

TO THE EDITOR
Ichthyosis with Confetti (IWC) is an autosomal dominant disorder of keratinization that is exceedingly rare, with approximately 40 cases reported. Although patients generally demonstrate ichthyosiform erythroderma at birth, the disorder is defined by the hundreds of confetti-like white spots that appear in childhood and grow in size and number over time (Choate and Milstone, 2015; Guerra et al., 2015). The histopathology of the red skin surrounding the macules shows perinuclear vacuolization, loss of the granular layer, and parakeratotic hyperkeratosis, whereas the white, revertant macules are histologically normal (Choate and Milstone, 2015).
The mutations identified in IWC to date arise de novo and cause frameshift deletions affecting the carboxyl tail of keratin 10 (K10) or keratin 1 (K1), causing Type I IWC (IWC-I) and Type II IWC (IWC-II), respectively (Choate et al., 2010; Choate et al., 2015). Prior investigation of 7 independent IWC-I probands identified distinct mutations causing entry into the same aberrant reading frame invariably replacing the endogenous glycine-rich tail of K10 with a polyarginine sequence (Choate et al., 2010; Guerra et al., 2015). In IWC-II, the K1 glycine tail is maintained, but the final 22 residues of the end domain are replaced with a novel 30 amino acid, non-repeating sequence (Choate et al., 2015).
Immunohistochemistry of K10 in IWC-I shows mislocalized K10 in aggregates within the nucleolus, with corresponding decrease in cytosolic intensity; K1 in IWC-II mislocalizes to the nucleus, along with perinuclear collapse of the cytokeratin network (Choate et al., 2010; Choate et al., 2015). In contrast, the white macules in both subtypes not only demonstrate correct cytosolic localization of their respective keratins, but also represent independent copy-neutral loss of heterozygosity (CN-LOH) events, in which the heterozygous mutant haplotype is lost without aberrations in chromosomal copy number. Via SNP genotyping, the LOH track was found to span the proximal q arm of chromosome 17 (IWC-I) or chromosome 12 (IWC-II) to the telomere, consistent with genetic reversion via mitotic recombination, a DNA break-induced event that is otherwise rare on a per cell basis (Choate et al., 2010; Choate et al., 2015; O’Keefe et al., 2010).
Given that nucleolar K10 is unique to IWC and all IWC-I patients invariably express poly-arginine K10, it was hypothesized that the mislocalization results from gain of the arginine-rich motif, as many RNA-binding proteins utilize their arginine-rich motif to interact with the phosphate backbone of RNA; mutant K10 would similarly associate with and aggregate within the ribosomal RNA-rich environment of the nucleolus (Choate et al., 2010; Draper, 1999). However, direct interaction between mutant K10 and ribosome components has not yet been observed, and the role of the poly-arginine tail in nucleolar localization and pathobiology of IWC remains unknown.
Here, we identify a 28-year-old Caucasian male with unaffected parents, who was noted at birth to have mild erythema, widespread scale, and mild palmoplantar keratoderma (IWC100). Other than mild ectropion, no dysmorphic features including nail deformities, nipple hypoplasia, or malformation of the auricle were noted, and no evidence of developmental delay was found (Hendrix et al., 1997). Thicker hyperkeratosis was noted over the extensor surfaces, axillae, and antecubital areas; fine scale covered the entire body including the scalp (Figure S1 online). Scales were fine, white, and loosely adherent in some spots, whereas they were larger and plate-like on the dorsal forearms and shins. Confetti-like white spots first appeared in his early twenties, but have remained limited to the forearms (Figure 1a, b) and are less than 5mm in diameter. Prior to the development of the white macules, FISH was performed for genetic testing for X-linked ichthyosis, but no STS deletion was detected. Histopathology of the affected skin shared many histological features with IWC-I, including acanthosis, parakeratosis, loss of the granular layer, and perinuclear vacuolization (Figure 1c). In contrast, the white spots showed normal histology (Figure 1d).
Figure 1. Clinical features and histopathology.
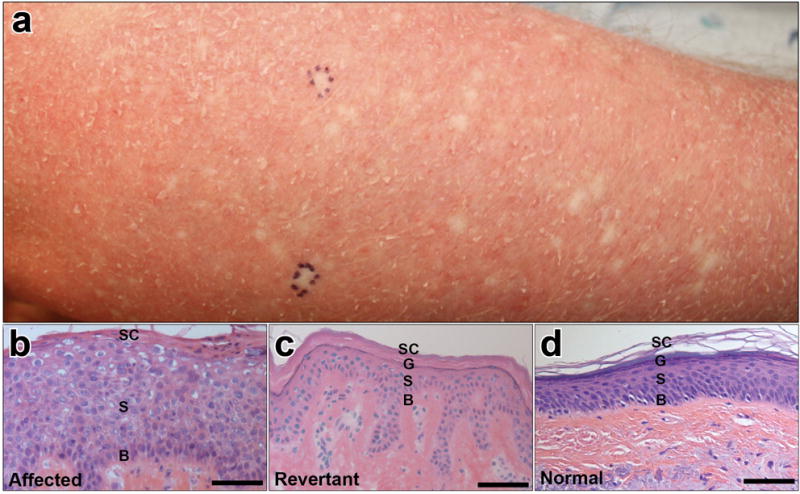
(a) Right forearm demonstrating marked erythema, fine white scale, and numerous small white spots, all less than 5mm in size. Pen markings designate 2 of 3 sites at which biopsies were performed. (b, c, d) 20X histology of red affected skin, revertant skin, and normal control, respectively, showing the basal layer (B), spinous layer (S), granular layer (G) where applicable, and stratum corneum (SC). The affected epidermis demonstrates acanthosis, perinuclear vacuolization, parakeratosis, and loss of the granular layer. Revertant skin shows normal histology. Scale bar: 150μm.
Given these findings, KRT10 was considered a candidate gene, and direct Sanger sequencing of blood DNA revealed a de novo c.1373G deletion in exon 6, which replaces the normal glycine-serine rich tail of K10 with a mutant polyalanine motif and extends the end domain by an additional 19 amino acids (Figure 2a and 2b). Most IWC KRT10 mutations reported thus far lead to a mutant polyarginine tail and this is the second report of a KRT10 mutation encoding a mutant polyalanine tail (Hotz et al., 2015), though the prior report neither investigated localization of the mutant protein nor mechanism of reversion.
Figure 2. A novel KRT10 mutation encoding a polyalanine motif causes IWC with white spots resulting from LOH.
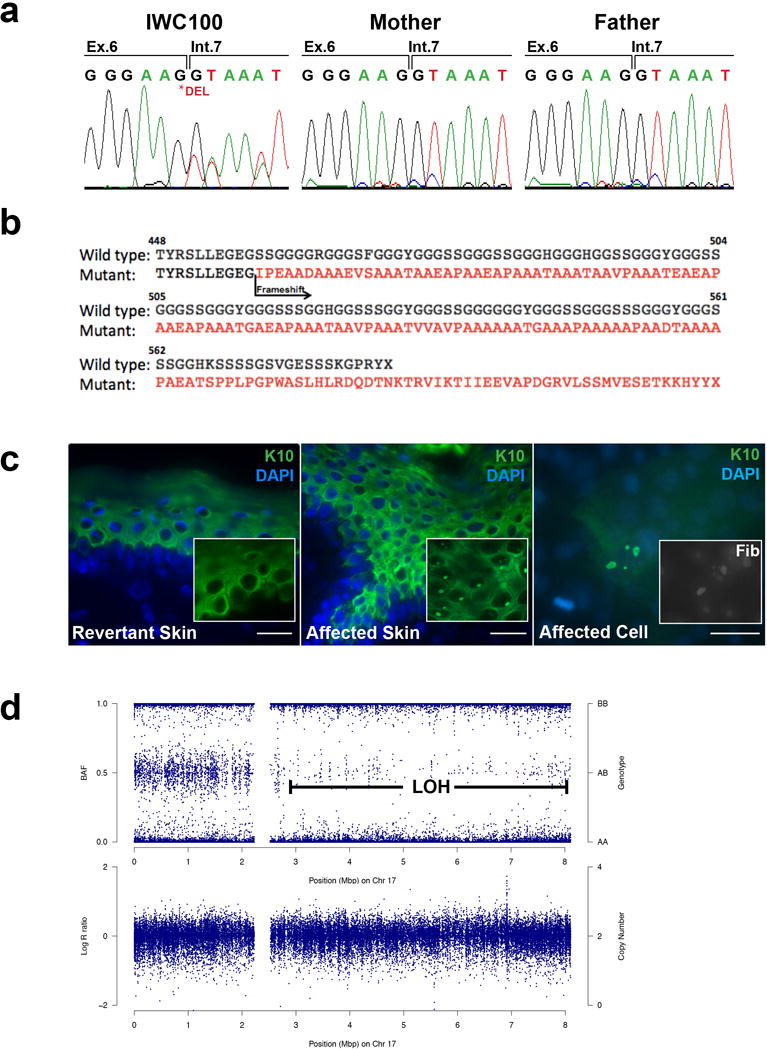
(a) Sanger sequencing of KRT10 in IWC100 identified a de novo c.1373Gdel mutation affecting the last base of exon 6. (b) This mutation causes entry into a mutant reading frame that abolishes the endogenous glycine-rich tail domain of K10 with an alanine rich motif that extends the carboxyl terminus by 19 additional amino acids. (c) Immunohistochemistry of KRT10 demonstrates nucleolar aggregates only in the affected (middle panel), but not revertant (left panel), skin. A higher power (63X) image is included as insert. Primary keratinocytes cultured from a biopsy also demonstrate nucleolar mislocalization of KRT10 that co-stain with fibrillarin, a nucleolar marker (right panel and insert). Scale bars = 100um. (d) LOH mapping demonstrates heterozygous genotype from 17pter to the proximal portion of the q arm, but converts to homozygosity downstream to the telomere without change in the logR ratio, indicating copy-neutral LOH.
To assess mutation pathogenesis, we first performed immunolocalization in skin from this subject, finding overall diminution of suprabasal K10 staining with evidence of filament network collapse and focal aggregates within the nuclei (Figure 2c). These findings were not identified in revertant or normal control skin and are similar to prior observations in polyarginine K10 mutant skin (Choate et al., 2010). Co-staining with the nucleolar marker fibrillarin revealed that K10 aggregates are within the nucleolus. Moreover, calcium-differentiated primary keratinocytes isolated from biopsies of affected skin demonstrated nucleolar aggregates that also co-stained with fibrillarin (Figure 2c). Immunolocalization of K1, the binding partner of K10, also demonstrated nuclear mislocalization, consistent with prior reports (Figure S2 online) (Choate et al., 2010).
To determine the genetic mechanism underlying the revertant mosaicism in this patient, we performed laser capture microdissection (LCM) of 3 individual white spots for isolation of DNA and SNP genotyping (Supplementary Materials and Methods and Figure S3 online). Like IWC-I patients with polyarginine K10, each revertant spot was found to harbor CN-LOH in the proximal q arm of chromosome 17 extending to the telomere, consistent with reversion via mitotic recombination (Figure 2d and Figure S3 online) (Choate et al., 2010; O’Keefe et al., 2010). For all 3 revertant spots, the region of crossover was estimated to fall between SNPs rs6505079 (chr17:28391158, GRCh38/hg38) and rs8078229 (chr17:28500820, GRCh38/hg38), based on binning by 500 SNPs and identifying the point of deviation from a heterozygous b-allele frequency (Figure S4 and Supplementary Materials and Methods online).
Polyalanine mutations have been previously reported in the V2 domain of K1 in patients with striate palmoplantar keratoderma (PPK) or ichthyosis hystrix, Curth-Macklin, though genetic reversion has not been found in either disorder (Richardson et al., 2006; Sprecher et al., 2001; Whittock et al., 2002). Polyalanine expansions are also associated with neurological and developmental disorders, including holoprosencephaly, synpolydactyly, and muscular dystrophy (Amiel et al., 2004; Moumne et al., 2008). Many of these polyalanine expansions result in intranuclear aggregation of their respective proteins, with longer tracts demonstrating more aggregates and more severe symptoms (Moumne et al., 2008). Mutations in PABPN1 and ARX encoding polyalanine motifs cause oculopharyngeal muscular dystrophy and Partington syndrome, respectively; in each, nuclear aggregates were found to promote cellular stress and death (Calado et al., 2000; Nasrallah et al., 2004). Polyalanine PABPN1 was further shown to sequester polyA mRNA by binding with high affinity to nascent poly(A) tails, potentially acting as a nuclear RNA trap to prevent proper cytoplasmic transcription of key proteins (Calado et al., 2000).
Although the mechanisms of genetic reversion in IWC are yet unknown, IWC is distinguished from other keratinopathies by keratin mislocalization to the nucleus. All prior reports of IWC-I have resulted in replacement of all or a portion of the K10 endogenous tail domain with a polyarginine motif which was thought relevant to reversion via mitotic recombination. Our discovery that IWC due to polyalanine frameshift mutation also leads to nuclear localization and genetic reversion via mitotic recombination, suggests the replacement of the K10 endogenous tail domain may be central not only to disease pathogenesis but also to genetic reversion via mitotic recombination.
Supplementary Material
Acknowledgments
We would like to thank Jonathan Levinsohn for plotting SNP genotyping data, and Jing Zhou and Rong-Hua Hu for technical assistance. This study was supported by a grant from the National Institute of Arthritis and Musculoskeletal and Skin Diseases/NIH (5K08AR056305 and 5R01AR062111) to K.A.C., and a Clinical Scientist Development Award from the Doris Duke Charitable Foundation to K.A.C. Y.H.L. was supported by the Medical Scientist Training Program at Yale University (NIH NIGMS GM007205) and is a recipient of the Clinical Mentorship Award from the Doris Duke Charitable Foundation.
Abbreviations
- IWC
ichthyosis with confetti
- K10
keratin 10
- K1
keratin 1
- CN-LOH
copy-neutral loss of heterozygosity
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest.
References
- Amiel J, Trochet D, Clement-Ziza M, Munnich A, Lyonnet S. Polyalanine expansions in human. Human molecular genetics. 2004;13(Spec No 2):R235–43. doi: 10.1093/hmg/ddh251. [DOI] [PubMed] [Google Scholar]
- Calado A, Tome FM, Brais B, Rouleau GA, Kuhn U, Wahle E, et al. Nuclear inclusions in oculopharyngeal muscular dystrophy consist of poly(A) binding protein 2 aggregates which sequester poly(A) RNA. Human molecular genetics. 2000;9:2321–8. doi: 10.1093/oxfordjournals.hmg.a018924. [DOI] [PubMed] [Google Scholar]
- Choate KA, Lu Y, Zhou J, Choi M, Elias PM, Farhi A, et al. Mitotic recombination in patients with ichthyosis causes reversion of dominant mutations in KRT10. Science. 2010;330:94–7. doi: 10.1126/science.1192280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choate KA, Lu Y, Zhou J, Elias PM, Zaidi S, Paller AS, et al. Frequent somatic reversion of KRT1 mutations in ichthyosis with confetti. The Journal of clinical investigation. 2015;125:1703–7. doi: 10.1172/JCI64415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choate KA, Milstone LM. Phenotypic expansion in ichthyosis with confetti. JAMA dermatology. 2015;151:15–6. doi: 10.1001/jamadermatol.2014.2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper DE. Themes in RNA-protein recognition. Journal of molecular biology. 1999;293:255–70. doi: 10.1006/jmbi.1999.2991. [DOI] [PubMed] [Google Scholar]
- Guerra L, Diociaiuti A, El Hachem M, Castiglia D, Zambruno G. Ichthyosis with confetti: clinics, molecular genetics and management. Orphanet journal of rare diseases. 2015;10:115. doi: 10.1186/s13023-015-0336-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix JD, Jr, Patterson JW, Greer KE. Skin cancer associated with ichthyosis: the MAUIE syndrome. Journal of the American Academy of Dermatology. 1997;37:1000–2. doi: 10.1016/s0190-9622(97)70084-7. [DOI] [PubMed] [Google Scholar]
- Hotz A, Oji V, Bourrat E, Jonca N, Mazereeuw-Hautier J, Betz RC, et al. Expanding the Clinical and Genetic Spectrum of KRT1, KRT2 and KRT10 Mutations in Keratinopathic Ichthyosis. Acta dermato-venereologica. 2015 doi: 10.2340/00015555-2299. [DOI] [PubMed] [Google Scholar]
- Moumne L, Dipietromaria A, Batista F, Kocer A, Fellous M, Pailhoux E, et al. Differential aggregation and functional impairment induced by polyalanine expansions in FOXL2, a transcription factor involved in cranio-facial and ovarian development. Human molecular genetics. 2008;17:1010–9. doi: 10.1093/hmg/ddm373. [DOI] [PubMed] [Google Scholar]
- Nasrallah IM, Minarcik JC, Golden JA. A polyalanine tract expansion in Arx forms intranuclear inclusions and results in increased cell death. The Journal of cell biology. 2004;167:411–6. doi: 10.1083/jcb.200408091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Keefe C, McDevitt MA, Maciejewski JP. Copy neutral loss of heterozygosity: a novel chromosomal lesion in myeloid malignancies. Blood. 2010;115:2731–9. doi: 10.1182/blood-2009-10-201848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson ES, Lee JB, Hyde PH, Richard G. A novel mutation and large size polymorphism affecting the V2 domain of keratin 1 in an African-American family with severe, diffuse palmoplantar keratoderma of the ichthyosis hystrix Curth-Macklin type. The Journal of investigative dermatology. 2006;126:79–84. doi: 10.1038/sj.jid.5700025. [DOI] [PubMed] [Google Scholar]
- Sprecher E, Ishida-Yamamoto A, Becker OM, Marekov L, Miller CJ, Steinert PM, et al. Evidence for novel functions of the keratin tail emerging from a mutation causing ichthyosis hystrix. The Journal of investigative dermatology. 2001;116:511–9. doi: 10.1046/j.1523-1747.2001.01292.x. [DOI] [PubMed] [Google Scholar]
- Whittock NV, Smith FJ, Wan H, Mallipeddi R, Griffiths WA, Dopping-Hepenstal P, et al. Frameshift mutation in the V2 domain of human keratin 1 results in striate palmoplantar keratoderma. The Journal of investigative dermatology. 2002;118:838–44. doi: 10.1046/j.1523-1747.2002.01750.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.