

Abstract
Oleanolic acid (OA) has neurotrophic effects on neurons, although its use as a neurological drug requires further research. In the present study, we investigated the effects of OA and OA derivatives on the neuronal differentiation of rat hippocampal neural progenitor cells. In addition, we investigated whether the class II histone deacetylase (HDAC) 5 mediates the gene expression induced by OA. We found that OA and OA derivatives induced the formation of neurite spines and the expression of synapse-related molecules. OA and OA derivatives stimulated HDAC5 phosphorylation, and concurrently the nuclear export of HDCA5 and the expression of HDAC5 target genes, indicating that OA and OA derivatives induce neural differentiation and synapse formation via a pathway that involves HDAC5 phosphorylation.
Keywords: HDAC5, neuronal differentiation, oleanolic acid
INTRODUCTION
Oleanolic acid (OA) is a naturally occurring triterpenoid compound that is found in food and plants, and has been isolated from various plant sources by many researchers (Pollier and Goossens, 2012). The biological activities of OA and other triterpenoids, immunomodulatory, anticancer, anti-inflammatory, anti-anxiety, antidepressant, memory enhancing, antinociceptive, and neuroprotective activities have been examined in a variety of experimental systems (Gangwal, 2013; Yin, 2015).
The association between OA and neural development has been suggested by previous studies. For example, synthetic oleanane triterpenoid enhances the neural differentiation of rat PC12 pheochromocytoma cells induced by nerve growth factor (Suh et al., 1999), and oleanolic acid enhances the action of nerve growth factor (NGF) (Li et al., 2003). Furthermore, OA protects against amyloid beta-protein-induced neurotoxicity in cultured neurons, and has anti-dementia effects in mice (Cho et al., 2009). Although the neuroprotective properties of OA have been investigated, the use of OA and OA derivatives as neurological drugs requires further molecular and biochemical research.
Recent studies have generated evidence that epigenetic regulation is closely involved in the pathophysiology of neurodegenerative disorders, and can therefore be considered for therapeutic approaches to these disorders (Coppede, 2014). Histone deacetylases (HDACs) are a family of enzymes that repress gene expression by removing acetyl groups from histones to produce a less accessible chromatin structure (Meier and Brehm, 2014). The expression of class II HDACs (4, 5, 6, 7, 9, and 10) is cell-type specific, suggesting that these molecules might be key regulators of neural development. HDAC5 is highly concentrated in the brain, with expression located in the forebrain regions, including the hippocampus, cortex, and amygdala (Broide et al., 2007). In the current study, we investigated the effects of OA and OA derivatives on HDAC5 because the subcellular localization of HDAC5 is associated with neural differentiation and maturation (Chawla et al., 2003). Phosphorylation of HDAC5 by kinases induces the transcription of HDAC5 target genes through the nuclear export of phosphorylated HDAC5 (Volmar and Wahlestedt, 2015).
In the present study, we examine the effects of OA and OA derivatives, including OA acetate (OAA) and OAA methyl ester (OAM), on the neural differentiation and synaptic formation of rat hippocampal neural progenitor cells. In addition, we investigate the effects of OA and OA derivatives on HDAC5 phosphorylation and target gene expression to assess their potential for clinical applications, including the promotion of neural differentiation.
MATERIALS AND METHODS
Rat hippocampal neurons: cell culture and drug treatment
Preparation of hippocampal neurons
Primary hippocampal neurons were prepared and processed as described previously (Son et al., 2012). Hippocampi from E16.5 Sprague-Dawley rat embryos were rapidly and aseptically dissected from each brain in ice-cold Ca2+- and Mg2+-free Hank’s balanced salt solution (HBSS; Gibco, USA), followed by removal of meninges and mincing of the remaining hippocampi into small pieces. The hippocampal tissue was then digested in 0.25% EDTA-trypsin (Welgene, Deagu, Korea) for 10 min at 37°C, and the digestion was stopped by neurobasal (NB) medium (Gibco) with 10% fetal bovine serum (FBS, Gibco), 75 mmol/L L-glutamine (Sigma Aldrich, USA) and 0.1% penicillin-streptomycin (Welgene). Tissue fragments were allowed to settle for 5 min, then the supernatant was transferred to a new tube and centrifuged at 1500 rpm for 5 min. The pellet was gently resuspended in culture medium and plated at 40,000–50,000 cells/cm2 on poly-L-lysine-(25 mg/ml in phosphate-buffered saline [PBS], Sigma Aldrich) and laminin-(10mg/ml in PBS, Invitrogen, USA) coated dishes and glass coverslips. Hippocampal cultures were grown for 1 day in NB medium containing 10% FBS, 75 mmol/L L-glutamine and 0.1% penicillin-streptomycin. The medium was changed the following day to NB supplemented with 0.02% B27 serum-free supplement (Gibco), 75 mmol/L L-glutamine, and 0.1% penicillin-streptomycin. This medium was replaced every 2 days. Cultures were maintained for 10–12 days at 37°C in a 5% CO2, 95% airhumidified incubator. The neurons were used after 10–14 days. Animal care and experiments were conducted in accordance with the 2004 Guide for the Care and Use of Laboratory Animals (Korea National Institute of Health) and Hanyang University Veterinary committee.
Drugs
OA, OAA, and oleanolic acid methyl ester (OME) were purchased from Sigma-Aldrich (USA), BOC Sciences (Shirley, USA), Extrasynthese (Genay, FRANCE), respectively. OAM was prepared from OME in the presence of pyridine by reacting with acetic anhydride overnight at room temperature. The chemical identity of OAM was confirmed by MS (HP 5989B, Agilent Technologies, USA), IR (FT/IR-4200, JASCO, USA), and NMR (GEMINI 2000, Varian, USA) spectral analysis. MS: 535.56 (M+Na)+, C33H52O4; IR(KBr): 2938, 1727, 1467, 1322, 1238, 1202, 1029, 827, 755; NMR(CDCl3) 1H: 2.01(s, CH3CO), 2.83(dd, J=11.0, 3.0, H-18β), 3.59(s, OCH3), 4.46(dd, J=6.6, 6.1, H-3α), 5.22(t, J=3.5, H-12). OA, OAA and OAM were solubilized in dimethyl sulfoxide (DMSO). The calcium/calmodulin-dependent protein kinase II (CaMKII) inhibitor, KN-62 (Sigma Aldrich), and the protein kinase D (PKD)-specific inhibitor, Gö6976 (Calbiochem, USA) were solubilized in DMSO, and the same volume of DMSO (to a final concentration 0.02%) was added into the medium of non-treated cultures as a control. Final (working) concentrations were as follows: OA (1.5 μM), OAA (2.5 μM), OAM (2.5 μM), KN-62 (30 μM), Gö6976 (1 μM).
MTT assay
Neuronal progenitor cells (NPCs) were plated at 1 × 104 per well in 96-well plate. Each well was treated with DMSO, OA, OAA, OAM at concentrations of 0.1 1, 5, 10 μM for 24 h. Cells were incubated for 20 min with MTT reagent (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, 500 μg/ml, Sigma Aldrich) in the dark. Medium was then removed and the formazan dye formed was extracted with 100% ethanol. Absorbance was read using an ELISA reader (Bio-Rad, USA) at 590 nm. For the quantitative analysis of cell viability, O.D. (optical density) was estimated from three independent cultures.
Neurite outgrowth assay
Immunocytochemistry
Cells were fixed in 4% paraformaldehyde in PBS for 20 min. The following primary antibodies were used: for neural differentiation, polyclonal anti-microtubule-associated protein-2 (MAP2) at 1:500 (Abcam, Cambridge, UK). For detection of primary antibodies, cells were incubated in PBS containing Alexa Fluor 488-conjugated secondary antibodies (1:200, Thermo Fisher Scientific, USA) for 2 h at room temperature. They were then mounted in Vectashield mounting medium containing DAPI (Vector Laboratories Inc., USA) and photographed using a confocal microscope (Leica Microsystems, Germany).
Measurement of neurite outgrowth and spine density
Images of MAP2+ cells were acquired using Z-stacks, which typically consisted of ten scans at high zoom at 1 μm steps in the z axis using the Leica TCS SP5 (Leica Microsystems). For the analysis of neurite spine density, we focused on the first-order neurites. Three neurite segments per cell were analyzed. The final value was averaged from 9–15 cells per experimental group and expressed as the number of spines per 10 μm. For analysis of neurite outgrowth of MAP2+ cells, the Z-trace feature was used to measure the three-dimensional length from 40 cells per experimental group. The length of neurite outgrowth was defined as the distance from the soma to the tip of the longest branch. For statistical analyses, at least ten random fields were measured for each experimental group.
HDAC5 subcellular localization: immunofluorescence study
After 3 days in vitro culture, hippocampal neurons grown on glass coverslips were transfected with a plasmid encoding a green fluorescent protein (GFP)-HDAC5 fusion protein (GFP-HDAC5-WT; Addgene plasmid #32211) using lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions (Yu et al., 2002). After 2 days transfection, the cells were fixed with 4% paraformaldehyde and immunostained with mouse monoclonal anti-GFP (1:300, Roche Applied Sciences, Penzberg, Germany) followed by 2 h incubation with Alexa Fluor 488-conjugated secondary antibodies (1:200, Thermo Fisher Scientific). The cells were then mounted in Vectashield mounting medium containing DAPI (Vector Laboratories Inc.). Images were captured using the Leica TCS SP5 (magnification: ×60, Leica Microsystems). To characterize the subcellular localizations of HDAC5, the intensity of GFP immunofluorescence in the cytoplasmic and nuclear compartments of transfected neurons was quantified using ImageJ software, as described previously (Cho et al., 2013). All experiments were replicated independently at least three times.
Western blot analysis
Whole-cell proteins were extracted, and Western blot analysis was performed, as previously described (Finsterwald et al., 2013), with the following antibodies: mouse anti-neuron-specific class III β-tubulin (Tuj1, 1:1000, Covance, USA), mouse anti-MAP2 (1:1000, Sigma Aldrich), rabbit anti-postsynaptic density protein 95 (PSD95, 1:1000, Cell Signaling, USA), rabbit anti-synapsin1 (SYN1, 1:1000, Cell Signaling), rabbit antisynaptophysin (SYP, 1:1000, Abcam), rabbit anti-HDAC5 (1:1000, Abcam), rabbit anti-phosphorylated HDAC5 (Ser259) (1:1000, Sigma Aldrich), rabbit anti-phosphorylated HDAC5 (Ser498) (1:1000, Abcam) rabbit anti-phosphorylated CaMKII (Thr286) (1:1000, Cell Signaling), rabbit anti-CaMKII (1:1000, Cell Signaling), rabbit anti-phosphorylated PKD (Ser744/748) (1:1000, Cell Signaling), rabbit anti-PKD (1:1000, Cell Signaling), mouse anti-myocyte enhancer factor-2 (MEF2D, 1:1000, BD Bioscience, USA), rabbit anti-brain-derived neurotrophic factor (BDNF, 1:1000, Santa Cruz, USA), rabbit anti-krüppel-like factor 6 (KLF6, 1:1000, Santa Cruz), mouse anti-β-actin (1:1000, Santa Cruz), and rabbit anti-lamin B1 (1:1000, Abcam) antibodies. The blots were then treated with anti-mouse or anti-rabbit IgG conjugated with peroxidase (1:2000, Santa Cruz) and bands were visualized using an enhanced chemi-luminescence (ECL) detection kit (Neuronex, Korea). The total densitometric value of each band was quantified using ImageJ software, normalized to the corresponding β-actin level, and expressed as fold change relative to the control value.
Reverse transcription polymerase chain reaction (RT-PCR) and quantitative real-time RT-PCR (q-PCR)
Total RNA was prepared from in vitro hippocampal neurons, as described previously (Son et al., 2012). β-actin was used as an internal control. DNA samples were resuspended in H2O and fractions used for real-time PCR (iCycler, Bio-Rad). DNA was amplified in duplicate using PCR in the presence of SYBR Green (Bioline, UK). Ct values for each sample were obtained using Sequence Detector 1.1 software. Each quantitative real-time RT-PCR (q-PCR) was performed in duplicate and repeated at least three times independently. Q-PCR data were analyzed using the ΔΔCt method. The primer sequences were: β-actin, forward (F): 5′-cggaaccgctcattgcc-3′, reverse (R): 5′-acccacactgtgcccatcta-3′; Bdnf, F: 5′-gtgacag tattagcgagtggg-3′, R: 5′-gggtagttcggcattgc-3′; Klf6, F: 5′-ttga aagcacagcgcactcac-3′, R: 5′-accggtatgctttcggaagtgtct-3′; Nr4a1, F: 5′-cggagatgccctgtatcc-3′, R: 5′-atggtgggcttgctgaac-3′.
Statistical analyses
All data were obtained from at least three independent experiments. Data are presented as mean ± stand error of the mean (SEM). Statistical significance was calculated using an unpaired Student’s t-test. *p < 0.05, **p < 0.01, ***p < 0.001.
RESULTS
The effects of OA, OAA and OAM on the differentiation of rat hippocampal neural progenitor cells, including the formation of neurite branches and spines
It has been reported that the OA increases neuronal differentiation of neural progenitors derived from the murine embryos in vitro (Ning et al., 2015). Therefore, we tested whether it stimulated the differentiation of neural progenitors derived from the embryonic hippocampus of rats. We first investigated cytotoxic effects of test compounds OA and two OA derivatives, OAA and OAM, dosed at various concentrations ranging from 0, 0.1, 1, 5, 10 μM using MTT assay which can estimate the number of viable cells (Berridge et al., 2005). All three compounds did not cause any significant changes in the number of viable neural progenitor cells, indicating that the test compounds do not exert significant toxic effects (Fig. 1A). In preliminary experiments, we determined that 1.5–2.5 μM was the optimal concentration as it provided maximal differentiation effects 72 h after treatment based on neuronal morphology and expression of molecules that are involved in neuronal functions (data not shown).
Fig. 1. OA-, OAA- and OAM-dependent activation of synaptic proteins and molecules.
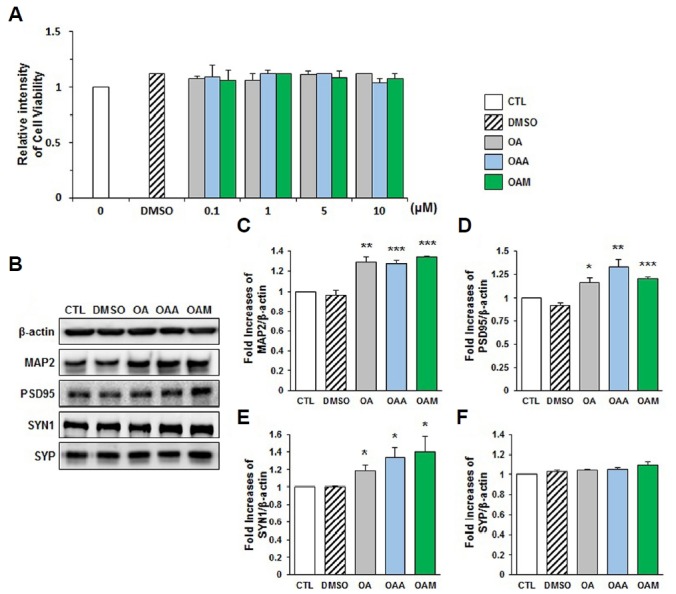
(A) In vitro effects of oleanolic acid (OA), oleanolic acid acetate (OAA), and oleanolic acetate methyl ester (OAM) on neural progenitor cell viability. MTT assay was performed as described in Materials and methods. Data were expressed as an arbitrary number as compared with the cell viability measured in the presence of DMSO control. (B–F) Hippocampal neurons were exposed to OA, OAA or OAM for 72 h. (B) Western blot analysis of OA-, OAA- and OAM-dependent expression of MAP2, PSD95, SYN1 and SYP in whole cellular extracts. (C–F) Quantification of OA-, OAA- and OAM-dependent expression of MAP2, PSD95, SYN1 and SYP normalized to the corresponding β-actin level (n = 3 independent experiments). Results are mean ± SEM. Student’s t-test *p < 0.05, **p < 0.01, ***p < 0.001 compared to control (CTL).
Neural progenitors were treated with OA (1.5 μM) for 72 h and examined the expression of microtubule associated protein-2 (MAP2), a neuronal marker (Harada et al., 2002; Soltani et al., 2005), and synaptic proteins in rat hippocampal neural progenitor cells (Fig. 1B). Western blots showed that the expression of MAP2 protein was indeed increased in the presence of OA (Figs. 1B and 1C), suggesting that the maturation of postmitotic neurons was enhanced by OA. We then investigated whether OA derivatives also increase the MAP2. Treatment of neural progenitor cells with OAA and OAM elicited higher expression of MAP2 protein than non-treated control cells 72 h after single treatment at concentration of 2.5 μM, respectively (Figs. 1B and 1C). Since MAP2 is induced by OA and OA derivatives, we investigated whether OA and OA derivatives increase the expression of synaptic molecules, including PSD95, SYN1 and SYP. PSD95 was significantly increased following treatment with OA, OAA and OAM (Fig. 1D). In addition, the increase in the level of SYN1 was detected in cells treated with OA and OA derivatives (Fig. 1E). The protein level of SYP remained unaltered in response to OA, OAA, and OAM treatments (Fig. 1F). These results indicate that, after 72 h treatment, OA (1.5 μM), OAA and OAM (2.5 μM) promote neural differentiation accompanied with expression of synaptic molecules in the similar time window.
We next examined whether OA, OAA and OAM affect neurite outgrowth from rat hippocampal neural progenitor cells. Neurite outgrowth was quantified by measuring the length and number of neurite branches extending from the soma of MAP2+ cells. Quantitative analysis revealed that treatment with OA, OAA or OAM for 72 h significantly increased both the length of neurites (Figs. 2A and 2B) and the number of neurite branches (Fig. 2C). To determine whether OA, OAA and OAM have a long-term effect on neural morphology, we examined the neurite spine density of neurons. Total spine density of the secondary branches of neurites from MAP2+ cells increased significantly following treatment with OA, OAA and OAM for 72 h (Figs. 2D and 2E). Taken together, these results indicate that OA, OAA and OAM promote neurite outgrowth and spine formation.
Fig. 2. OA, OAA and OAM increase neurite length and spine density.
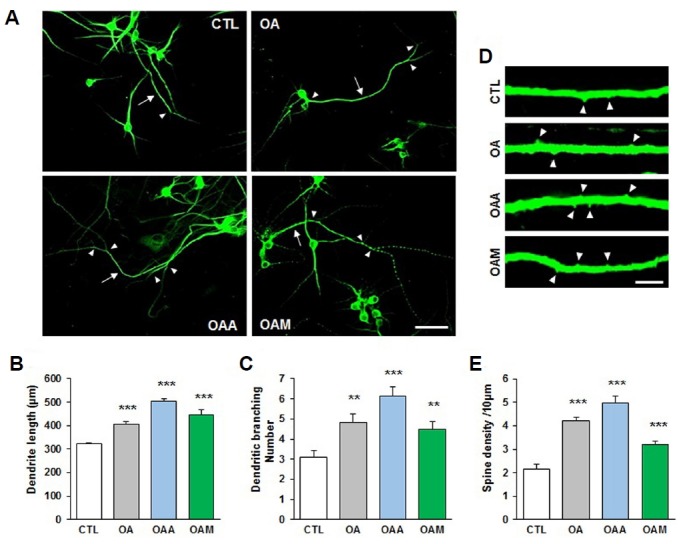
(A) Hippocampal primary neurons were exposed to OA, OAA or OAM for 72 h. The neurite length (arrow) and number of branches (arrowheads) were counted in neurons immunostained for MAP2. Images were captured at a magnification of ×60, using a confocal microscope. Scale bar, 50 μm. (B, C) Treatment with OA, OAA and OAM significantly increased neurite length and number of neurite branches (n = 10–16 neurons/condition, two independent cultures). (D) Representative images are shown of high-magnification Z-stack projections of neurite spine density. Arrowheads indicate the density of neurite spines. Scale bar, 5 μm. (E) The density of neurite spines was significantly increased by treatment with OA, OAA and OAM (n = 15–22 neurons/condition, two independent cultures). Results are mean ± SEM. Student’s t-test *p < 0.05, **p < 0.01, ***p < 0.001 compared to control (CTL).
OA, OAA and OAM induce HDAC5 phosphorylation
HDAC5 has been implicated in neural differentiation both in vitro and in vivo (Huang et al., 2012). Nuclear export of phosphorylated HDAC5 has been demonstrated as a transcriptional regulatory mechanism in mature cells of the brain, including hippocampal neurons (Schneider et al., 2008). To examine the role of HDAC5 in OA-induced signaling in rat hippocampal neurons, we investigated the phosphorylation of HDAC5 Ser259 and Ser498 residues in response to stimu lation with OA, OAA and OAM. The exposure of cultured hippocampal neurons to OA, OAA or OAM induced HDAC5 phosphorylation at Ser259 and Ser498 (Fig. 3A–3C). Previous work has shown that the phosphorylation of HDAC5 is regulated by CaMKII activity in neurons (Chawla et al., 2003) and by PKD in other cell types (Ha et al., 2008). To determine whether HDAC5 phosphorylation is mediated by CaMKII or PKD in hippocampal neurons in response to OA, OAA and OAM, we investigated the phosphorylation of CaMKII and PKD in the presence of OA, OAA and OAM, and showed that OA, OAA and OAM stimulate the phosphorylation of both PKD and CaMKII (Figs. 3A, 3D, 3E).
Fig. 3. OA, OAA and OAM stimulate HDAC5 phosphorylation through PKD-dependent pathways.
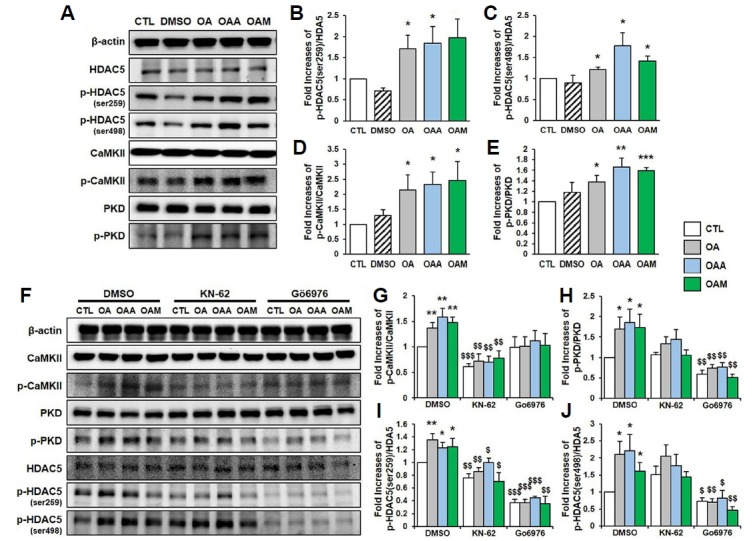
(A) Hippocampal neurons were exposed to OA, OAA or OAM for 6 h. The expression of p-HDAC5 (Ser259), p-HDAC5 (Ser498), p-CaMKII and p-PKD, and the levels of total HDAC5, CaMKII, PKD and β-actin were determined by Western blot analysis. (B–E) Quantification of the activation of p-HDAC5 (Ser259), p-HDAC5 (Ser498), p-CaMKII and p-PKD, and the levels of total HDAC5, CaMKII, and PKD normalized to the corresponding β-actin level (n = 3 independent experiments). (F) Hippocampal neurons were pretreated with DMSO, KN-62 (30 μM) or Gö6976 (1 μM) for 30 min, and then exposed to OA, OAA or OAM for 6 h. The activation of p-HDAC5 (Ser259), p-HDAC5 (Ser498), p-CaMKII and p-PKD, and the levels of total HDAC5, CaMKII, PKD and β-actin in whole cellular extracts were determined by Western blot analysis. (G–J) Quantification of the activation of p-CaMKII, p-PKD, p-HDAC5 (Ser259) and p-HDAC5 (Ser498), and the levels of total CaMKII, PKD and HDAC5 normalized to the corresponding β-actin level (n = 4 independent experiments). Results are mean ± SEM. Student’s t-test *p < 0.05, **p < 0.01, ***p < 0.001 compared to control (CTL). $p < 0.05, $$p < 0.01, $$$p < 0.001 compared to of the group that received DMSO treatment (DMSO).
To determine whether the phosphorylation of HDAC5 in response to OA, OAA and OAM is mediated by CaMKII or PKD in hippocampal neurons, we used the CaMKII inhibitor KN-62 and the PKD-specific inhibitor Gö6976. As expected, KN-62 and Gö6976 completely abolished the phosphorylation of CaMKII and PKD, respectively, induced by OA, OAA and OAM (Figs. 3F–3H). HDAC5 phosphorylation at Ser259 and Ser498 residues induced by OA, OAA or OAM was completely abolished by the addition of Gö6976 (Figs. 3F, 3I, and 3J). HDAC5 phosphorylation at S259 was significantly blocked by the CaMKII inhibitor KN-62 (Figs. 3F and 3I). However, KN-62 had lesser effect on HDAC5 phosphorylation at Ser498 (Figs. 3F and 3J). These results indicate that OA, OAA and OAM induce HDAC5 phosphorylation via a PKD-dependent pathway.
OA, OAA and OAM induce the nuclear export of HDAC5
As HDAC5 is phosphorylated and then exported out of the cell nucleus, we investigated whether the cytoplasmic localization of HDAC5 is induced by treatment with OA, OAA and OAM. We infected hippocampal neurons with plasmids expressing a construct of HDAC5 fused with GFP (GFP-HDAC5-WT), and studied the subcellular localization of HDAC5. GFP-HDAC5-WT is targeted predominantly to the nucleus of hippocampal neurons under basal conditions (Fig. 4A). After 6 h of treatment with OA, OAA, or OAM, however, GFP-HDAC5-WT was translocated to the cytoplasm (Figs. 4A and 4B).
Fig. 4. Subcellular localization of HDAC5 following stimulation with OA, OAA or OAM in rat hippocampal neurons.
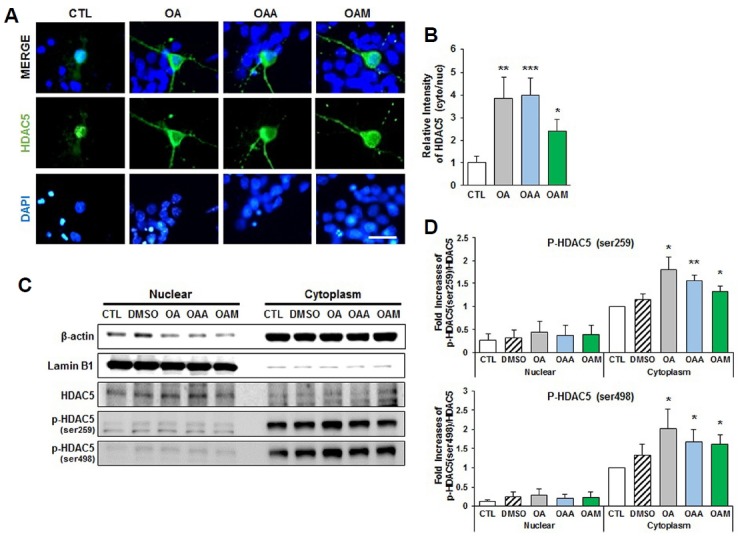
(A, B) Representative fields of GFP fluorescence in hippocampal neurons treated with OA, OAA or OAM, expressing GFP-HDAC5-WT. Cells were counterstained with DAPI (blue). Images were captured at a magnification of ×60, using a fluorescence microscope. Scale bar, 25 μm. (B) The kinetics of nuclear export of HDAC5; a quantitative analysis of the GFP immunofluorescence in the nucleus (nuc) and cytosol (cyt), expressed as a ratio of control (untreated) cells (CTL; n = 8–12 neurons/condition, two independent cultures). (C, D) Hippocampal neurons were exposed to OA, OAA or OAM for 6 h. Accumulation of p-HDAC5 (Ser259) and p-HDAC5 (Ser498) was shown in the cytosol of neurons. Nuclear extracts were normalized to lamin B1; cytoplasm extracts were normalized to β-actin (n = 4 independent experiments). Results are mean ± SEM. Student’s t-test, *p < 0.05, **p < 0.01, ***p < 0.001 compared to control (CTL).
We further analyzed the effects of OA, OAA and OAM on the nuclear and cytoplasmic distribution of endogenous HDAC5 using a biochemical fractionation approach. Hippocampal primary neurons were fractionated into cytoplasmic and nuclear fractions for blotting experiments. Following treatment with OA, OAA, or OAM, we observed a significant increase in p-HDAC5 in the cytoplasmic fraction compared to control (Figs. 4C and 4D). Taken together, these results reveal that OA, OAA and OAM stimulate the phosphorylation of HDAC5 and subsequently the translocation of HDAC5 to the cytoplasm.
OA, OAA and OAM regulate gene expression
Previous studies reported that HDAC5 represses the activities of the transcription factor MEF2 (Lu et al., 2000; Schneider et al., 2008). The nuclear export of HDAC5 induces a shifting of the chromatin state to one that favors histone acetylation (Cho et al., 2013). Therefore, as we have shown that OA, OAA and OAM induce the phosphorylation of HDAC5 and trigger nuclear export, we investigated whether MEF2-dependent gene expression is also increased in the presence of OA, OAA and OAM.
The treatment of rat hippocampal neurons with OA, OAA and OAM enhanced the mRNA expression of Bdnf, Klf6 and Nr4a1 (also known as Nur77), the genes regulated by MEF2 (Flavell et al., 2008; Gao et al., 2010; Salma and McDermott, 2012) (Figs. 5A–5C). In addition, OA, OAA and OAM increase the phosphorylation of HDAC5 (Figs. 5D–5I). Given that OA-induced HDAC5 phosphorylation is mediated by PKD, we examined whether the upregulation of BDNF and KLF6 requires the PKD activity. We found that OA, OAA and OAM increased BDNF and KLF6 protein levels, which was abolished by Gö6976 (Figs. 5J–5L), suggesting the involvement of PKD in the activation of MEF2-dependent transcription.
Fig. 5. OA, OAA and OAM induce MEF2 target gene expression via HDAC5 dissociation in rat hippocampal neurons.
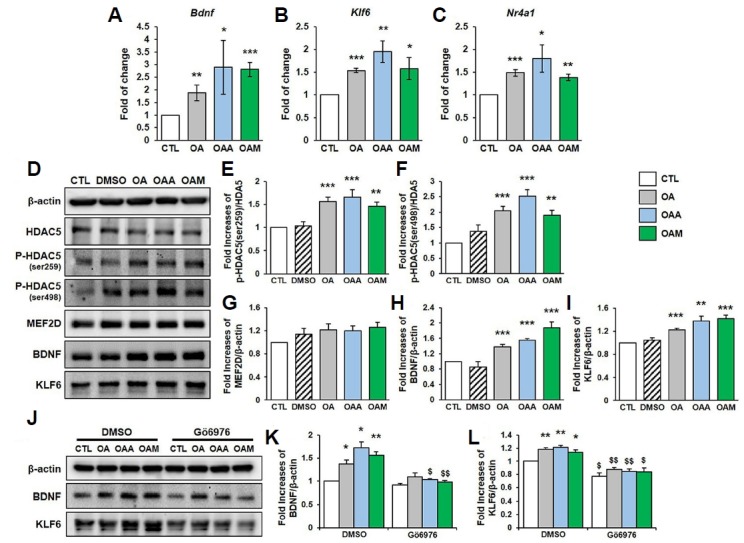
(A–C) The expression of Bdnf, Klf6 and Nr4a1 mRNA levels in hippocampal neurons after treatment with OA, OAA or OAM for 72 h. Results of quantitative PCR were normalized to the level of β-actin and are shown as fold changes relative to control neurons (n = 3 independent cultures). (D–I) Hippocampal neurons were treated with OA, OAA or OAM for 72 h. HDAC5 phosphorylation, and expression of MEF2D, BDNF and KLF6 proteins were analyzed by Western blot (n = 4 independent experiments). (J–L) Hippocampal neurons were pretreated with DMSO or Gö6976 (1 μM) for 30 min, and then exposed to OA, OAA or OAM for 72 h. The expression of BDNF, KLF6 and β-actin in whole cellular extracts were determined by Western blot analysis (n = 3 independent experiments). Results are mean ± SEM. Student’s t-test, *p < 0.05, **p < 0.01, ***p < 0.001 compared to control (CTL). $p < 0.05, $$p < 0.01, $$$p < 0.001 compared to of the group that received DMSO treatment (DMSO).
The MEF2 protein level was not altered by OA, OAA and OAM (Fig. 5G). These results indicate that OA, OAA and OAM induce the phosphorylation of HDAC5 followed by the nuclear export of p-HDAC5, which leads to the derepression of gene expression as a consequence of treatment with OA, OAA and OAM. Taken together, these data demonstrate that OA, OAA and OAM promote the phosphorylation of HDAC5 and the export of HDAC5 from the nucleus, resulting in the transactivation of target genes, possibly via MEF2.
DISCUSSION
OA increases neural differentiation in the hippocampus (Ning et al., 2015), but relatively little is known about the molecular mechanisms underlying this event. In the present study, we used the well-established model system of multipotent neural progenitor cells to provide novel insights into the effects of OA on neural differentiation and the connection between epigenetic regulation and neural gene expression. First, treatment of neural progenitor cells with OA results in the induction of neural differentiation, accompanied by increased expression of synaptic proteins and molecules, and the formation of neurite spines. Second, OA regulates the nuclear export of HDAC5 and thereby represses its activity, resulting in the up-regulation of MEF2 target genes, including Bdnf, Klf6 and Nr4a1. We observed a significant regulation of HDAC5 phosphorylation and gene expression levels in response to OA, OAA and OAM, which suggests that epigenetic regulation plays a crucial role in the actions of OA in neurons.
Our findings reveal that OA regulates the transient nuclear export of HDAC5, and this probably occurs via a molecular mechanism involving PKD-dependent phosphorylation of HDAC5 at two critical sites, S259 and S498. Importantly, our previous findings reveal that the phosphorylation of HDAC5 at S259 and S498 is critical for the ability of ketamine to derepress gene expression and produce behavioral improvement. Therefore, the observation of the coordinated actions of HDAC5 within the hippocampus in response to OA provides new insight into the biochemical and molecular actions of OA.
In the present study, we demonstrate that OA, OAA and OAM upregulate KLF6, a neural survival factor (Salma and McDermott, 2012) and key downstream component of HDAC5, raising the possibility that OA and OA derivatives have a physiological role in neural cell death. We also demonstrated that OA, OAA and OAM upregulate BDNF, also implicated in neural survival (Ghosh et al., 1994; Patterson et al., 1996) as well as in activity-dependent synapse modification that fulfills a crucial role in the refinement of neural circuitry (Flavell et al., 2006; Turrigiano, 2008). Therefore, OA and OA derivatives might have the potential to be used as therapeutic agents for neurological defects that are characterized by neural cell death.
We previously reported that the administration of ketamine activates HDAC5-dependent gene expression in hippocampal neurons (Choi et al., 2015), and ketamine-mediated increases in neurite spine density are dependent on HDAC5 derepression. Consistent with this observation that HDAC5 regulates neurite morphogenesis, OA and OA derivatives increase the neurite spine density and the expression of synapse-related proteins in hippocampal neurons. Clearly, the regulation of HDAC5 activity and downstream target gene expression in response to OA suggest that OA has the ability to influence synaptic function as well as neural survival.
The mechanism by which the OA regulates PKD activation and neuronal differentiation remains to be elucidated. Ursolic acid, a pentacyclic triterpene acid that is structurally similar to OA, induces the influx of calcium through T- and L-type voltage-dependent calcium channels in cardiac muscle cells and stimulates glucose uptake through a cross linking the phosphatidylinositol-4,5-bisphosphate 3-kinase and mitogen-activated protein kinase pathways with Ca2+-CaMKII network in skeletal muscle (Castro et al., 2015). Taking this into account, we consider that OA and its derivatives may similarly induce calcium influx and in turn activate signaling cascade such as PKD-involving pathways to elevate nuclear activity in neurons.
Given that reduced neurite spine formation and abnormal neurite morphology are implicated in mood disorders (Law et al., 2004), the increased neurite spine density and branching in response to OA and OA derivatives suggest that OA compounds might be applicable for treating the structural deficits observed in the pathophysiology of some mood disorders, including depression (DuPont et al., 1996). Previous studies showed that OA reduces chronic unpredictable stress-induced anhedonic and anxiogenic behaviors (Yi et al., 2014), which is consistent with our results, that OA can, in fact, induce neural differentiation. Although OA-induced neural differentiation is observed in our in vitro system, further studies are required to determine if OA mediates this effect and serves as a differentiation factor in vivo.
Taken together, our results suggest that OA, OAA and OAM can enhance the differentiation and synaptic formation of neural progenitor cells, and these compounds might provide neurotrophic support for neurite formation and synaptic connectivity during neural differentiation in the hippocampus. Additional studies are needed to clarify the mechanisms underlying the phosphorylation of HDAC5 following treatment with OA, OAA and OAM.
ACKNOWLEDGEMENTS
This research was supported by a National Research Foundation of Korea (NRF) grant (No. 2016R1A2B2006474) funded by the Ministry of Education, Science and Technology (MEST), Republic of Korea and the research fund of Hanyang University(HY-2017) awarded to HS.
REFERENCES
- Berridge M.V., Herst P.M., Tan A.S. Tetrazolium dyes as tools in cell biology: new insights into their cellular reduction. Biotechnol Annu Rev. 2005;11:127–152. doi: 10.1016/S1387-2656(05)11004-7. [DOI] [PubMed] [Google Scholar]
- Broide R.S., Redwine J.M., Aftahi N., Young W., Bloom F.E., Winrow C.J. Distribution of histone deacetylases 1–11 in the rat brain. J Mol Neurosci. 2007;31:47–58. doi: 10.1007/BF02686117. [DOI] [PubMed] [Google Scholar]
- Castro A.J., Frederico M.J., Cazarolli L.H., Mendes C.P., Bretanha L.C., Schmidt E.C., Bouzon Z.L., de Medeiros Pinto V.A., da Fonte Ramos C., Pizzolatti M.G., et al. The mechanism of action of ursolic acid as insulin secretagogue and insulinomimetic is mediated by cross-talk between calcium and kinases to regulate glucose balance. Biochim Biophys Acta. 2015;1850:51–61. doi: 10.1016/j.bbagen.2014.10.001. [DOI] [PubMed] [Google Scholar]
- Chawla S., Vanhoutte P., Arnold F.J., Huang C.L., Bading H. Neuronal activity-dependent nucleocytoplasmic shuttling of HDAC4 and HDAC5. J Neurochem. 2003;85:151–159. doi: 10.1046/j.1471-4159.2003.01648.x. [DOI] [PubMed] [Google Scholar]
- Cho S.O., Ban J.Y., Kim J.Y., Jeong H.Y., Lee I.S., Song K.S., Bae K., Seong Y.H. Aralia cordata protects against amyloid beta protein (25–35)-induced neurotoxicity in cultured neurons and has antidementia activities in mice. J Pharmacol Sci. 2009;111:22–32. doi: 10.1254/jphs.08271fp. [DOI] [PubMed] [Google Scholar]
- Cho Y., Sloutsky R., Naegle K.M., Cavalli V. Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell. 2013;155:894–908. doi: 10.1016/j.cell.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M., Lee S.H., Wang S.E., Ko S.Y., Song M., Choi J.S., Kim Y.S., Duman R.S., Son H. Ketamine produces antidepressant-like effects through phosphorylation-dependent nuclear export of histone deacetylase 5 (HDAC5) in rats. Proc Natl Acad Sci USA. 2015;112:15755–15760. doi: 10.1073/pnas.1513913112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppede F. The potential of epigenetic therapies in neurodegenerative diseases. Front Genet. 2014;5:220. doi: 10.3389/fgene.2014.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPont R.L., Rice D.P., Miller L.S., Shiraki S.S., Rowland C.R., Harwood H.J. Economic costs of anxiety disorders. Anxiety. 1996;2:167–172. doi: 10.1002/(SICI)1522-7154(1996)2:4<167::AID-ANXI2>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Finsterwald C., Carrard A., Martin J.L. Role of saltinducible kinase 1 in the activation of MEF2-dependent transcription by BDNF. PloS one. 2013;8:e54545. doi: 10.1371/journal.pone.0054545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavell S.W., Cowan C.W., Kim T.K., Greer P.L., Lin Y., Paradis S., Griffith E.C., Hu L.S., Chen C., Greenberg M.E. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science. 2006;311:1008–1012. doi: 10.1126/science.1122511. [DOI] [PubMed] [Google Scholar]
- Flavell S.W., Kim T.K., Gray J.M., Harmin D.A., Hemberg M., Hong E.J., Markenscoff-Papadimitriou E., Bear D.M., Greenberg M.E. Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron. 2008;60:1022–1038. doi: 10.1016/j.neuron.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangwal A. Neuropharmacological effects of triterpenoids. Phytopharmacology. 2013;4:354–372. [Google Scholar]
- Gao J., Wang W.Y., Mao Y.W., Graff J., Guan J.S., Pan L., Mak G., Kim D., Su S.C., Tsai L.H. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature. 2010;466:1105–1109. doi: 10.1038/nature09271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A., Carnahan J., Greenberg M.E. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- Ha C.H., Wang W., Jhun B.S., Wong C., Hausser A., Pfizenmaier K., McKinsey T.A., Olson E.N., Jin Z.G. Protein kinase D-dependent phosphorylation and nuclear export of histone deacetylase 5 mediates vascular endothelial growth factor-induced gene expression and angiogenesis. J Biol Chem. 2008;283:14590–14599. doi: 10.1074/jbc.M800264200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada A., Teng J., Takei Y., Oguchi K., Hirokawa N. MAP2 is required for dendrite elongation, PKA anchoring in dendrites, and proper PKA signal transduction. J Cell Biol. 2002;158:541–549. doi: 10.1083/jcb.200110134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.Y., Liu D.D., Chang H.F., Chen W.F., Hsu H.R., Kuo J.S., Wang M.J. Histone deacetylase inhibition mediates urocortin-induced antiproliferation and neuronal differentiation in neural stem cells. Stem Cells. 2012;30:2760–2773. doi: 10.1002/stem.1226. [DOI] [PubMed] [Google Scholar]
- Law A.J., Weickert C.S., Hyde T.M., Kleinman J.E., Harrison P.J. Reduced spinophilin but not microtubule-associated protein 2 expression in the hippocampal formation in schizophrenia and mood disorders: molecular evidence for a pathology of dendritic spines. Am J Psychiatry. 2004;161:1848–1855. doi: 10.1176/ajp.161.10.1848. [DOI] [PubMed] [Google Scholar]
- Li Y., Ishibashi M., Satake M., Chen X., Oshima Y., Ohizumi Y. Sterol and triterpenoid constituents of Verbena littoralis with NGF-potentiating activity. J Nat Prod. 2003;66:696–698. doi: 10.1021/np020577p. [DOI] [PubMed] [Google Scholar]
- Lu J., McKinsey T.A., Nicol R.L., Olson E.N. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc Natl Acad Sci USA. 2000;97:4070–4075. doi: 10.1073/pnas.080064097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier K., Brehm A. Chromatin regulation: how complex does it get? Epigenetics. 2014;9:1485–1495. doi: 10.4161/15592294.2014.971580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning Y., Huang J., Kalionis B., Bian Q., Dong J., Wu J., Tai X., Xia S., Shen Z. Oleanolic Acid Induces Differentiation of Neural Stem Cells to Neurons: An Involvement of Transcription Factor Nkx-2.5. Stem Cells Int. 2015;2015:672312. doi: 10.1155/2015/672312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson S.L., Abel T., Deuel T.A., Martin K.C., Rose J.C., Kandel E.R. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Pollier J., Goossens A. Oleanolic acid. Phytochemistry. 2012;77:10–15. doi: 10.1016/j.phytochem.2011.12.022. [DOI] [PubMed] [Google Scholar]
- Salma J., McDermott J.C. Suppression of a MEF2-KLF6 survival pathway by PKA signaling promotes apoptosis in embryonic hippocampal neurons. J Neurosci. 2012;32:2790–2803. doi: 10.1523/JNEUROSCI.3609-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider J.W., Gao Z., Li S., Farooqi M., Tang T.S., Bezprozvanny I., Frantz D.E., Hsieh J. Small-molecule activation of neuronal cell fate. Nat Chem Biol. 2008;4:408–410. doi: 10.1038/nchembio.95. [DOI] [PubMed] [Google Scholar]
- Soltani M.H., Pichardo R., Song Z., Sangha N., Camacho F., Satyamoorthy K., Sangueza O.P., Setaluri V. Microtubule-associated protein 2, a marker of neuronal differentiation, induces mitotic defects, inhibits growth of melanoma cells, and predicts metastatic potential of cutaneous melanoma. Am J Pathol. 2005;166:1841–1850. doi: 10.1016/S0002-9440(10)62493-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son H., Banasr M., Choi M., Chae S.Y., Licznerski P., Lee B., Voleti B., Li N., Lepack A., Fournier N.M., et al. Neuritin produces antidepressant actions and blocks the neuronal and behavioral deficits caused by chronic stress. Proc Natl Acad Sci USA. 2012;109:11378–11383. doi: 10.1073/pnas.1201191109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh N., Wang Y., Honda T., Gribble G.W., Dmitrovsky E., Hickey W.F., Maue R.A., Place A.E., Porter D.M., Spinella M.J., et al. A novel synthetic oleanane triterpenoid, 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, with potent differentiating, antiproliferative, and anti-inflammatory activity. Cancer Res. 1999;59:336–341. [PubMed] [Google Scholar]
- Turrigiano G.G. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volmar C.-H., Wahlestedt C. Histone deacetylases (HDACs) and brain function. Neuroepigenetics. 2015;1:20–27. [Google Scholar]
- Yi L.T., Li J., Liu B.B., Luo L., Liu Q., Geng D. BDNF-ERK-CREB signalling mediates the role of miR-132 in the regulation of the effects of oleanolic acid in male mice. J Psychiatry Neurosci. 2014;39:348–359. doi: 10.1503/jpn.130169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin M.C. Inhibitory effects and actions of pentacyclic triterpenes upon glycation. BioMedicine. 2015;5:13. doi: 10.7603/s40681-015-0013-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z., Zhang W., Kone B.C. Histone deacetylases augment cytokine induction of the iNOS gene. J Am Soc Nephrol. 2002;13:2009–2017. doi: 10.1097/01.asn.0000024253.59665.f1. [DOI] [PubMed] [Google Scholar]