamHI and the resulting 1034 bp fragment was purified from agarose gel and used to replace the corresponding SnaBI–BamHI fragment of the AvaII-containing fd-tet derivative described above for the construction of ftac88.
Figure 6.
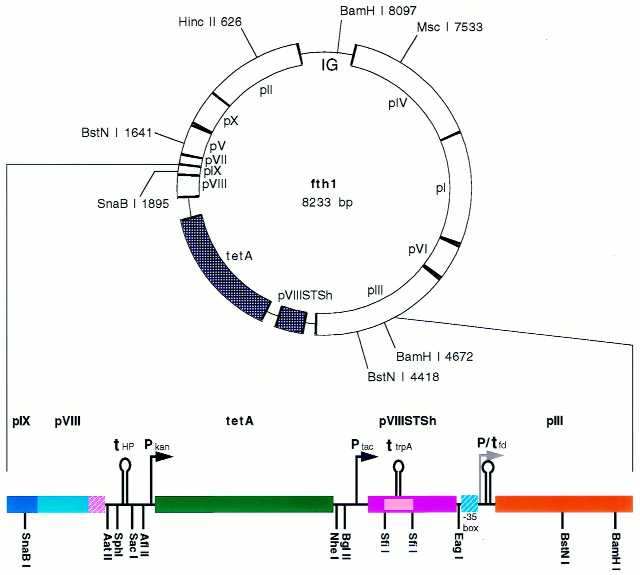
The fth1 vector. The general scheme of the fth1 vector is shown to scale illustrating the phage genes (opened segments), the reconstituted intergenic region (IG) and the genes introduced between the wild-type pVIII and pIII genes (stippled segments). Below, detail of the pIX to the middle of pIII is provided (not to scale). The hatched segments represent the modified wild-type pVIII 3′ end (violet) and the –35 box of the pIII promoter (cyan) (see also Fig. 7). For details see text.
Figure 7.
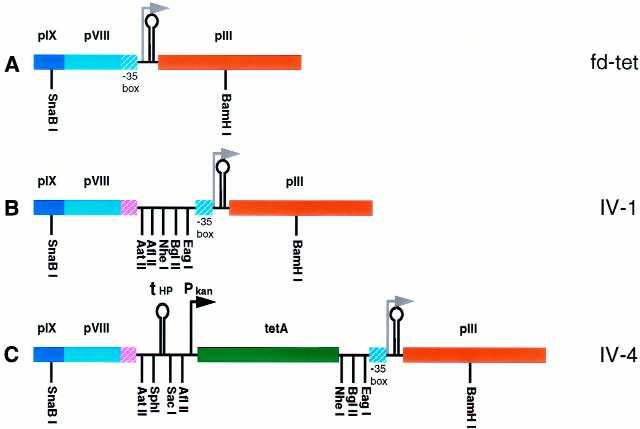
Intermediate vectors constructed for the formation of fth1 vector. (A) Detail of the segment of fd-tet containing the pIX, pVIII and pIII genes. The hatched cyan segment in pVIII indicates the –35 box of the pIII promoter situated in the 3′ region of the wild-type pVIII open reading frame. (B) IV-1. The –35 box of the pIII promoter was separated from the wild-type pVIII gene by inserting a small segment reconstituting the 3′ region (hatched violet) and introducing multiple cloning sites. (C) IV-4. A novel HP terminator (tHP) is situated downstream to the wild-type pVIII gene terminating its transcription followed by the tetracycline resistance gene (tetA) under the control of the kan promoter (Pkan).
Next, the IV-2 was constructed by introducing into the IV-1, downstream to the wild-type pVIII gene, the novel HP transcription terminator followed by the kanamycin resistance gene. This was performed in order to (i) functionally separate the two phage-transcriptional units and (ii) to enable the reconstitution of the intergenic region while maintaining an alternative antibiotic selectable marker (i.e. kanamycin). The kanamycin resistance gene was PCR amplified from M13K07 using the oligonucleotides ON13 and ON14. Both oligonucleotides contain 5′ extensions enabling the incorporation of additional flanking sequences. The 3′ addition contained the NheI restriction site and the 5′ extension contained additional cloning sites adjacent to the kanamycin promoter and part of the HP terminator. The resulting 944 bp fragment was purified from agarose gel and used as a template for PCR amplification using the oligonucleotides ON13 and ON15. The latter has a 5′ extension containing the second part of the HP terminator and additional cloning sites ending with the AatII site. Thus, the resulting 984 bp fragment contains the entire HP terminator bracketed between multiple cloning sites followed by the kanamycin resistance gene. This product was digested with NheI and AatII and the resulting fragment was purified from agarose gel. The purified fragment was ligated with NheI/AatII-digested IV-1 vector after removing the 20 bp stuffer by applying the vector digest on a chroma-spin™ column, ultimately generating IV-2.
The IV-2 was used to reconstitute the intergenic region containing the origins of replication. This was performed by PCR amplifying the intergenic region from fd phage using the oligonucleotides ON16 and ON17. The resulting 1081 bp fragment containing the MscI–DrdI segment was digested with MscI and DrdI restriction enzymes. The 679 bp MscI–DrdI product was purified from agarose gel and used to exchange the MscI–DrdI fragment in IV-2 containing the tet fragment, generating IV-3.
The open reading frame only of the kanamycin resistance gene was replaced by the open reading frame of the tetracycline resistance gene, thus constructing the IV-4 vector (Fig. 7C). This was performed using the ‘SOEing’ PCR. The oligonucleotides ON1 and ON18 were used to PCR amplify from IV-2 the 463 bp fragment containing the upstream sequence to the ATG initiating codon of the kanamycin gene. The oligonucleotides ON19 and ON20 were used to PCR amplify from fd-tet the 1265 bp fragment containing the open reading frame of the tetracycline resistance gene. The two fragments were mixed and used for PCR amplification of the 1698 bp fragment using the oligonucleotides ON1 and ON20. The PCR was designed to flank the resulting 1698 bp fragment between NheI and SacI restriction sites enabling the digestion of this fragment with the corresponding enzymes. After purifying the digested fragment from agarose gel, it was used to replace the corresponding NheI–SacI fragment of IV-3.
The final step was to generate the synthetic recombinant pVIII gene using a panel of overlapping oligonucleotides (ON21–ON31). Part of the panel was used as templates and the other part as 5′-extended primers for PCR amplification, thus generating separate small pieces of DNA. These small fragments were sequentially mixed and used as a combined template for the ‘SOEing’ PCR, ultimately generating the recombinant pVIIISTSh gene that subsequently was introduced between the BglII/EagI sites of IV-4 producing the fth1 vector.
Introduction of the GV4H3 epitope into ftac88 and fth1 vectors
The vectors ftac88 and fth1 were digested with SfiI restriction enzyme, and the 49 bp stuffer containing the trpA terminator (for orientation see Fig. 3A) was removed by applying the DNA digest on a chroma-spin™ column. The purified linear vector was ligated with GV4H3 epitope encoding linker produced by annealing the two complementary oligonucleotides ON32 and ON33.
Dot blot analysis of GV4H3 epitope presenting phages
The phages were applied via a vacuum manifold to a nitrocellulose membrane filter. After blocking [5% evaporated spray dried skim milk, 1.5% fat in Tris-buffered saline (TBS)] for 1 h, the membrane was washed briefly with TBS and incubated overnight with 1 µg/ml of GV4H3 mAb in TBS/5% milk at 4°C with gentle rocking. After washing, the membrane was incubated with goat anti-mouse IgG/HRP conjugate diluted 1:5000 in TBS/5% milk for 1 h at room temperature. The positive signals were detected by ECL (Amersham International plc) immunodetection.
RESULTS
The fd-tet vector of Smith and co-workers (38) was used as a starting point for our studies.
The fd-tet expression vector
Zacher et al. (38) constructed the fd-tet phage, which contains the tetracycline resistance gene as a selectable marker (Fig. 1B). The tetracycline resistance fragment was obtained from the transposon Tn10 (a 2775 bp fragment flanked by two BglII sites), which was introduced into the BamHI site of the intergenic region containing the origins of replication of the fd genome. The fd-tet vector has the following advantages: (i) fd-tet phages confer selectable tetracycline resistance to infected or transfected host bacteria; (ii) the tet fragment introduces unique restriction sites that can be exploited for cloning foreign DNA sequences.
Thus, fd-tet was used, for example, as a cloning vector by exploiting the unique HindIII site, situated in the tet fragment, to clone HindIII-digested phage λ DNA (38). Furthermore, Zhong et al. (43) have reported the introduction of a second pVIII gene in the same region generating a type 88 vector designated f88.4, and the successful production of pVIII mosaic phages.
Foreign DNA, however, cloned into the intergenic region of filamentous phages, tends to be unstable (19,44,45). Occasionally, parts of the cloned DNA are deleted at a rate that tends to be directly correlated with the size of the inserted fragment. Therefore, the question of the genetic stability of DNA inserts cloned into the intergenic region was examined.
Genetic instability in fd-tet vector
Purified fd-tet dsDNA generated two distinct eletrophoretic patterns on agarose gels when isolated from two different E.coli strains (K802 and K91KAN). The DNA isolated from K802 exhibited the supercoiled and relaxed forms of the fd-tet genome (Fig. 2A, lane 1), whereas the DNA isolated from K91KAN contained the supercoiled and relaxed forms of two distinct DNAs (Fig. 2A, lane 2). One DNA corresponded to the fd-tet genome while the second compared well with the parent fd genome (Fig. 2A, lane 3). The two purified fd-tet preparations and fd were then digested with the MscI restriction enzyme expected to generate full-length linear DNA for both fd and fd-tet (Fig. 1). Digestion of fd-tet from K802 and fd each generated only one band with the expected linear sizes of 9183 and 6414 bp respectively (Fig. 2B, lanes 1 and 3). However, digestion of fd-tet derived from K91KAN generated two bands, one indistinguishable in mobility from the linear fd-tet, and the second corresponding in size to linear fd (Fig. 2B, lane 2). This indicates that for a substantial amount of DNA, a deletion of ∼2.8 kb occurred when the fd-tet genome was produced in K91KAN bacteria.
Figure 2.
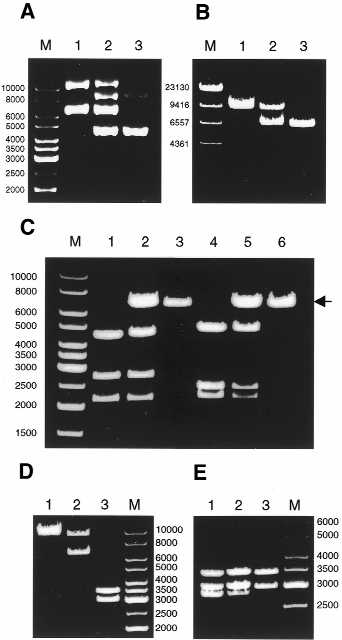
Analysis of the 2.8 kb deletion in fd-tet. (A) Uncut dsDNA of fd-tet was isolated from K802 (lane 1) or K91KAN (lane 2) and resolved on 0.8% agarose gel. M, DNA ladder (in bp). Uncut double-stranded DNA of wild-type fd was used for comparison (lane 3). (B) MscI digests of fd-tet derived from K802 (lane 1) or K91KAN (lane 2). MscI digest of fd was used for comparison (lane 3). M, λ HindIII digest. Note that the lower band in lane 2, corresponding to the deleted product, is at least as intense as the upper band corresponding to the linear full-length fd-tet. (C) SnaBI/AvaI digests of fd-tet derived from K802 (lane 1) or K91KAN (lane 2). SnaBI/AvaI digest of fd was used for comparison (lane 3). BspHI/HindIII digests of fd-tet derived from K802 (lane 4) or K91KAN (lane 5). BspHI/HindIII digest of fd DNA was used for comparison (lane 6). The arrow indicates the deleted products (lanes 2 and 5) corresponding in size to the linear full-length fd (lanes 3 and 6). (D) BamHI digests of fd-tet DNA derived from K802 (lane 1) or K91KAN (lane 2). BamHI digest of fd was used for comparison (lane 3). Note that the pattern obtained in lane 2 is identical to the MscI digest in lane 2 of (B). (E) BstYI digests of fd-tet derived from K802 (lane 1) or K91KAN (lane 2). BstYI digest of fd was used for comparison (lane 3). Note the relative drop in intensity for the lowest band in lane 2 of (E). M in gels (C–E) corresponds to DNA ladder (in bp). Agarose concentration in all gels was 0.8%.
Two points, therefore, must be addressed. (i) What DNA sequences are lost through the deletion process? (ii) How are the differences in the bacterial strains related to the apparent genetic instability?
Deletion of the tet fragment
As the deleted DNA was 2.8 kb, we postulated that it might correspond to the tet-fragment (2775 bp) residing in the intergenic space of fd-tet (Fig. 1). Therefore, we compared the SnaBI/AvaI digests of fd-tet DNA derived from both bacterial strains as before. The SnaBI/AvaI double digestion of fd-tet isolated from K802 generated three fragments with the expected sizes: 4381, 2690 and 2112 bp (Fig. 2C, lane 1). The double digestion of the K91KAN-derived fd-tet DNA, however, generated four DNA fragments (Fig. 2C, lane 2); the additional fragment corresponding to the deletion product similar in size to a single cut full-length 6414 bp fd (Fig. 2C, lane 3, compare also with Fig. 2B). This indicates that the SnaBI–AvaI fragment of the tet insert in the intergenic region was deleted from fd-tet.
This was further supported by BspHI/HindIII digestion of fd-tet derived from both bacteria. The BspHI/HindIII digestion of fd-tet DNA isolated from K802 generated the expected three fragments of 4676, 2366 and 2141 bp (Fig. 2C, lane 4). However, the BspHI/HindIII double digestion of fd-tet DNA isolated from K91KAN also generated a fourth fragment (∼6.4 kb) corresponding in size to single cut full-length fd [Fig. 2C, lane 5 (arrow)] derived from the deletion product. This not only confirms the selective deletion of the tet fragment but extends the boundaries of the deletion beyond the SnaBI site to at least the BspHI site (Fig. 1).
Is the deletion a precise reversion to fd? If so, one would reconstitute the original BamHI site used to clone the tet fragment, and thus generate two restriction fragments (3425 and 2989 bp as in the fd BamHI digest; Fig. 2D lane 3) at the expense of the linearized deletion product (for example, see the MscI digest in Fig. 2B, lane 2). As can be seen in Figure 2D (lane 2), BamHI digestion of fd-tet isolated from K91KAN produced a pattern similar to that shown for the MscI digest. Two bands corresponding to the linearized fd-tet (9183 bp) and the full-length linearized deletion product (∼6.4 kb) were generated. This thus illustrates that the deletion does not precisely regenerate the second BamHI site of the fd parent molecule.
Thus, the next question we addressed was whether or not the excision sites lay within or outside the tet fragment. For this, the BstYI digest illustrated in Figure 2E was performed (two BstYI sites, created during the fd-tet construction, precisely bracket the tet fragment; Fig. 1). The gel pattern obtained for the BstYI digest of fd-tet derived from K802 had three fragments (3425, 2983 and 2775 bp) identical in size to those generated when K91KAN-derived DNA was used (Fig. 2E, compare lanes 1 and 2). This demonstrates that at least one of the two BstYI sites was retained in the deleted product. Therefore, sequences immediately adjacent to the original BamHI site must also be preserved in the deleted fd-tet, a fact that may be relevant to the mechanism of the deletion process (Discussion).
The deletion is recA independent and F+ dependent
Table 3 illustrates that in a variety of E.coli strains tested, the tet deletions (described above) were detected only in the F+ strains. Note, for example, that the deletion occurs only in the DH5αF′ E.coli strain and not in its isogenic strain DH5α. Therefore, the deletion event is F+ dependent and recA independent (both bacteria are recA–).
Table 3. The deletion is F+ dependent and recA independent.
|
Escherichia coli strain |
recA |
F-episome |
Deletion |
| K802 | + | – | – |
| K91KAN | + | + | + |
| DH5αa | – | – | – |
| DH5αF′a | – | + | + |
| JM109 | - | + | + |
| XL1-Blue | – | + | + |
| MC1061b | + | – | – |
| NM554b | – | – | – |
aDH5α and DH5αF′ are isogenic strains; DH5αF′ contains the F-episome.
bMC1061 and NM554 are isogenic strains; NM554 is recA–.
When using filamentous phages, working with F+ bacterial strains cannot be avoided. Therefore, introducing the recombinant pVIII gene in a region prone to F+ dependent deletions is potentially problematic. We therefore explored the suitability of the non-coding region between the wild-type pVIII gene and the pIII gene as a site for genetic manipulation and introduction of a second pVIII gene.
Construction of the ftac88 vector
Designing a phage display vector with the aim of introducing a second pVIII gene in the non-coding region between the wild-type pVIII and pIII genes presents two obstacles that must be considered: (i) unique restriction sites for cloning are unavailable in this region and (ii) this region contains an essential overlapping promoter and rho-independent transcription terminator that are inseparable (Fig. 1C) (33).
The terminator terminates the transcription of upstream genes ending with the pVIII gene, whereas the promoter initiates the transcription of downstream genes starting with the pIII gene. Therefore, the strategy adopted to insert the recombinant second pVIII gene was to duplicate this promoter/terminator as well. Ultimately, we constructed a type 88 expression vector (designated ftac88) as follows.
The first step was to generate a modified pVIII containing unique restriction sites in order to introduce foreign DNA sequences. Such a modified pVIII gene, designated pVIIISTS, was produced by ‘SOEing’ PCR (42) mutagenesis in which the GAC codon for residue Asp, at position 4 of mature wild-type PVIII protein, was replaced by an insert of 62 bp (‘STS’ insert; Fig. 3A). This insert contained two SfiI sites that bracketed two translation stop-codons followed by the trpA transcription terminator. Thus, the transcription of the pVIIISTS gene terminates prematurely, rendering the expression of this gene silent. In order to express this pVIII gene, one must exchange the SfiI flanked stuffer for a functional in-frame insert of DNA. The PCR was designed to generate a fragment corresponding to the SnaBI–BamHI segment of fd-tet (Fig. 1C) that was cloned into the pGEM-T vector, designated pGEM-T(p8STS) (Fig. 3B).
Next, a wild-type pVIII gene was cloned into the SnaBI site of pGEM-T(p8STS). This was achieved by PCR of the wild-type pVIII gene segment from fd-tet starting from the upstream SnaBI site and continuing just beyond the overlapping promoter/terminator. The downstream antisense primer incorporated three unique restriction sites as well. The PCR product was cloned into the blunt ends of SnaBI cut pGEM-T(p8STS), thus generating the desired pGEM-T(p88STS). As is illustrated in Figure 3C, the SnaBI–BamHI fragment of pGEM-T(p88STS) contains two pVIII genes, duplicated promoter/terminator elements and several unique cloning sites.
The SnaBI site in the tetR gene of fd-tet was removed (Fig. 1C). For this, fd-tet genome was digested with XbaI/BstXI to remove the 725 bp fragment containing the SnaBI site, which was replaced by a linker containing an AvaII site. This intermediate modified fd-tet was then digested with SnaBI/BamHI and the wild-type segment was exchanged for the correspondingly cut fragment derived from pGEM-T(p88STS) described above. Furthermore, a tac promoter was introduced between the XhoI and SacI sites to produce the type 88 vector designated ftac88 shown in Figure 4.
In order to evaluate whether or not the ftac88 vector can produce mosaic phages, a linker encoding the linear peptide APAGFAIL [the epitope corresponding to the anti-gp120 mAb GV4H3 (41)] was inserted in-frame between the two SfiI sites, eliminating the trpA terminator. As demonstrated by dot blot analysis (Fig. 5A), this construct [ftac88(4H3)] produced phages that were recognized by the GV4H3 mAb.
Figure 5.
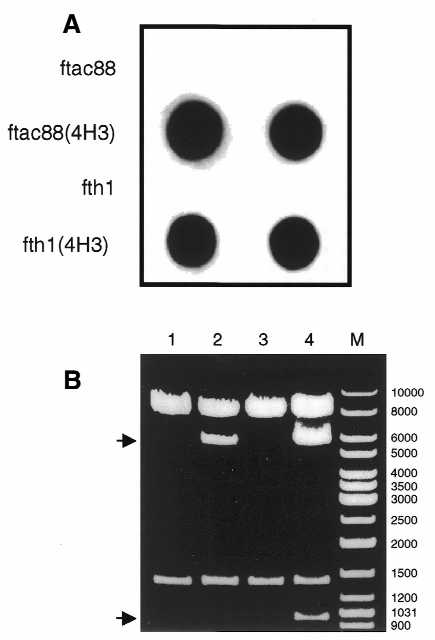
(A) Binding of mAb GV4H3 to phages expressing the GV4H3 epitope. Equal amounts of phages were applied in duplicate to a nitrocellulose membrane filter and reacted with the GV4H3 mAb. The GV4H3 epitope displaying phages, ftac88(4H3) and fth1(4H3), showed strong signals. Phages ftac88 and fth1 were used as negative controls. (B) Deletion of the recombinant pVIII gene in ftac88(4H3). SnaBI/BamHI digests of ftac88 derived from DH5α (lane 1) or DH5αF′ (lane 2) were run on 0.8% agarose gel. SnaBI/BamHI digests of ftac88(4H3) isolated from DH5α (lane 3) or DH5αF′ (lane 4) are also shown. M, DNA ladder mix. The upper arrow indicates the tet insert deleted product and the lower arrow indicates the recombinant pVIII gene deleted fragment.
Genetic instability in ftac88
The orientation of the wild-type pVIII and the modified pVIIISTS genes in the ftac88 vector is a direct tandem repeat. In view of the fact that the deletion of the tet fragment in fd-tet (described above) is recA independent and F+ dependent, we examined the genetic stability of the modified pVIIISTS gene in DH5αF′. For this, ftac88 DNA preparations from DH5α and DH5αF′ bacteria were compared.
Double digestion of ftac88 with SnaBI and BamHI released the expected 1378 bp fragment, regardless of the source of the DNA (Fig. 5B, lanes 1 and 2; for orientation see Fig. 4). The deletion of the tet fragment still occurred in DH5αF′ (Fig. 5B, lane 2; indicated by the upper arrow). Thus, one can conclude that the modified pVIIISTS gene is genetically stable in ftac88.
However, it was important to further investigate the genetic stability of the modified pVIIISTS gene when it contained a peptide coding sequence. For this, the ftac88 DNA construct expressing the GV4H3 epitope (above) was prepared from DH5α and DH5αF′ bacteria and used for the SnaBI/BamHI double digestion as above. Here, the situation was surprising. Digestion of the DNA derived from DH5α released as expected a 1376 bp fragment (Fig. 5B, lane 3). However, SnaBI/BamHI digestion of the same DNA prepared from DH5αF′ generated an additional smaller fragment of ∼950 bp (Fig. 5B, lane 4). This indicates that a deletion of ∼400 bp occurred. This deletion corresponded to selective loss of the recombinant pVIII gene and reconstitution of the wild-type pVIII–pIII configuration including the original GAC Asp4-codon as confirmed by sequence analysis (not shown). Therefore, it was concluded that the modified pVIIISTS gene, when containing coding sequences, becomes genetically unstable in DH5αF′. This suggests that the stability of the ftac88 observed in DH5αF′ is rendered due to the trpA terminator which could imply that the deletion process may require transcribed regions.
Construction of fth1 vector
In light of the above experience in designing, constructing and testing the ftac88 vector, we redesigned a novel type 88 vector to overcome the problems of genetic instability and include other improvements. The attributes of this vector, designated fth1 (Fig. 6; the sequence has been deposited in GenBank as accession no. AF362081) are listed below.
(i) The second pVIII gene containing the 62 bp STS insert was constructed using synthetic oligonucleotides to generate a PVIII protein of identical amino acid sequence yet coded for with alternative codon usage. This obviated the formation of tandem repeats, a configuration that is prone to deletion events as illustrated in the analysis of ftac88 (above). The homologous gene was designated pVIIISTSh. The pVIIISTSh gene was cloned, as in ftac88, between the wild-type pVIII and pIII genes.
(ii) To avoid the generation of additional tandem repeats, an alternative HP terminator (46) was introduced into the 3′ region of the wild-type pVIII gene, 10 codons upstream to the pVIII stop codon. Moreover, the severed 3′ region coding for the C-terminal domain of the wild-type PVIII protein was reconstituted using alternative codon usage [so to reduce homology within the –35 box of the promoter of the pIII gene residing in this region (33)].
(iii) The open reading frame of the tetracycline resistance gene was cloned between the two pVIII genes downstream to a kanamycin promoter (derived from M13K07 phage). This was done to avoid using the bi-directional tetracycline promoter (Fig. 1C) which would otherwise generate an antisense transcript of the wild-type pVIII gene were it used in this new position. The exclusion of the tetracycline repressor is advantageous as the tetracycline resistance gene thus becomes constitutively expressed, making pre-incubation with low levels of tetracycline to induce resistance unnecessary.
(iv) The original intergenic space containing the ori sequences was reconstituted. Previously, the tet fragment used by Smith (47) to construct fd-tet was inserted into the fd ori(–) rendering this vector and its derivatives low copy number and thus producing relatively low phage titers. Reconstituting the intergenic region in fth1 provided two advantages: the copy number of the DNA was greatly increased and the titers of secreted phage increased as well (see below).
The construction of fth1 (Fig. 7) was accomplished using ‘SOEing’ PCR, as described above, enabling the introduction of multiple cloning sites and novel genes into the non-coding region between the wild-type pVIII and pIII genes of fd-tet.
The first step was to modify fd-tet by the introduction of a short segment of DNA, 10 codons upstream from the stop codon of the wild-type pVIII gene (Fig. 7B). This segment not only provided novel restriction sites but also separated the promoter/terminator adjacent to the pIII gene from the wild-type pVIII gene by reconstituting the severed last 10 codons of the pVIII open reading frame (IV-1; Fig. 7B).
Subsequently, additional intermediate vectors were produced (IV-2 and IV-3, see Materials and Methods) which eventually led to IV-4 (Fig. 7C). In the latter, the novel HP terminator was incorporated downstream to the reconstituted wild-type pVIII gene followed by the tetracycline resistance open reading frame driven by the kanamycin promoter. Moreover, the ori(–) in the intergenic region was also reconstituted in IV-4.
The final vector was produced by cloning the synthetic pVIIISTSh gene, driven by the tac promoter, into the BglII/EagI-digested IV-4 vector to give fth1 (see detailed map, Fig. 6).
In order to evaluate fth1 as a display vector the GV4H3 epitope was introduced between the SfiI sites. This construct, called fth1(4H3), produced mosaic phages presenting the GV4H3 epitope that were recognized by the GV4H3 mAb (Fig. 5A).
The stability of the tetracycline resistance gene and the modified pVIIISTSh gene was tested using restriction analyses as before. Digestion of fth1 and fth1(4H3) DNA, derived from DH5α or DH5αF′, with BstNI generated identical fragments, is illustrated in Figure 8. BstNI sites appear twice in both fd (Fig. 1) and fth1 (Fig. 6). BstNI digestion of fd generated the expected two fragments of 5456 and 952 bp (Fig. 8, lane 1). The latter contains the non-coding region between the pVIII and pIII genes. As this region in fth1 contains the tetracycline resistance and the recombinant pVIIISTSh genes, the 952 bp fragment was shifted to generate the 2777 and 2775 bp bands in BstNI digests of fth1 and fth1(4H3), respectively, derived from DH5α and DH5αF′ (Fig. 8, lane 2–5). Any deletions of either the tetracycline resistance gene or the recombinant pVIII gene or their parts would generate DNA fragments ranging in size from 952 to 2777 bp. No such fragments could be detected, illustrating that the fth1 vector is genetically stable.
Figure 8.
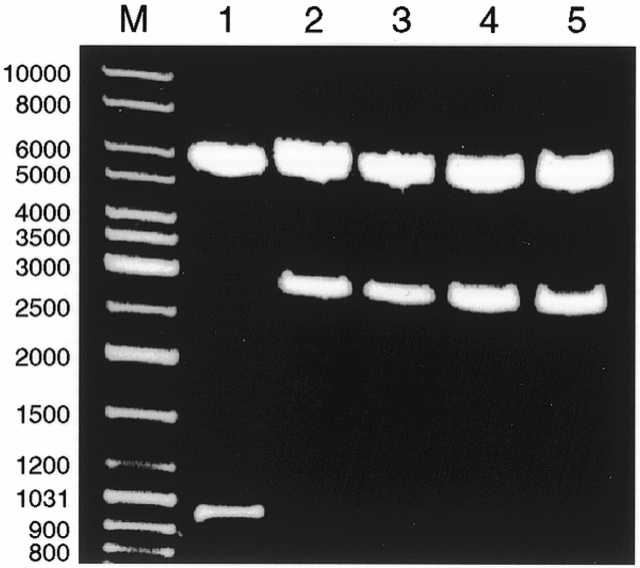
The genetic stability of fth1. BstNI digests of fth1 derived from DH5α (lane 2) or DH5αF′ (lane 3). BstNI digests of fth1(4H3) derived from DH5α (lane 4) or DH5αF′ (lane 5). BstNI digest of fd was used for comparison (lane 1). M, DNA ladder mix. Agarose gel concentration was 0.8%.
The functionality of the reconstituted ori(–) was directly apparent. In several independent purifications of fth1 DNA, we routinely obtained 0.5–1 mg DNA/liter of bacteria, as opposed to the typical yields of 50 µg DNA/liter of ftac88-harboring bacteria. Similarly, a 100-fold increase is typically seen in phage titers for which 1012 phages/ml is measured for fth1 as compared to 1010 phages/ml achieved for fd-tet and its derivatives.
Thus, the fth1 vector provides a convenient and stable type 88 phage display expression system.
DISCUSSION
We have observed that, in F+ bacterial strains, a 2.8 kb fragment of DNA can be excised from the region of the foreign tet insert, which Smith and co-workers (38) originally introduced into the B-stem–loop of the ori(–) structure. The loss of the insert’s AvaI site, which overlaps the original remnant BamHI site, illustrates that downstream aspects of the insert are not preserved in the deleted form. The loss of the BspHI site argues that no more than 18 bp of the insert can be retained at the other extreme. Preservation of at least one BstYI site, yet lack of reconstitution of the original BamHI site, further stresses the close but imprecise correspondence of the deleted fragment with the original tet insert.
As the insert is flanked by the inverted repeats that create the B-stem–loop, and since these repeats are retained in the deleted form of the phage, one might postulate that the mechanism generating the deletion involves homology and base paring. Clearly, however, this cannot be homologous recombination, as that would require direct repeats of homologous stretches of over 50 bases and, of course, a functional RecA protein in order to enable a deletion event (48,49). The deletion events in our experiments occurred in recA– bacterial strains. An alternative mechanism, the ‘slippage mechanism’ proposed by Feschenko and Lovett (50), also requires direct repeats and therefore cannot explain the deletion.
One might imagine that before the conversion of the ssDNA of fd-tet to a double-stranded form, the inverted repeats, flanking the tet fragment align to form a B-stem–loop of sorts. This stem–loop would be greatly distorted by a grossly extended ‘bubble’ (where the BamHI site had been), consisting of the 2.8 kb insert. During replication, the polymerase might be able to ‘skip’ from one side of the bubble to the other, thus omitting the entire insert and generating the deletion. Assuming that this is a very rare event, one must still address the fact that in K91Kan cells, >50% of the DNA isolated was of the deleted form. Moreover, why is the deletion F+ dependent?
The matter of copy number might be explained due to two selective advantages that are realized in the deleted form. The mere fact that the genome is 2.8 kb shorter than fd-tet should speed up the replication. More important may be the fact that the deletion most probably partially reconstitutes the ori(–) structure. The ramifications of a markedly more functional ori(–) would substantially enable more effective production of dsDNA.
Another possibility may be related to the packaging of phages for secretion. The A-stem–loop structure, situated adjacent to the B-stem–loop, is the packaging signal of the phage (33). It stands to reason that the tet insert may sterically hinder the packaging of the recombinant phages, a burden that would be removed in the deleted form.
However, the major phenomenon encountered here is the fact that the deletion is detectable only in the F+ bacteria. This might be due to the fact that even rare events can be markedly amplified when superinfection of bacteria can take place. Obviously, for such superinfection the bacteria must be compatible, a situation only satisfied in F+ bacteria. Therefore, it is proposed that the deletion leads to shorter and more efficient replicating forms of the phage, albeit a rare event. Subsequently, however, such deleted phages can be markedly amplified in F+ bacteria via super infection, thus enabling accumulation of very substantial amounts of the deleted form.
Despite the above, the intergenic region of the phage has been used to generate an effective type 88 vector in the past such as the f88.4 vector (43). In f88.4, the recombinant pVIII gene was introduced into the tet fragment of fd-tet. However, one would expect with such a vector the gradual appearance of deleted forms of the phages and loss of their peptide inserts displayed by the chimeric pVIII protein. Indeed, such ‘contaminating‘, fast-growing, tetracycline-sensitive phages have been reported by Bonnycastle et al. (18) when they used the f88.4 vector.
An alternative cloning site for the second pVIII gene was tested, namely the non-coding region between the pVIII and pIII genes. The ftac88 described in this report illustrated that substantial amounts of deletions of the recombinant pVIII continued to occur. In contrast, however, to the situation of the deletion of the tet insert, the deletion of the modified pVIII gene entails reasonably long direct repeats. Thus, whereas homologous recombination is not an option (experiments were performed in recA– bacteria), the slippage mechanism, described by Feschenko and Lovett (50), might be responsible for the selective loss of the second pVIII gene in ftac88. This would entail the slipped misalignment of the nascent strand during DNA replication. During replication of the repeated DNA, the nascent strand becomes displaced from its template and pairs with the other copy on the template strand. Then, replication is resumed and the slipped misalignment can lead to either deletion or expansion within tandem repeat arrays. Therefore, deletions due to the slippage mechanism are homology dependent, a requirement satisfied in ftac88.
The selective loss of the recombinant pVIII gene further supports the slippage mechanism. This mechanism requires the stabilization of the slipped misalignment structure (50,51). The longer the displaced nascent strand, the more stabilized the slipped misalignment structure becomes. The recombinant pVIII gene contains a foreign sequence in its 5′ region. This severs the repeat into two parts, a short sequence upstream and a long downstream sequence. The long sequence therefore enables the stabilization of the slipped misaligned structure more efficiently than the short one. Therefore, excision sites in the long downstream identical sequence should predominate, thus eliminating upstream sequences including the foreign DNA insert.
The actual deletion event may be a very rare one, typical of the slippage mechanism. One can postulate that deleted DNA products could be amplified due to F+-dependent superinfection (see above).
Therefore, the last aspect that must be considered is why the pVIIISTS form is stable as opposed to those constructs in which the stuffer containing the trpA terminator is exchanged for a peptide coding sequence (such as the GV4H3 epitope). The stability rendered by the presence of the trpA transcriptional terminator indicates that transcription might be a positive and necessary factor. The trpA terminator stops the transcription of the recombinant pVIII gene prematurely and consequently no deletion is observed. Transcription through the second homologous DNA repeat appears to be a necessary prerequisite that provides the opportunity to form the misaligned structure that leads to subsequent deletion during DNA replication. Such a mechanism may explain how misalignment can exist between the first and second repeats at the moment when the first repeat is being replicated yet the second downstream repeat is still in the closed dsDNA configuration. In order to mispair the nascent strand to the closed double-stranded repeat, the latter would have to be opened. In the absence of RecA protein, which catalyzes such reactions (52), transcription might be an efficient mode for opening dsDNA. However, as opening dsDNA during transcription is transient, this mode would be efficient only when the DNA repeats are in reasonably close proximity. Indeed, the deletion frequency, reported for the slippage mechanism, is correlated directly with the distance separating the DNA repeats (53,54). Moreover, this mechanism would require that the wild-type pVIII gene be the first to be replicated. Indeed, the most extensive replication during infection is that of the + strand, proceeding from the intergenic region through the wild-type pVIII gene to the recombinant pVIII gene (55).
As a result of the above analyses and the experiences gained through the construction and characterization of the ftac88 vector, we ultimately designed an improved novel vector that corrects for the problems we encountered. This vector has been designated fth1 and has been found to be a functional and genetically stable phage display vector.
Acknowledgments
ACKNOWLEDGEMENTS
This study was supported by research grants provided by the Israeli Ministry of Science and the Israel Science Foundation.
References
- 1.Scott J.K. and Smith,G.P. (1990) Searching for peptide ligands with an epitope library. Science, 249, 386–390. [DOI] [PubMed] [Google Scholar]
- 2.Dower W.J. (1992) Phage power. Curr. Biol., 2, 251–253. [DOI] [PubMed] [Google Scholar]
- 3.Lane D.P. and Stephen,C.W. (1993) Epitope mapping using bacteriophage peptide libraries. Curr. Opin. Immunol., 5, 268–271. [DOI] [PubMed] [Google Scholar]
- 4.Cortese R., Felici,F., Galfre,G., Luzzago,A., Monaci,P. and Nicosia,A. (1994) Epitope discovery using peptide libraries displayed on phage. Trends Biotechnol., 12, 262–267. [DOI] [PubMed] [Google Scholar]
- 5.Cortese R., Monaci,P., Nicosia,A., Luzzago,A., Felici,F., Galfre,G., Pessi,A., Tramontano,A. and Sollazzo,M. (1995) Identification of biologically active peptides using random libraries displayed on phage. Curr. Opin. Biotechnol., 6, 73–80. [DOI] [PubMed] [Google Scholar]
- 6.Cortese R., Monaci,P., Luzzago,A., Santini,C., Bartoli,F., Cortese,I., Fortugno,P., Galfre,G., Nicosia,A. and Felici,F. (1996) Selection of biologically active peptides by phage display of random peptide libraries. Curr. Opin. Biotechnol., 7, 616–621. [DOI] [PubMed] [Google Scholar]
- 7.Parmley S.F. and Smith,G.P. (1988) Antibody-selectable filamentous fd phage vectors: affinity purification of target genes. Gene, 73, 305–318. [DOI] [PubMed] [Google Scholar]
- 8.Koiunen E., Wang,B. and Ruoslahti,E. (1995) Phage libraries displaying cyclic peptides with different ring sizes: ligand specificities of the RGD-directed integrins. Bio-Technology, 13, 265–270. [DOI] [PubMed] [Google Scholar]
- 9.Wrighton N.C., Farrell,F.X., Chang,R., Kashyap,A.K., Barbone,F.P., Mulcahy,L.S., Johnson,D.L., Barrett,R.W., Jolliffe,L.K. and Dower,W.J. (1996) Small peptides as potent mimetics of the protein hormone erythropoietin. Science, 273, 458–463. [DOI] [PubMed] [Google Scholar]
- 10.Sparks A.B., Quillam,L.A., Thorn,J.M., Der,C.H. and Kay,B.K. (1994) Identification and characterization of Src SH3 ligands from phage-displayed random peptide libraries. J. Biol. Chem., 269, 23853–23856. [PubMed] [Google Scholar]
- 11.Rasqualini R. and Ruoslahti,E. (1996) Organ targeting in vivo using phage display peptide libraries. Nature, 380, 364–366. [DOI] [PubMed] [Google Scholar]
- 12.Matthews D.J. and Wells,J.A. (1993) Substrate phage: selection of protease substrates by monovalent phage display. Science, 260, 1113–1116. [DOI] [PubMed] [Google Scholar]
- 13.Schmitz R., Baumann,G. and Gram,H. (1996) Catalytic specificity of phosphotyrosine kinases Blk, Lyn, c-Src and Syk as assessed by phage display. J. Mol. Biol., 260, 664–677. [DOI] [PubMed] [Google Scholar]
- 14.Cwirla S.E., Peters,E.A., Barrett,R.W. and Dower,W.J. (1990) Peptides on phage: a vast library of peptides for identifying ligands. Proc. Natl Acad. Sci. USA, 87, 6378–6382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Felici F., Castagnoli,L., Musacchio,A., Jappelli,R. and Cesareni,G. (1991) Selection of antibody ligands from a large library of oligopeptides expressed on a multivalent exposition vector. J. Mol. Biol., 222, 301–310. [DOI] [PubMed] [Google Scholar]
- 16.Luzzago A., Felici,F., Tramontano,A., Pessi,A. and Cortese,R. (1993) Mimicking of discontinuous epitopes by phage displayed peptides, I. Epitope mapping of human H ferritin using a phage library of constrained peptides. Gene, 128, 51–57. [DOI] [PubMed] [Google Scholar]
- 17.Hoess R., Brinkmann,U., Handel,T. and Pastan,I. (1993) Identification of a peptide which binds to the carbohydrate-specific monoclonal antibody B3. Gene, 128, 43–49. [DOI] [PubMed] [Google Scholar]
- 18.Bonnycastle L.L.C., Mehroke,J.S., Rashed,M., Gong,X. and Scott,J.K. (1996) Probing the basis of antibody reactivity with a panel of constrained peptide libraries displayed by filamentous phage. J. Mol. Biol., 258, 747–762. [DOI] [PubMed] [Google Scholar]
- 19.Model P. and Russel,M. (1988) Filamentous bacteriophage. In Calendar,R. (ed.), The Bacteriophages. Plenum Press, New York and London, Vol. 2, p. 375.
- 20.Goldsmith M.E. and Konigsberg,W.H. (1977) Adsorption protein of bacteriophage fd: isolation, molecular properties, and location in virus. Biochemistry, 16, 2686–2694. [DOI] [PubMed] [Google Scholar]
- 21.Gray C.W., Brown,R.S. and Marvin,D.A. (1981) Adsorption complex of filamentous fd virus. J. Mol. Biol., 146, 621–627. [DOI] [PubMed] [Google Scholar]
- 22.Holliger P. and Riechmann,L. (1997) A conserved infection pathway for filamentous bacteriophages is suggested by the structure of the membrane penetration domain of the minor coat protein g3p from phage fd. Structure, 5, 265–275. [DOI] [PubMed] [Google Scholar]
- 23.Bluthner M., Bautz,E.K.F. and Bautz,F.A. (1996) Mapping of epitopes recognized by PM/Sc1 autoantibodies with gene-fragment phage display libraries. J. Immunol. Methods, 198, 187–198. [DOI] [PubMed] [Google Scholar]
- 24.Kay B.K., Adey,N.B., He,Y.-S., Manfredi,J.P., Mataragnon,A.H. and Fowlkes,D.M. (1993) An M13 phage library displaying 38-amino-acid peptides as a source of novel sequences with affinity to selected targets. Gene, 128, 59–65. [DOI] [PubMed] [Google Scholar]
- 25.McCafferty J., Griffiths,A.D., Winter,G. and Chiswell,D.J. (1990) Phage antibodies: filamentous phage displaying antibody variable domains. Nature, 348, 552–554. [DOI] [PubMed] [Google Scholar]
- 26.McCafferty J., Jackson,R.H. and Chiswell,D.J. (1992) Phage enzymes: expression and affinity chromatography of functional alkaline phosphatase on the surface of bacteriophage. Protein Eng., 4, 955–961. [DOI] [PubMed] [Google Scholar]
- 27.Marvin D.A., Hale,R.D., Nave,C. and Citterich,M.H. (1994) Molecular model and structural comparisons of native and mutant class I filamentous bacteriophages Ff (fd, f1, M13), If1 and IKe. J. Mol. Biol., 235, 260–286. [DOI] [PubMed] [Google Scholar]
- 28.Sugimoto K., Sugisaki,H., Okamoto,T. and Takanami,M. (1977) Studies on bacteriophage fd DNA. IV. The sequence of messenger RNA for the major coat protein gene. J. Mol. Biol., 111, 487–507. [DOI] [PubMed] [Google Scholar]
- 29.Greenwood J., Willis,A.E. and Perham,R.N. (1991) Multiple display of foreign peptides on a filamentous bacteriophage. J. Mol. Biol., 220, 821–827. [DOI] [PubMed] [Google Scholar]
- 30.Iannolo G., Minenkova,O., Petruzzelli,R. and Cesareni,G. (1995) Modifying filamentous phage capsid: limits in the size of the major capsid protein. J. Mol. Biol., 248, 835–844. [DOI] [PubMed] [Google Scholar]
- 31.Willis A.E., Perham,R.N. and Wraith,D. (1993) Immunological properties of foreign peptides in multiple display on a filamentous bacteriophage. Gene, 128, 79–83. [DOI] [PubMed] [Google Scholar]
- 32.Smith G.P. (1993) Surface display and peptide libraries. Gene, 128, 1–2. [PubMed] [Google Scholar]
- 33.Van Wezenbeek P.M.G.F., Hulsebos,T.J.M. and Schoenmakers,J.G.G. (1980) Nucleotide sequence of the filamentous bacteriophage M13 DNA genome: comparison with phage fd. Gene, 11, 129–148. [DOI] [PubMed] [Google Scholar]
- 34.Kim M.H., Hines,J.C. and Ray,D.S. (1981) Viable deletions of the M13 complementary strand origin. Proc. Natl Acad. Sci. USA, 78, 6784–6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dotto G.P., Horiuchi,K. and Zinder,N.D. (1984) The functional origin of bacteriophage f1 DNA replication: its signal and domains. J. Mol. Biol., 172, 507–521. [DOI] [PubMed] [Google Scholar]
- 36.Messing J. (1983) New M13 vectors for cloning. Methods Enzymol., 101, 20–78. [DOI] [PubMed] [Google Scholar]
- 37.Moses P.B., Boeke,J.D., Horiuchi,K. and Zinder,N.D. (1980) Restructuring the bacteriophage f1 genome: expression of gene VIII in the intergenic space. Virology, 104, 267–278. [DOI] [PubMed] [Google Scholar]
- 38.Zacher A.N., Stock,C.A., Golden,J.W. and Smith,G.P. (1980) A new filamentous phage cloning vector: fd-tet. Gene, 9, 127–140. [DOI] [PubMed] [Google Scholar]
- 39.Krebber C., Spada,S., Desplancq,D. and Pluckthun,A. (1995) Co-selection of cognate-antigen pairs by selectively-infective phages. FEBS Lett., 377, 227–231. [DOI] [PubMed] [Google Scholar]
- 40.Stern B. and Gershoni,J.M. (1998) Construction and use of a 20-mer phage display epitope library. In Cabilly,S. (ed.), Methods in Molecular Biology: Combinatorial Peptide Library Protocols. Humana Press, Totowa, NJ, p. 137. [DOI] [PubMed]
- 41.Denisova G., Stern,B., Raviv,D., Zwickel,J., Smorodinsky,N.I. and Gershoni,J.M. (1996) Humoral immune response to immunocomplexed HIV envelop glycoprotein 120. AIDS Res. Hum. Retroviruses, 12, 901–909. [DOI] [PubMed] [Google Scholar]
- 42.Horton R.M., Cai,Z., Ho,S.N. and Pease,L.R. (1990) Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques, 8, 528–535. [PubMed] [Google Scholar]
- 43.Zhong G., Smith,G.P., Berry,J. and Brunham,R.C. (1994) Conformational mimicry of a chlamydial neutralization epitope on filamentous phage. J. Biol. Chem., 269, 24183–24188. [PubMed] [Google Scholar]
- 44.Sambrook T., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
- 45.Vieira J. and Messing,J. (1987) Production of single-stranded plasmid DNA. Methods Enzymol., 153, 3–11. [DOI] [PubMed] [Google Scholar]
- 46.Nohno T., Saito,T. and Hong,J.-S. (1986) Cloning and complete sequence of the Escherichia coli glutamine permease operon (glnHPQ). Mol. Gen. Genet., 205, 260–269. [DOI] [PubMed] [Google Scholar]
- 47.Smith G.P. (1988) Filamentous phage assembly: morphogenetically defective mutants that do not kill the host. Virology, 167, 156–165. [DOI] [PubMed] [Google Scholar]
- 48.Clark A.J. (1973) Recombination deficient mutants of Escherichia coli and other bacteria. Annu. Rev. Genet., 7, 67–86. [DOI] [PubMed] [Google Scholar]
- 49.Matfield M., Badawi,R. and Brammar,W.J. (1985) Rec-dependent and Rec-independent recombination of plasmid-borne duplications in Escherichia coli K12. Mol. Gen. Genet., 199, 518–523. [DOI] [PubMed] [Google Scholar]
- 50.Feschenko V.V. and Lovett,S.T. (1998) Slipped misalignment mechanisms of deletion formation: analysis of deletion endpoints. J. Mol. Biol., 276, 559–569. [DOI] [PubMed] [Google Scholar]
- 51.Lovett S.T. and Feschenko,V.V. (1996) Stabilization of diverged tandem repeats by mismatch repair: evidence for deletion formation via a misaligned replication intermediate. Proc. Natl Acad. Sci. USA, 93, 7120–7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.West S.C. (1992) Enzymes and molecular mechanisms of genetic recombination. Annu. Rev. Biochem., 61, 603–640. [DOI] [PubMed] [Google Scholar]
- 53.Lovett S.T., Gluckman,T.J., Simon,P.J., Sutera,V.A.,Jr and Drapkin,P.T. (1994) Recombination between repeats in Escherichia coli by a recA-independent, proximity-sensitive mechanism. Mol. Gen. Genet., 245, 294–300. [DOI] [PubMed] [Google Scholar]
- 54.Bi X. and Liu,L.F. (1994) recA-independent and recA-dependent intramolecular plasmid recombination. J. Mol. Biol., 235, 414–423. [DOI] [PubMed] [Google Scholar]
- 55.Horiuchi K. and Zinder,N.D. (1976) Origin and direction of synthesis of bacteriophage f1 DNA. Proc. Natl Acad. Sci. USA, 73, 2341–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]