

Abstract
Autophagy is a catabolic process by which cells can dispose of damaged content and intracellular microorganisms. Recent evidence implicates autophagy as a crucial repair process necessary to recover from critical illness-induced organ failure. Withholding parenteral nutrition in the acute phase of critical illness activates autophagy and enhances recovery. Several registered drugs have autophagy-stimulating properties, but all lack specificity and none has been investigated in critically ill patients for this purpose.
Keywords: Critical illness, Sepsis, Autophagy, Nutrition, Insulin, Multiple organ dysfunction syndrome
Main text
In critical illness, a variety of stressors may induce cellular damage and lead to organ failure. After treatment of the primary condition leading to intensive care unit (ICU) admission and general ICU care, the majority of patients will recover. However, despite maximal care, approximately 10–30% of patients do not swiftly recover and enter a phase of prolonged critical illness, characterized by a prolonged dependency on vital organ support and associated with a high risk of mortality and long-term debilities [1, 2]. The underlying reasons why certain patients quickly recover whereas others remain ICU-dependent are incompletely understood. Despite the severe organ failure, frank necrosis or apoptosis are uncommon, and in patients surviving this condition, (partial) recovery is possible, even when organs with poor regenerative capacity are involved. This suggests that the cells from critically ill patients dispose of repair mechanisms that allow—at least partially—reversal of the organ dysfunction. Recent evidence implicates macroautophagy, hereafter referred to as autophagy, as a crucial cellular repair process that is essential to recover from critical illness-induced organ failure.
Autophagy, literally meaning “self-eating”, is a catabolic process by which intracellular content is delivered to the lysosome via an intermediate organelle, the autophagosome [1]. Autophagosomes are intracellular vesicles formed out of isolation membranes in the cytoplasm, which elongate around its substrate to finally form a vesicle (Fig. 1). Substrates may be recruited to growing autophagosomes by tagging molecules such as ubiquitin. Mature autophagosomes fuse with lysosomes, after which the engulfed content is degraded and recycled. Autophagy can be activated by starvation, exercise, and a variety of stress signals. Nutrients, growth factors and insulin suppress autophagy. Importantly, autophagy is the only process that is able to remove macromolecular damage, such as damaged organelles and potentially toxic protein aggregates, and intracellular microorganisms. As such, it serves an important homeostatic role, which is illustrated by the severe phenotypes that develop when autophagy is selectively and tissue-specifically knocked out in adult animals. Both insufficient and excessive autophagy have been associated with pathology. Whereas autophagy was originally mainly considered as a negative process, recent evidence has identified autophagy activation as adaptive in many disease states, including critical illness-induced vital organ failure and muscle weakness [1–3].
Fig. 1.
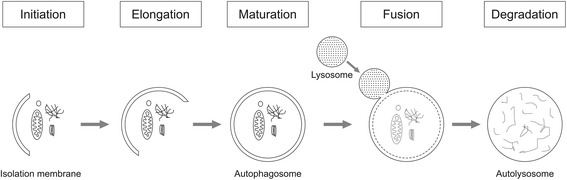
Overview of autophagy. Autophagy initiates with the formation of an isolation membrane (also called phagophore) in the cytoplasm. Isolation membranes elongate to finally form a double-membrane vesicle, the autophagosome. During the elongation process, intracellular cargo (e.g., damaged organelles, protein aggregates, invaded microorganisms) is recruited to the isolation membrane to be engulfed. After maturation of the autophagosome, the vesicle fuses with a lysosome, resulting in formation of an autolysosome, in which the sequestered content is degraded and thereafter recycled to the cytoplasm. The figure is original for this article
Early observational studies attributed sepsis-induced organ damage to the concomitant appearance of autophagosomes. However, without interfering with the process, causality is unclear. Recently, a considerable number of studies have provided clear evidence of potentially insufficient autophagy activation in relation to the degree of cellular stress. A pioneer study identified hallmarks of insufficient autophagy activation in liver and muscle of prolonged critically ill patients, with accumulation of autophagic substrate in the presence of a reduced number of autophagic vacuoles [1]. Similar observations were performed in a critically ill animal model, in which the degree of autophagy activation inversely correlated with the degree of respective organ dysfunction and mortality risk, which supports the functional relevance of activated autophagy [4]. Likewise, in patients with prolonged critical illness, the degree of muscular autophagy inversely correlated with the incidence of ICU-acquired muscle weakness [2]. Moreover, administration of the autophagy inducer rapamycin to burn-injured critically ill rabbits activated autophagy and protected against organ dysfunction and bone loss [4, 5]. Recently, a considerable number of studies have confirmed a protective role of autophagy against various types of organ failure in critically ill animal models. Indeed, in rodents, active autophagy protected against ischemia-reperfusion injury in various organs as well as against sepsis- or endotoxin-induced pulmonary, renal, cardiac, hepatic, neuronal, and immune dysfunction/injury and against sepsis-induced mortality [6–13].
The necessary activation of autophagy in response to severe stress may be affected by artificial feeding and insulin therapy. Human and animal studies have demonstrated significant autophagy suppression by early parenteral nutrition (PN) in critical illness, which coincided with more organ damage and—in patients—a prolonged dependency on vital organ support [2, 14]. Among the different macronutrients, amino acids especially strongly suppressed autophagy, more than glucose or lipids [14]. Although speculative, this may explain why amino acid administration, and not glucose or lipids, statistically explained the harm evoked by early PN in a multicenter clinical study [15]. Although insulin is a known suppressor of autophagy, tissular glucose overload evoked by severe hyperglycemia may also impair autophagy. Hence, lowering blood glucose with insulin may influence autophagy in both directions and the net effect remains unclear. Whereas a study on human samples found a reduction in autophagic vacuoles following this therapy in liver, other hepatic autophagy markers and muscular autophagy were unaffected [1]. An animal study demonstrated improved autophagy in liver and kidney by preventing severe hyperglycemia with insulin [4]. Importantly, in both human and animal studies, early PN was administered and in this context, maintaining normoglycemia with insulin protected against organ damage [1, 4]. Hence, with early PN, the net balance between genesis and removal of cellular damage was in favor of preventing severe hyperglycemia with insulin, even if the net effect on autophagy remains unclear.
Hence, autophagy activation emerges as a potentially important therapeutic target in critical illness. Withholding PN in the acute phase of critical illness was found to stimulate autophagy and improve organ recovery [2]. However, prolonged starvation may come at a price. In an animal study, feeding-induced suppression of autophagy could be overcome by administration of the autophagy inducer rapamycin, which decreased morbidity [4]. However, rapamycin also has potent immune-suppressive effects, which precludes its widespread use in critically ill patients. Likewise, several other registered drugs have been identified as potential autophagy inducers [3]; however, these lack specificity and none has been investigated in critically ill patients for this purpose. Future research should aim at identifying novel, more specific autophagy inducers that are suitable for study in ICU patients.
Acknowledgements
Not applicable.
Funding
Not applicable.
Availability of data and materials
Not applicable.
Abbreviations
- ICU
Intensive care unit
- PN
Parenteral nutrition
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The author declares that he has no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Vanhorebeek I, Gunst J, Derde S, Derese I, Boussemaere M, Guiza F, Martinet W, et al. Insufficient activation of autophagy allows cellular damage to accumulate in critically ill patients. J Clin Endocrinol Metab. 2011;96:E633–645. doi: 10.1210/jc.2010-2563. [DOI] [PubMed] [Google Scholar]
- 2.Hermans G, Casaer MP, Clerckx B, Guiza F, Vanhullebusch T, Derde S, et al. Effect of tolerating macronutrient deficit on the development of intensive-care unit acquired weakness: a subanalysis of the EPaNIC trial. Lancet Respir Med. 2013;1:621–29. doi: 10.1016/S2213-2600(13)70183-8. [DOI] [PubMed] [Google Scholar]
- 3.Levine B, Packer M, Codogno P. Development of autophagy inducers in clinical medicine. J Clin Invest. 2015;125:14–24. doi: 10.1172/JCI73938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gunst J, Derese I, Aertgeerts A, Ververs EJ, Wauters A, Van den Berghe G, et al. Insufficient autophagy contributes to mitochondrial dysfunction, organ failure, and adverse outcome in an animal model of critical illness. Crit Care Med. 2013;41:182–94. doi: 10.1097/CCM.0b013e3182676657. [DOI] [PubMed] [Google Scholar]
- 5.Owen HC, Vanhees I, Gunst J, Van Cromphaut S, Van den Berghe G. Critical illness-induced bone loss is related to deficient autophagy and histone hypomethylation. Intensive Care Med Exp. 2015;3:52. doi: 10.1186/s40635-015-0052-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu S, Hartleben B, Kretz O, Wiech T, Igarashi P, Mizushima N, et al. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy. 2012;8:826–37. doi: 10.4161/auto.19419. [DOI] [PubMed] [Google Scholar]
- 7.Papadakis M, Hadley G, Xilouri M, Hoyte LC, Nagel S, McMenamin MM, et al. Tsc1 (hamartin) confers neuroprotection against ischemia by inducing autophagy. Nat Med. 2013;19:351–57. doi: 10.1038/nm.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lo S, Yuan SS, Hsu C, Cheng YJ, Chang YF, Hsueh HW, et al. Lc3 over-expression improves survival and attenuates lung injury through increasing autophagosomal clearance in septic mice. Ann Surg. 2013;257:352–63. doi: 10.1097/SLA.0b013e318269d0e2. [DOI] [PubMed] [Google Scholar]
- 9.Mei S, Livingston M, Hao J, Li L, Mei C, Dong Z. Autophagy is activated to protect against endotoxic acute kidney injury. Sci Rep. 2016;6:22171. doi: 10.1038/srep22171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsieh CH, Pai PY, Hsueh HW, Yuan SS, Hsieh YC. Complete induction of autophagy is essential for cardioprotection in sepsis. Ann Surg. 2011;253:1190–200. doi: 10.1097/SLA.0b013e318214b67e. [DOI] [PubMed] [Google Scholar]
- 11.Lalazar G, Ilyas G, Malik SA, Liu K, Zhao E, Amir M, et al. Autophagy confers resistance to lipopolysaccharide-induced mouse hepatocyte injury. Am J Physiol Gastrointest Liver Physiol. 2016;311:G377–386. doi: 10.1152/ajpgi.00124.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y, Wang F, Luo Y. Ginsenoside Rg1 protects against sepsis-associated encephalopathy through beclin 1-independent autophagy in mice. J Surg Res. 2017;207:181–89. doi: 10.1016/j.jss.2016.08.080. [DOI] [PubMed] [Google Scholar]
- 13.Oami T, Watanabe E, Hatano M, Sunahara S, Fujimura L, Sakamoto A, et al. Suppression of T cell autophagy results in decreased viability and function of T cells through accelerated apoptosis in a murine sepsis model. Crit Care Med. 2017;45:e77–85. doi: 10.1097/CCM.0000000000002016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Derde S, Vanhorebeek I, Guiza F, Derese I, Gunst J, Fahrenkrog B, et al. Early parenteral nutrition evokes a phenotype of autophagy deficiency in liver and skeletal muscle of critically ill rabbits. Endocrinology. 2012;153:2267–76. doi: 10.1210/en.2011-2068. [DOI] [PubMed] [Google Scholar]
- 15.Vanhorebeek I, Verbruggen S, Casaer MP, Gunst J, Wouters PJ, Hanot J, et al. Effect of early supplemental parenteral nutrition in the paediatric ICU: a preplanned observational study of post-randomization treatments in the PEPaNIC trial. Lancet Respir Med. 2017;5:475–83. doi: 10.1016/S2213-2600(17)30186-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.