

Abstract
Inflammatory myopathy is a rare autoimmune muscle disorder. Treatment typically focuses on skeletal muscle weakness or inflammation within muscle, as well as complications of respiratory failure secondary to respiratory muscle weakness. Impaired respiratory muscle function contributes to increased dyspnea and reduced exercise capacity in pulmonary hypertension (PH), a debilitating condition that has few treatment options. The initiation and progression of PH is associated with inflammation and inflammatory cell recruitment and it is established that hypoxia-induced mitogenic factor (HIMF, also known as resistin-like molecule α), activates macrophages in PH. However, the relationship between HIMF and inflammatory myoblasts remains unclear. This study investigated the signaling pathway involved in interleukin-18 (IL-18) expression and its relationship with HIMF in cultured myoblasts. We found that HIMF increased IL-18 production in myoblasts and that secreted IL-18 promoted tube formation of the endothelial progenitor cells. We used the mouse xenograft model and the chick chorioallantoic membrane assay to further explore the role of HIMF in inflammatory myoblasts and angiogenesis in vivo. Thus, our study focused on the mechanism by which HIMF mediates IL-18 expression in myoblasts through angiogenesis in vitro and in vivo. Our findings provide an insight into HIMF functioning in inflammatory myoblasts.
Introduction
Inflammation in myoblasts is associated with different conditions, such as hypoxia, strong muscle contraction, and inflammaging, a chronic low-grade inflammatory state that is associated with functional impairments, loss of strength, and myopathy1. Inflammatory myopathies, commonly referred to as myositis, are a rare and heterogeneous group of acquired autoimmune muscle disorders2. Previous research has shown that cytokines appear to play an important role in the development of inflammatory myopathy3.
Inflammation has also been associated with angiogenesis. Several studies have used anti-angiogenic strategies to suppress inflammation4, 5. Under pathological conditions, angiogenesis stimulates vascular endothelial growth factor (VEGF) expression in the smooth muscle cell phenotype6. Interleukin-18 (IL-18) is a pro-inflammatory cytokine that belongs to the IL-1 family, and has been identified in autoimmune disorders7. High levels of serum IL-18 have been observed in patients with inflammatory myopathy8. Moreover, vascular IL-18 has been found to correlate with tissue macrophages and increased levels are found in injured, aging vascular smooth muscle cells9. However, the role of IL-18 in inflammatory skeletal myoblasts remains to be elucidated.
Hypoxia-induced mitogenic factor (HIMF), also known as resistin-like molecule α, is a cysteine-rich secreted protein and pleiotropic cytokine and arises from activated macrophages of the inflammatory responses and exerts mitogenic property in inflammation and angiogenesis10. Previous research has reported that HIMF regulates calcium mobilization in pulmonary smooth muscle cells11, 12. HIMF also has significant angiogenic properties and contributes to the extension of pulmonary arterial pressure13. However, the regulation of HIMF in response to inflammatory myopathy remains unclear. In the present study, we describe intracellular signals showing that HIMF upregulates the expression of IL-18 through the 3-phosphoinositide-dependent protein kinase-1 (PDK1)/ phosphatidylinositol 3-kinase (PI3K)/Akt/ activator protein 1 (AP-1) signaling pathway in myoblasts. These results provide an insight into the mechanism of HIMF functioning in inflammatory myoblasts.
Results
HIMF induces angiogenesis through IL-18 expression in murine myoblasts
Since HIMF has been previously indicated the important role in pulmonary inflammation and angiogenesis14, we therefore used murine myoblasts to investigate the effects of HIMF in inflammatory responses and angiogenesis. Myoblastic cell lines (C2C12 and G7) were treated with HIMF for 24 h in dose-dependent concentrations, and an increase in IL-18 was derived from HIMF-induced protein and mRNA levels (Fig. 1A and B). Treatment of C2C12 cell line with HIMF at different time intervals induced IL-18 protein and mRNA expression in time-dependent manners (Supplementary Figure S1). These results suggested the kinetics of IL-18 release from muscle cells in response to HIMF exposure. HIMF also induced IL-18 secretion from murine myoblasts (Fig. 1C). Conditioned medium (CM) was prepared and collected from the HIMF-treated C2C12 myoblast cell line, to explore the tube formation ability of EPCs in angiogenesis. To investigate the relationship between IL-18 and angiogenesis, CM from HIMF-treated myoblasts was pretreated with anti-IL-18 or anti-VEGF antibodies for 30 min in response to EPC tube formation (Fig. 1D). The numbers of tube branches and the total tube lengths were calculated and quantified (Fig. 1E). The data indicated that HIMF induced IL-18 expression in myoblasts that could potentially correlate with angiogenesis.
Figure 1.
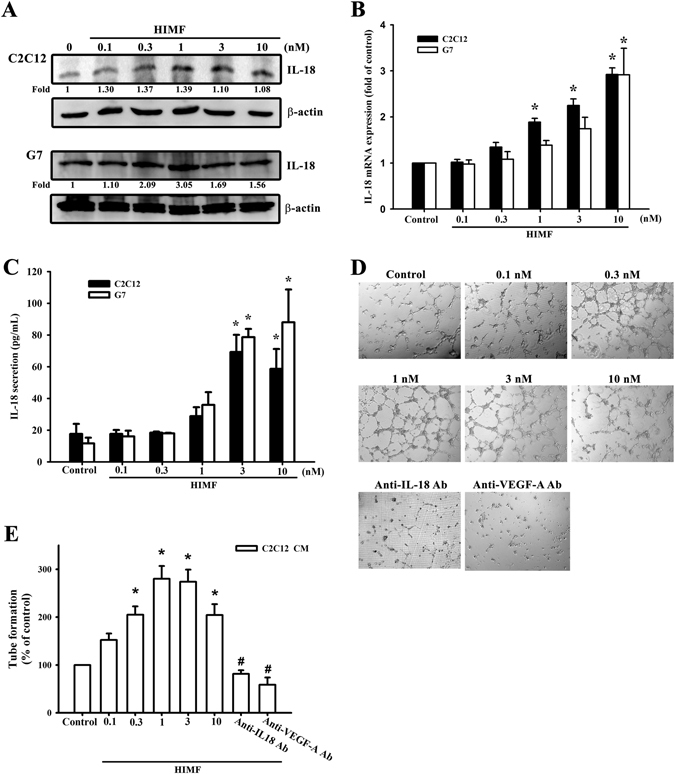
HIMF induces IL-18 expression and EPC tube formation. (A) HIMF induced a dose-dependent increase of IL-18 protein expression in myoblastic cell lines. (B) IL-18 mRNA levels were increased by HIMF in two cell lines. (C) Secreted IL-18 production from HIMF-treated myoblast conditioned medium was increased in a dose-dependent manner. (D) EPC tube formation was increased by conditioned medium from HIMF-treated myoblasts with dose-dependent manners but inhibited by anti-IL18 or anti-VEGF-A antibodies. (E) Quantification of tube formation. Results are expressed as the means ± SEM of four independent experiments. *p < 0.05 as compared with the control group. # p < 0.05 as compared with HIMF-treated group.
PDK1/PI3K/Akt signaling is required for the HIMF-promoted IL-18 expression
PDK1 has been reported to play a critical role in transducing inhibitory signals on eosinophil effector function15. We measured the signaling pathway of PDK1 phosphorylated site at Ser 241 in a time-dependent manner (Fig. 2A), and the involvement of PDK1 by using PDK1 small-interfering RNAs (siRNAs) in Western blots (Fig. 2B). Next, we detected the involvement of PDK1 in HIMF-induced IL-18 mRNA levels (Fig. 2C) and in HIMF-secreted IL-18 protein expression (Fig. 2D). We also explored the involvement of PDK1 phosphorylation in HIMF-induced IL-18 protein expression (Fig. 2E). In addition, to further confirm the signaling pathway of PDK1/PI3K, we performed an off-target assay by using constitutive active PI3K. The C2C12 cells co-transfected with a constitutive active PI3K plasmid and PDK1 siRNA for 24 h rescued PDK1 siRNA-decreased IL-18 mRNA levels, suggesting PI3K is a downstream signal of PDK1 (Supplementary Fig. S7). A recent study has demonstrated that the PI3K/Akt signaling pathway is associated with PDK1 in inflammatory diseases16. We continued to explore the signaling pathways of PI3K and Akt phosphorylated site at Tyr 458 and Ser 473, respectively (Figs 3A and 4A), and their involvement by using PI3K or Akt siRNA in Western blots (Figs 3B and 4B). Pretreatment of cells with PI3K or the Akt inhibitor inhibited HIMF-increased IL-18 mRNA levels and their production (Fig. 3C,D and 4C,D).We also determined that HIMF-increased Akt phosphorylation decreased in response to pretreatment with PKD1 or the PI3K inhibitor (Fig. 4E). To demonstrate the inhibition of PI3K and Akt on IL-18, we measured PI3K and Akt phosphorylation in the inhibitors and siRNAs, respectively (Figs 3E and 4F). To further confirm the signaling pathway of PI3K/Akt, C2C12 cells co-transfected with a constitutive active Akt plasmid and PI3K siRNA for 24 h rescued PI3K siRNA-decreased IL-18 mRNA levels, suggesting Akt is a downstream signal of PI3K (Supplementary Fig. S7). Above results indicate that the PDK1/PI3K/Akt signaling pathways are involved in HIMF-induced IL-18 production.
Figure 2.
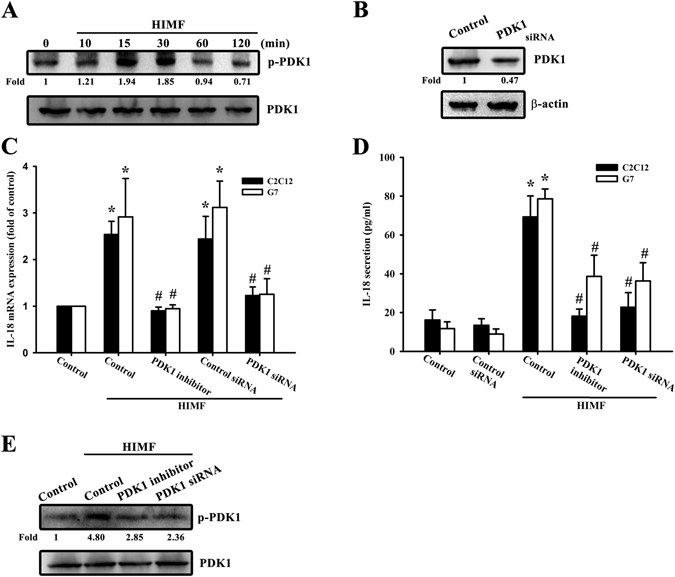
PDK1 is involved in HIMF-induced IL-18 production. (A) Cells were incubated with HIMF (3 nM) for indicated time intervals and cell lysates were immunoblotted with an antibody specific for phospho-PDK1. (B) Cells were transfected with a PDK1 siRNA for 24 h, and PDK1 protein expression was analyzed. (C) HIMF-induced IL-18 mRNA was inhibited by a PDK1 inhibitor or PDK1 siRNA. (D) Cells were pretreated for 30 min with a PDK1 inhibitor or transfected with siRNA against PDK1 for 24 h, then stimulated with HIMF for 24 h, and the conditioned media was collected and examined by ELISA. (E) HIMF-induced PDK1 phosphorylation was inhibited by a PDK1 inhibitor or PDK1 siRNA. Results are expressed as the means ± SEM of four independent experiments. *p < 0.05 as compared with the control group. # p < 0.05 as compared with HIMF-treated group.
Figure 3.
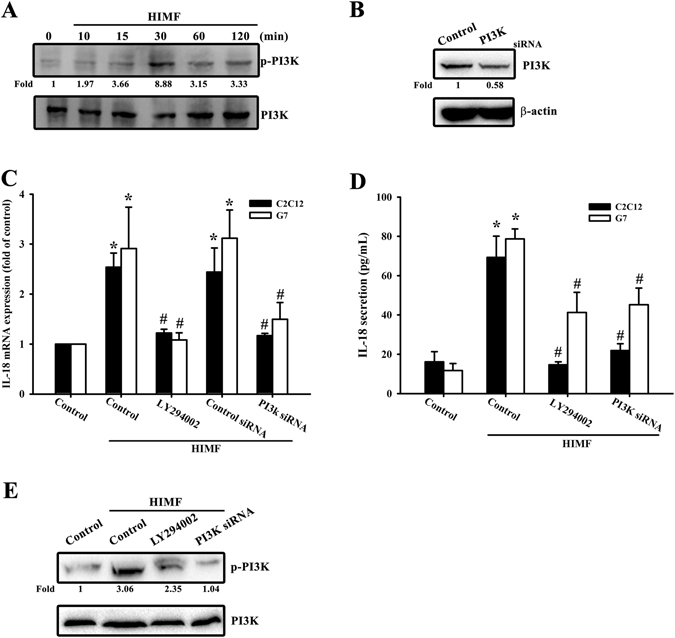
PI3K is involved in HIMF-induced IL-18 production. (A) Cells were incubated with HIMF (3 nM) for indicated time intervals and cell lysates were immunoblotted with an antibody specific for phospho-PI3K. (B) Cells were transfected with a PI3K siRNA for 24 h, and PI3K protein expression was analyzed. (C) HIMF-induced IL-18 mRNA levels were inhibited by LY294002 or a PI3K siRNA. (D) Cells were pretreated for 30 min with LY294002 or transfected with a PI3K siRNA for 24 h then stimulated with HIMF for 24 h, and the conditioned media was collected and examined by ELISA. (E) HIMF-induced PI3K phosphorylation was inhibited by LY294002 or PI3K siRNA. Results are expressed as the means ± SEM of four independent experiments. *p < 0.05 as compared with the control group. # p < 0.05 as compared with HIMF-treated group.
Figure 4.
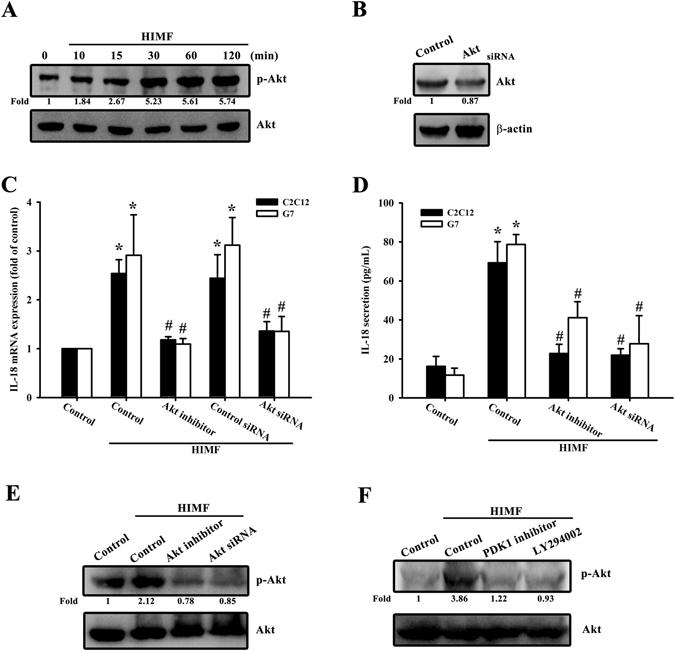
Akt is involved in HIMF-induced IL-18 production. (A) Cells were incubated with HIMF (3 nM) for indicated time intervals and cell lysates were immunoblotted with an antibody specific for phospho-Akt. (B) Cells were transfected with Akt siRNA for 24 h, and Akt protein expression was analyzed. (C) HIMF-induced IL-18 mRNA was inhibited by the Akt inhibitor or Akt siRNA. (D) Cells were pretreated for 30 min with the Akt inhibitor or transfected with Akt siRNA for 24 h, then stimulated with HIMF for 24 h, and the conditioned media was collected and examined by ELISA. (E) HIMF-induced Akt phosphorylation was inhibited by an Akt inhibitor or Akt siRNA. (F) Cells were treated with the PDK1 inhibitor or LY294002 for 30 min then stimulated with HIMF for 120 min, and phospho-Akt protein expression was analyzed. Results are expressed as the means ± SEM of four independent experiments. *p < 0.05 as compared with the control group. #p < 0.05 as compared with HIMF-treated group.
Involvement of c-Jun in HIMF-increased IL-18 production
To further understand the signaling mechanisms underlying HIMF-induced IL-18 production, we sought to determine which transcription factor might be implicated. AP-1 has been widely reported to be the most important transcription factor in inflammatory diseases17. Figure 5B showed that HIMF-increased IL-18 expression was inhibited by Tanshinone IIA which has been reported to suppress AP-1 activity with c-Jun and c-Fos DNA complex formation. To clarify the composition of AP-1 dimers being activated by HIMF exposure, we performed a co-immunoprecipitation manner followed by immunoblot analysis. After co-immunoprecipitation with anti-c-Fos antibodies for 24 h, the blotting expression of c-Jun was activated by HIMF exposure (Supplementary Fig. S2). Thus, we demonstrated c-Jun phosphorylated site at Ser 73 in a time-dependent manner (Fig. 5A), and cells transfected with c-Jun siRNA were confirmed (Fig. 5B upper panel). The role of c-Jun was involved in IL-18 secreted protein expression (Fig. 5B lower panel), mRNA levels (Fig. 5C), and protein expression (Fig. 5D) by pretreating cells with Tanshinone IIA or co-transfecting cells with c-Jun siRNA followed by HIMF treatment. To further indicate the signaling pathway of Akt/c-Jun, C2C12 cells co-transfected with a constitutive active c-Jun plasmid and Akt siRNA for 24 h rescued Akt siRNA-decreased IL-18 mRNA levels, suggesting c-Jun is a downstream signal of Akt (Supplementary Fig. S7). In order to confirm the upstream molecules engaged in HIMF-increased AP-1 signaling pathways, we used a murine AP-1 promoter-luciferase construct. HIMF markedly increased AP-1 promoter activity in myoblasts, which was reversed by pretreating cells with the PDK1 inhibitor LY294002, an Akt inhibitor, and Tanshinone IIA (Fig. 5E), and by co-transfection with PDK1, PI3K, Akt, and c-Jun siRNAs (Fig. 5F). We measured c-Jun phosphorylation through various inhibitors to confirm the upstream signals of AP-1 (Fig. 5G). In addition, we also performed IL-18 mRNA levels in which the effects of inhibitors and siRNA per se in the absence of HIMF (Supplementary Fig. S3). To demonstrate whether c-Jun could directly bind to the IL-18 target gene in the nucleus, we performed the ChIP assay and used the TFSEARCH website to examine the c-Jun binding site in the IL-18 promoter region. HIMF-induced binding of c-Jun to IL-18 was attenuated by the PDK1 inhibitor Ly294002 and the Akt inhibitor (Fig. 5H). These data suggest that HIMF-enhanced IL-18 expression requires c-Jun involvement in the signaling transduction.
Figure 5.
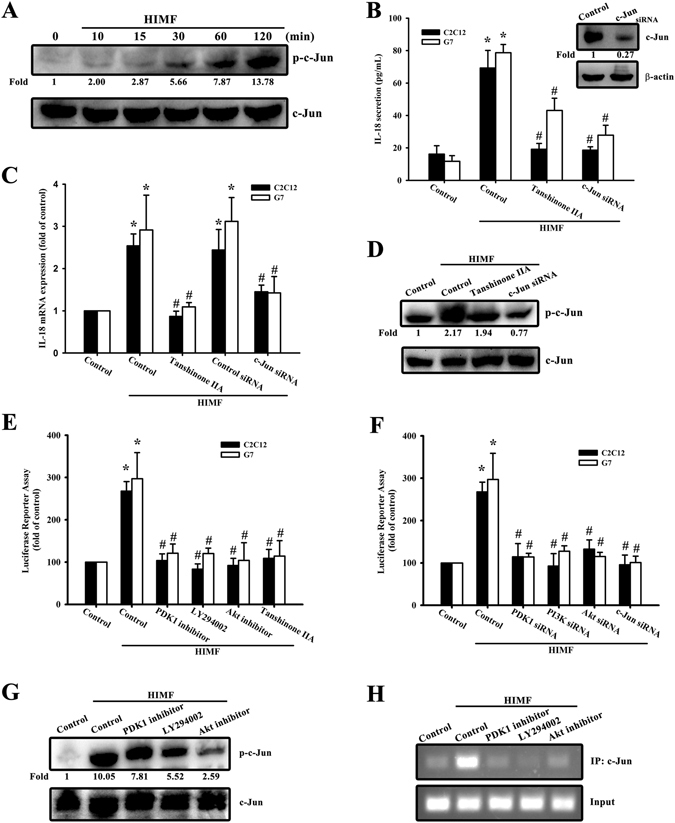
HIMF induces IL-18 production through AP-1. (A) Cells were incubated with HIMF (3 nM) for indicated time intervals and cell lysates were immunoblotted with an antibody specific for phospho-c-Jun. (B) Cells were transfected with siRNA against c-Jun for 24 h, and c-Jun protein expression was analyzed (upper panel). Cells were treated for 30 min with Tanshinone IIA or transfected with a c-Jun siRNA for 24 h, then stimulated with HIMF for 24 h, and the conditioned media was collected and examined by ELISA (lower panel). (C) HIMF-induced IL-18 mRNA was inhibited by Tanshinone IIA or a c-Jun siRNA. (D) HIMF-induced c-Jun phosphorylation was inhibited by a Tanshinone IIA or c-Jun siRNA. (E) Cells were treated with the PDK1 inhibitor, LY294002, an Akt inhibitor, or Tanshinone IIA for 30 min, then incubated with HIMF for 24 h. AP-1-luciferase activity was measured, and the results were normalized to β-galactosidase activity. (F) Cells were co-transfected with either AP-1-luciferase plasmid or siRNA against PDK1, PI3K, Akt, or c-Jun for 24 h, then stimulated with HIMF. AP-1-luciferase activity was measured, and the results were normalized to β-galactosidase activity. (G) Cells were treated with the PDK1 inhibitor, LY294002, or an Akt inhibitor for 30 min, then stimulated with HIMF for 120 min, and phospho-c-Jun protein expression was analyzed. (H) The recruitment of c-Jun to the IL-18 promoter was analyzed using the ChIP assay. Results are expressed as the means ± SEM of four independent experiments. *p < 0.05 as compared with the control group. # p < 0.05 as compared with HIMF-treated group.
HIMF promotes IL-18-dependent angiogenesis in vivo
Our results show that secreted IL-18 expression from myoblasts CM induces EPC tube formation in vitro. To illustrate this angiogenesis in vivo, we used the mouse xenograft model. We treated myoblasts with anti-IL-18 antibody for 1 h followed by HIMF treatment. CM from HIMF-treated myoblasts was injected subcutaneously into the flanks of nude mice and incubated for 10 days. After sacrifice, the plugs were photographed and measured for hemoglobin concentration (Fig. 6A and B). The sections were immunostained with vascular markers, CD31 and VEGF (Fig. 6C). We then performed another CAM assay to determine the effect of HIMF on angiogenesis (Fig. 6D), and quantified the vessels under microscope (Fig. 6E). On the other hand, to further confirm the involvement of those signals in angiogenesis in vivo, CM with kinase inhibitors followed by HIMF treatment were used in Matrigel plugs and CAM assay (Supplementary Fig. S4). Our in vivo angiogenesis results demonstrate that HIMF-induced IL-18 secretion from myoblasts promotes angiogenesis, which might be linked to the upstream signaling pathway.
Figure 6.
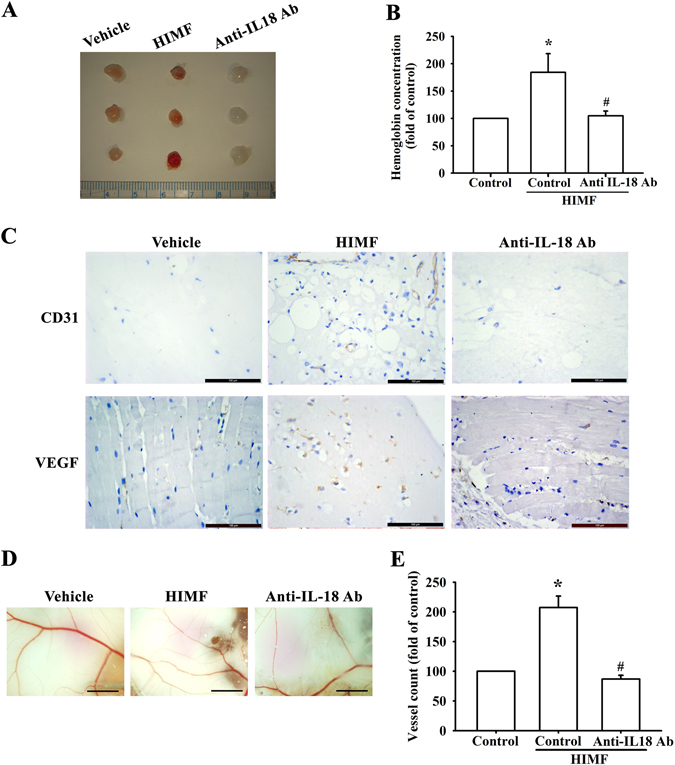
HIMF promotes VEGF expression and angiogenesis in vivo. (A) Matrigel plugs containing myoblast conditioned medium (CM) of PBS (vehicle), HIMF (10 nM), or anti-IL-18 antibody combined with HIMF were subcutaneously injected into the flanks of nude mice. After 7 days, the plugs were photographed. (B) Hemoglobin levels from the plugs were quantified and normalized to control group. (C) Specimens from the plugs were immunostained with antibodies against CD31 and VEGF. Scale bar: 100 μm. (D) Data from a CAM assay using 5-day-old fertilized chick embryos (6 eggs/group): PBS (vehicle), CM with HIMF (10 nM) treatment, or CM with anti-IL-18 antibody followed by HIMF treatment were resuspended in Matrigel and placed onto the CAMs, which were allowed to develop for another 3 days. The CAMs were then examined by microscopy and photographed. Scale bar: 2 mm. (E) CAM vessels were calculated and quantified. Results are expressed as the means ± SEM of four independent experiments. *p < 0.05 as compared with the control group. #p < 0.05 as compared with HIMF-treated group.
Discussion
Inflammatory myopathy is a type of autoimmune muscle disorder. Treatment of myopathy commonly focuses on the effects in skeletal muscles in the C2C12 myoblast model2. A recent investigation into muscle differentiation and regeneration using the dermatomyositis autoantigen transcription intermediary factor 1γ (TIF1γ) demonstrated that premature silencing of this protein in cultured myoblasts accelerates myosin heavy chain expression (a later marker of muscle differentiation) and myoblast fusion18. In this study, we sought to determine how HIMF might associate with inflammatory cell infiltration in response to muscle inflammation, since it has been reported that HIMF induces endothelial smooth muscle inflammation through Th2 cell infiltration in pulmonary hypertension19 and increases pro-inflammatory and smooth muscle expression in lung fibroblasts20. However, the role of HIMF in skeletal muscle inflammation is poorly understood. Our results demonstrate that HIMF induces skeletal muscle inflammation in C2C12 and G7 cell lines, suggesting that HIMF might act as a pro-inflammatory cytokine to induce IL-18 expression related to inflammatory skeletal muscle cells.
IL-18 is a potential pro-inflammatory cytokine. Although the involvement of IL-18 has been implicated in various diseases, especially autoimmune diseases, such as allergy and systemic lupus erythematosus7, 21, only a few studies have demonstrated IL-18 regulation is involved in inflammatory myopathy22. We show that HIMF induces IL-18 expression in murine myoblasts through PDK1, which acts as a downstream effector in the PI3K/Akt/c-Jun signaling pathway and has been implicated in the regulation of neutrophil migration. These results not only provide evidence for a role of HIMF in inflammatory myopathy, but also they elucidate the mechanism of the signaling pathway.
HIMF-induced IL-18 expression through the PDK1/PI3K/Akt/c-Jun signaling pathways highlights the importance of the signaling connection between the transcription factor and target gene in the nucleus. Although there are many reports demonstrating the binding of AP-1 and IL-1β or other inflammatory cytokines, there is scant evidence on the binding of AP-1 and IL-1823, 24. Our findings demonstrate that HIMF augments the binding of AP-1 to the IL-18 promoter and reveal that the c-Jun binding site is located at positions −153 to −147 in the murine IL-18 promoter region. As shown in our results, HIMF-enhanced binding of AP-1 to the IL-18 promoter is antagonized by the PDK1, PI3K, or Akt inhibitors, which suggests that HIMF transcriptionally upregulates IL-18 production via the c-Jun transcription factor. Although our co-immunoprecipitation results indicate that HIMF activated the composition of c-Jun/c-Fos hetrodimers, the specific mechanism by which c-Fos involved in HIMF-mediated IL-18 production needs to be further studied by using c-Fos siRNA.
In addition to playing a role in pro-inflammation, IL-18 is a potent angiogenic mediator. VEGF, a hallmark of angiogenesis, is upregulated by pro-inflammatory cytokines or growth factors in diabetic retinopathy25 and therefore is involved in both inflammation and angiogenesis. Increased IL-18 expression has been found to be associated with angiogenic activity in diabetic retinopathy26. A previous study also demonstrated the correlation between IL-18 and VEGF in patient with diabetic retinopathy, suggesting that IL-18 may contribute to retinal angiogenesis by acting together with VEGF27. The role of IL-18 in angiogenesis has been further explored in recent studies into neurovascular damage and breast cancer28, 29. In addition, hypoxia-induced angiogenesis has recently become a critical topic for investigation not only in tumor cells but also in autoimmune diseases involving skeletal muscle30, 31. Here, we hypothesize that HIMF-induced IL-18 expression could be associated with angiogenesis. Since previous evidence has shown that IL18 promotes angiogenesis through the release of VEGF and other angiogenesis factor in lung cells32, our in vitro results of EPC tube formation implied that both IL-18 and VEGF-A were involved in HIMF-induced angiogenesis. Thus, the dominant angiogenesis of VEGF-A is more important than IL-18 and whether HIMF mediated rise in IL18 production caused increased angiogenesis through direct effects of IL18 on endothelial progenitor cells or through indirect effects of VEGF and other angiogenesis factors needs to be further investigated.
In conclusion, we found that HIMF increased IL-18 secretion in myoblasts and promoted EPC tube formation and angiogenesis via the PDK1/PI3K/Akt/c-Jun signaling pathway. Our evidence indicates a possible new target for drug development relating to the angiogenic mechanism by which HIMF may play a role in the pathology of inflammatory myopathies.
Methods
Materials
Rabbit polyclonal antibody specific for phosphate-p85, a heterodimer of PI3K, p-Akt, and p-PDK1 were purchased from Cell Signaling Technology (Danvers, MA, USA). Rabbit polyclonal antibodies specific for PDK1, PI3K, Akt, c-Jun, β-actin, and mouse polyclonal antibodies specific for IL-18 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Recombinant human HIMF was purchased from PeproTech (Rocky Hill, NJ). Neutralized monoclonal anti-IL-18 and anti-VEGF-A antibodies were both purchased from R&D Systems (Minneapolis, MN, USA). PI3K inhibitors (Ly294002), the Akt inhibitor, and c-Jun inhibitors (Tanshinone IIA) were purchased from Sigma-Aldrich (St. Louis, MO). Those siRNAs against PDK1, p85, Akt, and c-Jun were purchased from Dharmacon Research (Lafayette, CO, USA). Constitutive active plasmids of PI3K and c-Jun were gifts from Lewis Cantley (Addgene plasmid # 1399) and from Axel Behrens (Addgene plasmid # 47443), respectively. Another constitutive active plasmid of Akt was a kind gift from Dr. W.C. Huang (China Medical University, Taiwan). Dulbecco’s Modified Eagle Medium (DMEM), fetal bovine serum (FBS), and all other cell culture reagents were purchased from Gibco Life Technologies (Grand Island, NY, USA).
Cell culture
Murine myoblast cell lines C2C12 and G7 were purchased from the American Type Culture Collection (Manassas, VA). Cell culture protocols were followed as described in previous studies33. C2C12 and G7 cells were cultured in DMEM supplemented with 10% FBS (Invitrogen, Carlsbad, CA) and antibiotics (100 U/mL penicillin G and 100 mg/mL streptomycin). Cultures were maintained in a humidified atmosphere of 5% CO2 at 37 °C.
Human circulating EPCs
EPCs were derived from healthy donors who gave informed consent before enrollment, and ethical approval was granted by the Institutional Review Board of the Mackay Medical College (New Taipei City, Taiwan; reference number: P1000002). All research was performed in accordance with the relevant guidelines and regulations. EPCs were isolated and purified as previously described34. Briefly, EPCs were seeded on gelatin-coated dishes containing MV2 medium, SupplementMix (PromoCell, Heidelberg, Germany) and 20% non-heat-inactivated defined FBS (HyClone, Logan, UT, USA) and incubated for 3 days; the medium was changed every 3 days. This study’s protocol was approved by the institutional review board and the ethics committee of Human Studies Committee of China Medical University Hospital.
Conditioned medium preparation
C2C12 (2 × 106) were grown overnight in 6-well in cell culture medium. After two washes with phosphate-buffered saline (PBS), cells were incubated in 0.5% FBS in DMEM medium for 16–18 h before different stimulations. After HIMF treatment for 24 h, CM was collected for all in vitro and in vivo experiments.
EPC tube formation
This assay was conducted as per previous protocols34. Briefly, EPCs (2 × 104/100 µL) were resuspended in MV2 serum-free medium according to different CM and neutralized antibodies, then added to the wells with Matrigel (BD Biosciences, Bedford, MA, USA). After 6–8 h of incubation at 37 °C, EPC tube formation was assessed by microscopy, and each well was photographed at 200 × magnification under a light microscope in four random fields. The numbers of tube branches were calculated and quantified using Angiogenesis Analyzer Image J software according to previously substantial reports35, 36.
Western blot analysis
Cellular lysates were prepared as described in previous studies37. Proteins were resolved through sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to Immobilon polyvinyl difluoride membranes (Millipore, Billerica, MA, USA). The blots were blocked with 4% non-fat milk for 1 h at room temperature then probed with rabbit anti-human antibodies against p-PDK1, p-PI3K, p-Akt, p-c-Jun, and mouse anti-human antibodies against IL-18, for 1 h at room temperature. After undergoing three washes, the blots were incubated with goat anti-rabbit or goat anti-mouse peroxidase-conjugated secondary antibody for 1 h at room temperature. Blots with horseradish peroxidase-labeled substrate were detected by enhanced chemiluminescence and visualized using a Fujifilm LAS-3000 chemiluminescence detection system (Fujifilm, Tokyo, Japan). All results are obtained from more than four independent experiments.
Quantitative real-time polymerase chain reaction
Total RNA was extracted from osteoblasts using a TRIzol kit (MDBio, Taipei, Taiwan). RNA quality and yield were determined by absorbance at 260-nm measurements performed with a Nanovue Spectrophotometer (GE Healthcare, Madison, WI). Complementary DNA was synthetized from 1 μg total RNA using a Moloney Murine Leukemia Virus Reverse Transcription kit (Invitrogen), following the manufacturer’s recommendations. Quantitative real-time polymerase chain reaction (qRT-PCR) analysis was carried out with a SYBR One-Step RT-PCR Master Mix (Applied Biosystems, Foster City, CA). All target gene primers were purchased from Applied Biosystems. qPCR assays were carried out in triplicate using a StepOnePlus sequence detection system (Applied Biosystems), according to the manufacturer’s instructions. All results are obtained from six independent experiments performed in duplicate.
Enzyme-linked immunosorbent assay
Myoblasts were cultured in 24-well culture plates. Cells were treated with HIMF and then incubated at 37 °C for 24 h. To examine the downstream signaling pathways responsive to HIMF treatment, cells were pretreated with various inhibitors for 30 minutes before the addition of HIMF (100 ng/mL). After incubation, the supernatant medium was collected and stored at −80 °C until the assay was performed. IL-18 content in the medium was assayed using an IL-18 enzyme-linked immunosorbent assay (ELISA) kit (R&D), according to the manufacturer’s instructions. All results are obtained from five independent experiments.
Transfection and reporter gene assay
Control ON-TARGETplus small-interfering RNAs (siRNAs) and ON-TARGETplus siRNAs against PDK1, p85, Akt, and c-Jun were purchased from Dharmacon Research (Lafayette, CO, USA). Transient transfection of siRNA (0.5 nM) was carried out using Dharma FECT 1 transfection reagent (Thermo, Waltham, MA, USA) for 24 h, according to the manufacturer’s instructions.
The AP-1 luciferase plasmid is originated from the background pLuc-MCS, as 5.7 kb in length, and it contains direct seven times repeats of the transcription recognition sequences, TGACTAA. For the reporter assay as described in previous studies38, cells were co-transfected with AP-1 luciferase (0.5 μg) and β-galactosidase expression vector (0.5 μg) for 24 h using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s protocol. All results are obtained from four independent experiments.
Chromatin immunoprecipitation (ChIP) assay
Chromatin immunoprecipitation was performed as detailed in a previous experiment39. DNA pellets were immunoprecipitated by anti-c-Jun purified and extracted by phenol-chloroform, then subjected to PCR resolved by 2% agarose gel electrophoresis and visualized under UV light. Primers Forward-TCGACCCGACTCTCAACTCT and Reverse-CGTGCAGAATTTTGAGCCTG were utilized for amplification across the murine IL-18 promoter region containing c-Jun binding site.
In vivo Matrigel plug assay
The Matrigel plug assay was performed as described in previous research34. Thirty 4-week-old male nude mice were randomized into 3 groups and subcutaneously injected with 0.15 mL Matrigel containing CM without HIMF (vehicle), CM with HIMF, or CM with anti-IL-18 antibody and HIMF, and the Matrigel:CM ratio was 1:1.5 for all groups. On day 10, the Matrigel plugs were harvested; some were fixed and embedded in paraffin and subsequently processed for immunostaining of VEGF and CD31, while others were evaluated by Drabkin’s method (Drabkin’s Reagent Kit; Sigma) to quantify hemoglobin content.
Immunohistochemistry
Paraffin-embedded plug sections were prepared, mounted on silane-coated slides, deparaffinized in xylene, rehydrated in a graded alcohol series, and washed in deionized water. After antigen retrieval (boiling in a microwave for 30 min in 10 mM sodium citrate, pH 6.0), intrinsic peroxidase activity was blocked by incubation with 3% hydrogen peroxide. Nonspecific antibody-binding sites were blocked using 3% bovine serum albumin (BSA) in PBS. Plug sections were then incubated with the primary antibody, VEGF or CD31 (Abcam, MA, USA), at 4 °C overnight. After 3 washes in PBS, the secondary antibody (biotin-labeled goat anti-rabbit IgG) was applied for 1 h at room temperature. Staining was detected with 3,3ʹ-diaminobenzidine tetrahydrochloride (DAB) and observed under a light microscope.
Chick chorioallantoic membrane assay
In vivo angiogenetic activity was determined using a chick chorioallantoic membrane (CAM) assay as described previously34. Briefly, 5-day-old fertilized chick embryos (6 eggs/group) were incubated at 37 °C in an 80% humidified atmosphere. On developmental day 8, CM without HIMF (vehicle), CM with HIMF, or CM with anti-IL-18 antibody and HIMF was separately re-suspended in Matrigel as per the Matrigel:CM ratio equal to 1:1.2 and placed onto the CAMs for another 3 days. The CAMs were then examined by microscopy and photographed. Angiogenesis was quantified by counting the number of blood vessel branches. All research was performed in accordance with the relevant guidelines and regulations. All animal work was performed in accordance with a protocol approved by the China Medical University’s (Taichung, Taiwan) Institutional Animal Care and Use Committee. This study’s protocol was approved by the institutional review board and the ethics committee of Human Studies Committee of China Medical University Hospital.
Statistical analysis
Data are presented as the mean ± standard error of the mean. Statistical analysis of comparisons between the experimental groups and control was performed by using the Student’s t test. For Statistical comparisons of multi-groups, we used a two-way analysis-of-variance with statistical significance (experiments versus control) and negative control as the fixed factors. Main and interaction effects were tested at a critical level of α = 0.05. In all cases, p < 0.05 was considered significant.
Electronic supplementary material
Acknowledgements
We thank Dr. W.C. Huang for providing the constitutive active plasmid of Akt. This work was supported by grants from the Ministry of Science and Technology of Taiwan (MOST 103-2628-B-039-002-MY3; MOST 104-2314-B-039-038) and China Medical University Hospital (DMR-106-006); and Dongyang People’s Hospital (2015-ZD004).
Author Contributions
C.-M. Su and C.-H. Tang were substantial contributions to study conception and design and drafted the article or revised it critically for important intellectual content. I-C. Wang and Y. Sun performed all experiments and analyzed the data. S.-C. Liu and L. Jin performed experiments and analysis of the revised manuscript. S.-W. Wang, H.-P. Lee, and W.-P. Tseng were all substantial contributions to acquisition of human data.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-07952-9
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Wen-Pei Tseng, Email: twp@cc.ncue.edu.tw.
Chih-Hsin Tang, Email: chtang@mail.cmu.edu.tw.
References
- 1.Draganidis D, et al. Inflammaging and Skeletal Muscle: Can Protein Intake Make a Difference? The Journal of nutrition. 2016 doi: 10.3945/jn.116.230912. [DOI] [PubMed] [Google Scholar]
- 2.Lilleker J, Murphy S, Cooper R. Selected aspects of the current management of myositis. Therapeutic advances in musculoskeletal disease. 2016;8:136–144. doi: 10.1177/1759720X16655126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Narazaki M, et al. Therapeutic effect of tocilizumab on two patients with polymyositis. Rheumatology. 2011;50:1344–1346. doi: 10.1093/rheumatology/ker152. [DOI] [PubMed] [Google Scholar]
- 4.Knod JL, et al. Angiogenesis and Vascular Endothelial Growth Factor-A Expression Associated with Inflammation in Pediatric Crohn’s Disease. Journal of gastrointestinal surgery: official journal of the Society for Surgery of the Alimentary Tract. 2016;20:624–630. doi: 10.1007/s11605-015-3002-1. [DOI] [PubMed] [Google Scholar]
- 5.Chernoguz A, et al. Pretreatment with anti-VEGF therapy may exacerbate inflammation in experimental acute colitis. Journal of pediatric surgery. 2012;47:347–354. doi: 10.1016/j.jpedsurg.2011.11.028. [DOI] [PubMed] [Google Scholar]
- 6.Butoi E, et al. Cross-talk between macrophages and smooth muscle cells impairs collagen and metalloprotease synthesis and promotes angiogenesis. Biochimica et biophysica acta. 2016;1863:1568–1578. doi: 10.1016/j.bbamcr.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 7.Sanders NL, Mishra A. Role of interleukin-18 in the pathophysiology of allergic diseases. Cytokine & growth factor reviews. 2016 doi: 10.1016/j.cytogfr.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gono T, et al. Interleukin-18 is a key mediator in dermatomyositis: potential contribution to development of interstitial lung disease. Rheumatology. 2010;49:1878–1881. doi: 10.1093/rheumatology/keq196. [DOI] [PubMed] [Google Scholar]
- 9.Rodriguez-Menocal L, et al. Macrophage-derived IL-18 and increased fibrinogen deposition are age-related inflammatory signatures of vascular remodeling. American journal of physiology. Heart and circulatory physiology. 2014;306:H641–653. doi: 10.1152/ajpheart.00641.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steppan CM, et al. A family of tissue-specific resistin-like molecules. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:502–506. doi: 10.1073/pnas.98.2.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fan, C. et al. S100A11 mediates hypoxia-induced mitogenic factor (HIMF)-induced smooth muscle cell migration, vesicular exocytosis, and nuclear activation. Molecular & cellular proteomics: MCP10, M110 000901, 10.1074/mcp.M110.000901 (2011). [DOI] [PMC free article] [PubMed]
- 12.Fan C, et al. Hypoxia-induced mitogenic factor/FIZZ1 induces intracellular calcium release through the PLC-IP(3) pathway. American journal of physiology. Lung cellular and molecular physiology. 2009;297:L263–270. doi: 10.1152/ajplung.90416.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teng X, Li D, Champion HC, Johns RA. FIZZ1/RELMalpha, a novel hypoxia-induced mitogenic factor in lung with vasoconstrictive and angiogenic properties. Circulation research. 2003;92:1065–1067. doi: 10.1161/01.RES.0000073999.07698.33. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, et al. FIZZ1 promotes airway remodeling through the PI3K/Akt signaling pathway in asthma. Experimental and therapeutic medicine. 2014;7:1265–1270. doi: 10.3892/etm.2014.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sturm EM, et al. Phosphoinositide-dependent protein kinase 1 (PDK1) mediates potent inhibitory effects on eosinophils. European journal of immunology. 2015;45:1548–1559. doi: 10.1002/eji.201445196. [DOI] [PubMed] [Google Scholar]
- 16.Kim E, et al. 1-(2,3-Dibenzimidazol-2-ylpropyl)-2-methoxybenzene Is a Syk Inhibitor with Anti-Inflammatory Properties. Molecules. 2016;21:508. doi: 10.3390/molecules21040508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uluckan O, Guinea-Viniegra J, Jimenez M, Wagner EF. Signalling in inflammatory skin disease by AP-1 (Fos/Jun) Clinical and experimental rheumatology. 2015;33:S44–49. [PubMed] [Google Scholar]
- 18.Mohassel P, Rosen P, Casciola-Rosen L, Pak K, Mammen AL. Expression of the dermatomyositis autoantigen transcription intermediary factor 1gamma in regenerating muscle. Arthritis & rheumatology. 2015;67:266–272. doi: 10.1002/art.38863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamaji-Kegan K, et al. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMalpha) increases lung inflammation and activates pulmonary microvascular endothelial cells via an IL-4-dependent mechanism. Journal of immunology. 2010;185:5539–5548. doi: 10.4049/jimmunol.0904021. [DOI] [PubMed] [Google Scholar]
- 20.Song L, et al. A therapeutic role for mesenchymal stem cells in acute lung injury independent of hypoxia-induced mitogenic factor. Journal of cellular and molecular medicine. 2012;16:376–385. doi: 10.1111/j.1582-4934.2011.01326.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jafari-Nakhjavani MR, Abedi-Azar S, Nejati B. Correlation of plasma interleukin-18 concentration and severity of renal involvement and disease activity in systemic lupus erythematosus. Journal of nephropathology. 2016;5:28–33. doi: 10.15171/jnp.2016.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nalbandian A, et al. Activation of the NLRP3 Inflammasome Is Associated with Valosin-Containing Protein Myopathy. Inflammation. 2016 doi: 10.1007/s10753-016-0449-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cahill CM, et al. Differential Expression of the Activator Protein 1 Transcription Factor Regulates Interleukin-1ss Induction of Interleukin 6 in the Developing Enterocyte. PloS one. 2016;11:e0145184. doi: 10.1371/journal.pone.0145184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmeck B, et al. Streptococcus pneumoniae induced c-Jun-N-terminal kinase- and AP-1 -dependent IL-8 release by lung epithelial BEAS-2B cells. Respiratory research. 2006;7:98. doi: 10.1186/1465-9921-7-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Capitao M, Soares R. Angiogenesis and Inflammation Crosstalk in Diabetic Retinopathy. Journal of cellular biochemistry. 2016;117:2443–2453. doi: 10.1002/jcb.25575. [DOI] [PubMed] [Google Scholar]
- 26.Skopinski P, et al. Increased interleukin-18 content and angiogenic activity of sera from diabetic (Type 2) patients with background retinopathy. Journal of diabetes and its complications. 2005;19:335–338. doi: 10.1016/j.jdiacomp.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 27.Song Z, et al. Increased intravitreous interleukin-18 correlated to vascular endothelial growth factor in patients with active proliferative diabetic retinopathy. Graefe’s archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2014;252:1229–1234. doi: 10.1007/s00417-014-2586-6. [DOI] [PubMed] [Google Scholar]
- 28.Johnson EA, Guignet MA, Dao TL, Hamilton TA, Kan RK. Interleukin-18 expression increases in response to neurovascular damage following soman-induced status epilepticus in rats. Journal of inflammation. 2015;12:43. doi: 10.1186/s12950-015-0089-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xing Y, et al. Inhibition of blood vessel formation in tumors by IL-18-polarized M1 macrophages. Genes to cells: devoted to molecular & cellular mechanisms. 2016;21:287–295. doi: 10.1111/gtc.12329. [DOI] [PubMed] [Google Scholar]
- 30.Deng W, Feng X, Li X, Wang D, Sun L. Hypoxia-inducible factor 1 in autoimmune diseases. Cellular immunology. 2016;303:7–15. doi: 10.1016/j.cellimm.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 31.Lindholm ME, Rundqvist H. Skeletal muscle hypoxia-inducible factor-1 and exercise. Experimental physiology. 2016;101:28–32. doi: 10.1113/EP085318. [DOI] [PubMed] [Google Scholar]
- 32.Rovina N, et al. VEGF and IL-18 in induced sputum of lung cancer patients. Cytokine. 2011;54:277–281. doi: 10.1016/j.cyto.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 33.Tseng WP, Su CM, Tang CH. FAK activation is required for TNF-alpha-induced IL-6 production in myoblasts. Journal of cellular physiology. 2010;223:389–396. doi: 10.1002/jcp.22047. [DOI] [PubMed] [Google Scholar]
- 34.Su CM, et al. Resistin Promotes Angiogenesis in Endothelial Progenitor Cells Through Inhibition of MicroRNA206: Potential Implications for Rheumatoid Arthritis. Stem cells. 2015;33:2243–2255. doi: 10.1002/stem.2024. [DOI] [PubMed] [Google Scholar]
- 35.Barooji YF, Rorvig-Lund A, Semsey S, Reihani SN, Bendix PM. Dynamics of membrane nanotubes coated with I-BAR. Scientific reports. 2016;6:30054. doi: 10.1038/srep30054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang Y, et al. Salidroside exerts angiogenic and cytoprotective effects on human bone marrow-derived endothelial progenitor cells via Akt/mTOR/p70S6K and MAPK signalling pathways. British journal of pharmacology. 2014;171:2440–2456. doi: 10.1111/bph.12611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Su CM, et al. Osteopontin Promotes Oncostatin M Production in Human Osteoblasts: Implication of Rheumatoid Arthritis Therapy. Journal of immunology. 2015;195:3355–3364. doi: 10.4049/jimmunol.1403191. [DOI] [PubMed] [Google Scholar]
- 38.Su CM, et al. A novel benzofuran derivative, ACDB, induces apoptosis of human chondrosarcoma cells through mitochondrial dysfunction and endoplasmic reticulum stress. Oncotarget. 2016;7:83530–83543. doi: 10.18632/oncotarget.13171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen CY, et al. CCN1 induces oncostatin M production in osteoblasts via integrin-dependent signal pathways. PloS one. 2014;9:e106632. doi: 10.1371/journal.pone.0106632. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.