

Abstract
Background
Clostridium difficile is an anaerobic Gram-positive spore-forming gut pathogen that causes antibiotic-associated diarrhea worldwide. A small number of C. difficile strains express the binary toxin (CDT), which is generally found in C. difficile 027 (ST1) and/or 078 (ST11) in clinic. However, we isolated a binary toxin-positive non-027, non-078 C. difficile LC693 that is associated with severe diarrhea in China. The genotype of this strain was determined as ST201. To understand the pathogenesis-basis of C. difficile ST201, the strain LC693 was chosen for whole genome sequencing, and its genome sequence was analyzed together with the other two ST201 strains VL-0104 and VL-0391 and compared to the epidemic 027/ST1 and 078/ST11 strains.
Results
The project finally generated an estimated genome size of approximately 4.07 Mbp for strain LC693. Genome size of the three ST201 strains ranged from 4.07 to 4.16 Mb, with an average GC content between 28.5 and 28.9%. Phylogenetic analysis demonstrated that the ST201 strains belonged to clade 3. The ST201 genomes contained more than 40 antibiotic resistance genes and 15 of them were predicted to be associated with vancomycin-resistance. The ST201 strains contained a larger PaLoc with a Tn6218 element inserted than the 027/ST1 and 078/ST11 strains, and encoded a truncated TcdC. In addition, the ST201 strains contained intact binary toxin coding and regulation genes which are highly homologous to the 027/ST1 strain. Genome comparison of the ST201 strains with the epidemic 027 and 078 strain identified 641 genes specific for C. difficile ST201, and a number of them were predicted as fitness and virulence associated genes. The presence of those genes also contributes to the pathogenesis of the ST201 strains.
Conclusions
In this study, the genomic characterization of three binary toxin-positive C. difficile ST201 strains in clade 3 was discussed and compared to the genomes of the epidemic 027 and the 078 strains. Our analysis identified a number fitness and virulence associated genes/loci in the ST201 genomes that contribute to the pathogenesis of C. difficile ST201.
Electronic supplementary material
The online version of this article (doi:10.1186/s13099-017-0191-z) contains supplementary material, which is available to authorized users.
Keywords: Clostridium difficile, ST201, Binary toxin-positive, Whole genome sequencing, Comparative genomic analysis
Background
Clostridium difficile infection (CDI) causes huge morbidities and mortalities, as well as great economical burdens throughout the world especially in Europe and North America [1, 2]. Clinical manifestations of CDI range from asymptomatic carriage, to mild or moderate diarrhea, to fulminant colitis [3]. The causative agent of CDI, C. difficile is an anaerobic Gram-positive, spore-forming, toxin-producing bacillus that generally colonizes the large intestine of humans and animals [4]. Six distinct phylogenetic clades (clades 1, 2, 3, 4, 5, and C–I) are determined within C. difficile, and representatives from most clades are associated with CDI in humans [5]. Prior to 2003, the emergence and prevalence of an epidemic C. difficile 027/ST1 with high-level fluoroquinolone resistance in clade 2 and efficient sporulation increases the severity and the harmfulness of CDI [4]. In addition to 027, other recently emerging ribotypes include 001, 017, and 078 [6], and the 078/ST11 strains appear to share the same genetic virulence characteristics as 027 and cause severe disease at a similar rate, but has also been associated with community-acquired infection [7, 8].
Toxin expression is considered as the key contribution factor to the development of CDI [9]. Two main toxins produced by C. difficile are TcdA and TcdB, which are generally encoded on a 19.6-kbp pathogenicity locus (PaLoc) [10, 11]. PaLoc also contains another three genes tcdC, tcdE, and tcdR implicated in regulating the expression of the toxins. Besides TcdA and TcdB, approximately 20% of C. difficle strains also express the binary toxin (CDT) that is encoded on a locus (CdtLoc) physically separated from the PaLoc [5, 12]. Although the detailed role of CDT in the development of human disease is not well understood, previous data have found that the patients infected with C. difficile producing CDT had higher fatality rate (approximately 60%) than those infected with CDT-deficient strains [13]. In clinic, the binary toxin-positive strains are generally 027/ST1 or 078/ST11, and both of them were rarely reported in China [14]. However, we isolated a binary toxin-positive C. difficile designated strain LC693 from the fecal sample of a patient with severe diarrhea in China, and the genotype of this strain was neither 027/ST1 nor 078/ST11 but determined as ST201 [14]. To understand the pathogenesis basis of this novel isolate, the strain was then chosen for whole genome sequencing. Comparative genomic analysis of the ST201 strains with the epidemic 027/ST1 strain R20291 and 078/ST11 strain M120 was performed to figure out fitness and virulence associated genes.
Methods
Bacterial strains
Clostridium difficile ST201 strain LC693 was isolated from the stool specimens from a 65-year-old man with fever, headache, diarrhea, and impaired consciousness. Detailed descriptions of the disease history and clinical diagnose of this man were noted in our previous report [14]. The isolate was determined to be positive for toxin A, toxin B, and binary toxin via PCR assay [15]. In addition to LC693, there are another two ST201 clinical strains whose whole genome sequences are publically available in GenBank: strain VL-0391 (ST201; clinical isolate, recovered date not available, Canada, GenBank Accession No. FALK01000000) and VL-0104 (ST201; clinical isolate, recovered date not available, Canada, GenBank Accession No. FAAJ01000000) [16].
Genome sequencing, assembly, and annotation
Prior to genomic DNA isolation, a single colony of the strain LC693 was selected from C. difficile agar (Sigma, St. Louis, USA) and inoculated in BHIS medium (Brain–heart infusion broth with 10% (w/v) l-cysteine) incubating under an anaerobic atmosphere at 37 °C for 12–24 h. Then the genomic DNA was extracted using QIAGEN Genomic-tip 500/G (QIAGEN, Hilden, Germany) following the manufactory instructions. Total DNA obtained was subjected to quality control by agarose gel electrophoresis and quantified by Qubit (Thermo Fisher Scientific, Waltham, USA). The genome of C. difficile L693 was sequenced with massively parallel sequencing (MPS) Illumina technology. A paired-end library with an insert size of 419 bp was sequenced using an Illumina MiSeq by PE300 strategy. Library construction and sequencing were performed at the Beijing Novogene Bioinformatics Technology Co., Ltd (Beijing, China). Quality control of both paired-end and mate-pair reads were performed using in-house program. After this step, Illumina PCR adapter reads and low quality reads were filtered. The filtered reads were assembled by SOAPdenovo [17, 18] to generate contigs. Contigs were then ordered and oriented by mapping them against the reference C. difficile 630 genome (GenBank Accession No. NC_009089) using Mauve [19, 20]. Ordered matching contigs were pasted together into a pseudochromosome using a contig linker NNNNNCATTCCATTCATTAATTAATTAATGAATGAATGNNNNN, and nonmatching contigs were tacked on the end in random order, as previous studies did [21, 22]. The LC693 pseudochromosome was then annotated via RAST Server program [23]. Predicted proteins were assigned into the COG database for functional classification [24]. This whole genome shotgun project has been deposited at DDBJ/ENA/GenBank under the Accession NCXL00000000. The version described in this paper is version NCXL01000000. Because there is no annotation information for the genome sequences of strain VL-0391 and VL-0104, therefore, their genome sequences were handled using the same strategy mentioned above.
Sequence analysis and comparative genomics
Prophages in the genome were predicted by PHAST [25]. Antibiotic resistance-associated genes and virulence-associated genes were determined by performing BLAST analysis of the genome sequence against the antibiotic resistance genes database (ARDB) [26] and the virulence factor database (VFDB) [27], respectively. For comparative analysis, genome sequences of C. difficile strains R20291 (027/ST1, recent epidemic and hypervirulent, clade 2) and M120 (078/ST11, hypervirulent, clade 5) as well as their annotations were retrieved from GenBank under Accession Numbers FN545816 and NC_017174, respectively. Sequence comparisons were performed using either BRIG software [28], progressive-Mauve procedure [29], or Easyfig software [30]. Single nucleotide polymorphisms (SNPs) between C. difficile genomes were also exported via progressive-Mauve [29]. The coding effects of SNPs were analyzed using a local Perl command reported before [31]. Orthologous proteins were differentiated via BLUSTCLUST (version 2.2.24) for amino acids with the identity ≥90% plus alignment coverage ≥90% and an e-value of 1e-6 as cut-off. Phylogenetic tree was constructed and graphically presented by MEGA 7.0 [32] based on the sequences of seven conserved house-keeping genes adk, atpA, dx, glyA, recA, sodA, and tpi, using neighbor-joining algorithm with 1000 bootstrapping.
Results
Phylogeny
Phylogenetic analysis based on conserved genes across the C. difficile genomes showed that the five C. difficile clinical isolates discussed in this study belonged to three different clades (Fig. 1). All ST201 strains were members of clade 3, while the epidemic 027/ST1 strain R20291 and 078/ST11 strain M120 belonged to clade 2 and clade 5, respectively. More interestingly, all 027/ST1 clinical strains were concentrated in clade 2 and the 078/ST11 strains were included in clade 5 (Fig. 1).
Fig. 1.
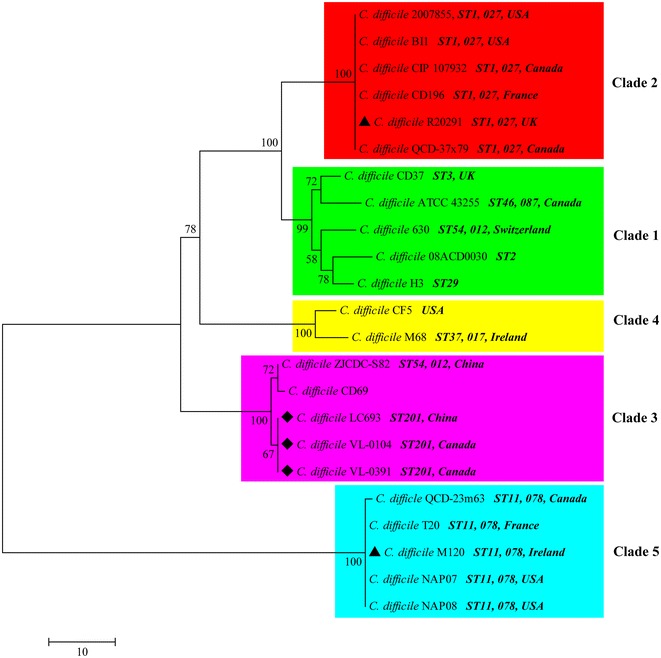
Evolutionary relationships of Clostridium difficile clinical strains. The evolutionary history was inferred using the neighbor-joining method. The optimal tree with the sum of branch length = 182.92187500 is shown. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the number of differences method and are in the units of the number of base differences per sequence. The analysis involved 23 nucleotide sequences. All positions containing gaps and missing data were eliminated. There were a total of 7050 positions in the final dataset. Evolutionary analyses were conducted in MEGA7
Overview of the C. difficile ST201 genomes
Whole genome sequencing strategy on C. difficile strain LC693 yielded a total of 1,413,333 reads with 106-fold coverage (Q20 98.43%, Q30 94.48%). Those reads were then used to the draft assemble, generating 146 contigs larger than 500 bp, of which the largest one was 150,334 bp in length. The contigs were then mapped to C. difficile 630 genome sequence to generate an estimated genome size of approximately 4.07 Mbp. This size was quite similar to another ST201 strain VL-0104, but was approximately 8.8 kb smaller than strain VL-0391 (Table 1). Genome sizes of the ST201 strains were located between the genome of the 078/ST11 strain M120 and the 027/ST1 strain R20291. The average GC contents of the ST201 genome sequences were also near, between 28.5 and 28.9%. Those contents were also similar to the 027/ST1 and 078/ST11 genome sequences. No plasmids were identified in the genome sequences discussed in this study (Table 1). According to annotation using RAST Server, the ST201 genomes carried 3921–3956 predicted open reading frames (ORFs), which corresponded to 3868–3833 putative coding DNA sequences (CDSs), 69–79 tRNAs and 9–41 rRNAs (Table 1).
Table 1.
General features of the C. difficile genomes
| Strain | LC693 | VL-0104 | VL-0391 | R20291 | M120 |
|---|---|---|---|---|---|
| Places of isolation | China | Canada | Canada | UK | UK |
| Ribotype | – | – | – | 027 | 078 |
| Sequence type | ST201 | ST201 | ST201 | ST1 | ST11 |
| Toxin profile | A + B + CDT+ | A + B + CDT+ | A + B + CDT+ | A + B + CDT+ | A + B + CDT+ |
| Genome completion | Draft | Draft | Draft | Complete | Complete |
| Genome size (bp) | 4,073,021 | 4,068,388 | 4,160,703 | 4,191,339 | 4,047,729 |
| GC% | 28.5 | 28.7 | 28.9 | 28.6 | 28.7 |
| Predicted CDSs | 3868 | 3833 | 3832 | 3508 | 3490 |
| Predicted tRNAs | 79 | 77 | 69 | 65 | 86 |
| Predicted rRNAs | 9 | 11 | 41 | 28 | 32 |
| Plasmid | 0 | 0 | 0 | 0 | 0 |
| Prophages | 7 (3a) | 7 (2a) | 8 (3a) | 4 (2a) | 4 (1a) |
aIndicates the number of intact prophages
Antibiotic resistance associated genes
The antibiotic resistance proteins were figured out by performing BLAST analysis of the CDSs predicted in the ST201 genomes against the ARDB database using a percent identity over 40% and an E value of 10−4. The prediction identified 40 (LC693 and VL-0104) to 41 (VL-0391) putative antibiotic resistance associated genes within the ST201 genomes (Table 2). Based on their functional predictions, a total of 15 genes conferred vancomycin-resistance to the three ST201 strains (nine mediated vancomycin-resistance only and another six mediated both vancomycin- and teicoplanin-resistance); 11 (LC693 and VL-0104) or 12 genes (VL-0391) mediated macrolide-resistance; the rest conferred resistance to other antibiotics to the ST201 strains: bacitracin (7 genes), streptogramin A (4 genes), deoxycholate (1 gene), fosfomycin (1 gene), tetracycline (1 gene) and fluoroquinolone (1 gene). It is worthy of note that broth microdilution test showed that the minimum inhibitory concentration of vancomycin to strain LC693 was 4 μg/ml. This result suggests that strain LC693 is resistant to vancomycin, according to EUCAST breakpoint (http://www.eucast.org/clinical_breakpoints/). Interestingly, all antibiotic resistance genes identified LC693 were homologous to those predicted in VL-0104 genome, and 39 of them were also homologous to those determined in VL-0391, with the exception of a vancomycin-resistance-associated gene (Table 2). In addition, most of the antibiotic resistance genes determined in the ST201 genomes were also found in the ST1 and ST11 genomes (Table 2).
Table 2.
Antibiotic resistance associated proteins predicted in the ST201 genomes
| Locus in LC693 | Locus in VL-0104 | Locus in VL-0391 | Length (aa) | Description | Resistance | Presence in R20291 | Presence in M120 |
|---|---|---|---|---|---|---|---|
| 0042 | 2926 | 1001 | 196 | Virginiamycin A acetyltransferase, which can inactivate the target drug | Streptogramin_a | + | + |
| 0115 | 2999 | 1075 | 197 | Virginiamycin A acetyltransferase, which can inactivate the target drug | Streptogramin_a | + | − |
| 0120 | 3004 | 1080 | 241 | VanA type vancomycin resistance operon genes, which can synthesize peptidoglycan with modified C-terminal d-Ala-d-Ala to d-alanine-d-lactate | Teicoplanin; vancomycin | + | + |
| 0267 | 3195 | 1228 | 65 | Major facilitator superfamily transporter. Multidrug resistance efflux pump | Deoxycholate; fosfomycin | − | + |
| 0402 | 3309 | 1347 | 228 | Resistance-nodulation-cell division transporter system. Multidrug resistance efflux pump. Macrolide-specific efflux system | Macrolide | + | + |
| 0410 | 3317 | 1355 | 228 | Resistance-nodulation-cell division transporter system. Multidrug resistance efflux pump. Macrolide-specific efflux system | Macrolide | + | + |
| 0412 | 3319 | 1357 | 223 | VanA type vancomycin resistance operon genes, which can synthesize peptidoglycan with modified C-terminal d-Ala-d-Ala to d-alanine-d-lactate | Teicoplanin; vancomycin | + | + |
| 0416 | 3323 | 1361 | 221 | Resistance-nodulation-cell division transporter system. Multidrug resistance efflux pump. Macrolide-specific efflux system | Macrolide1094 | + | + |
| 0576 | 3486 | 1519 | 222 | VanB type vancomycin resistance operon genes, which can synthesize peptidoglycan with modified C-terminal d-Ala-d-Ala to d-alanine-d-lactate | Vancomycin | + | + |
| 0580 | 3490 | 1523 | 607 | Ribosomal protection protein, which protects ribosome from the translation inhibition of tetracycline | Tetracycline | + | + |
| 0601 | 3511 | 1544 | 67 | VanG type vancomycin resistance operon genes, which can synthesize peptidoglycan with modified C-terminal d-Ala-d-Ala to d-alanine-d-serine | Vancomycin | + | + |
| 0699 | 3617 | 1658 | 210 | Virginiamycin A acetyltransferase, which can inactivate the target drug | Streptogramin_a | + | + |
| 0805 | 3681 | 1722 | 230 | VanG type vancomycin resistance operon genes, which can synthesize peptidoglycan with modified C-terminal d-Ala-d-Ala to d-alanine-d-serine | Vancomycin | + | + |
| 0820 | 3697 | 1738 | 238 | VanG type vancomycin resistance operon genes, which can synthesize peptidoglycan with modified C-terminal d-Ala-d-Ala to d-alanine-d-serine | Vancomycin | + | + |
| 1054 | 0094 | 1967 | 227 | Resistance-nodulation-cell division transporter system. Multidrug resistance efflux pump. Macrolide-specific efflux system | Macrolide | + | + |
| 1094 | 0134 | 2007 | 238 | VanG type vancomycin resistance operon genes, which can synthesize peptidoglycan with modified C-terminal d-Ala-d-Ala to d-alanine-d-serine | Vancomycin | + | + |
| 1247 | 0352 | 2230 | 234 | VanA type vancomycin resistance operon genes, which can synthesize peptidoglycan with modified C-terminal d-Ala-d-Ala to d-alanine-d-lactate | Teicoplanin; vancomycin | + | + |
| 1500 | 0582 | 2467 | 283 | Undecaprenyl pyrophosphate phosphatase, which consists in the sequestration of Undecaprenyl pyrophosphate | Bacitracin | + | + |
| 1568 | 0651 | 2535 | 250 | Resistance-nodulation-cell division transporter system. Multidrug resistance efflux pump. Macrolide-specific efflux system | Macrolide | + | + |
| 1685 | 0767 | − | 238 | VanG type vancomycin resistance operon genes, which can synthesize peptidoglycan with modified C-terminal d-Ala-d-Ala to d-alanine-d-serine | Vancomycin | − | − |
| 1799 | 0854 | 2700 | 110 | ABC transporter system, bacitracin efflux pump | Bacitracin | − | − |
| 1869 | 0925 | 3336 | 371 | VanA type vancomycin resistance operon genes, which can synthesize peptidoglycan with modified C-terminal d-Ala-d-Ala to d-alanine-d-lactate | Teicoplanin; vancomycin | + | − |
| 1870 | 0926 | 3337 | 232 | VanA type vancomycin resistance operon genes, which can synthesize peptidoglycan with modified C-terminal d-Ala-d-Ala to d-alanine-d-lactate | Teicoplanin; vancomycin | + | − |
| 1962 | 1044 | 2900 | 276 | VanG type vancomycin resistance operon genes, which can synthesize peptidoglycan with modified C-terminal d-Ala-d-Ala to d-alanine-d-serine | Vancomycin | + | − |
| 2234 | 1252 | 3136 | 228 | Resistance-nodulation-cell division transporter system. Multidrug resistance efflux pump. Macrolide-specific efflux system | Macrolide | + | + |
| 2251 | 1268 | 3166 | 230 | VanG type vancomycin resistance operon genes, which can synthesize peptidoglycan with modified C-terminal d-Ala-d-Ala to d-alanine-d-serine | Vancomycin | + | + |
| 2272 | 1289 | 3187 | 288 | Pentapeptide repeat family, which protects DNA gyrase from the inhibition of quinolones | Fluoroquinolone | + | + |
| 2466 | 1548 | 3530 | 305 | ABC transporter system, bacitracin efflux pump | Bacitracin | + | + |
| 2642 | 1673 | 3662 | 227 | Resistance-nodulation-cell division transporter system. Multidrug resistance efflux pump. Macrolide-specific efflux system | Macrolide | + | + |
| 2655 | 1998 | 3676 | 234 | ABC transporter system, bacitracin efflux pump | Bacitracin | + | + |
| 2658 | 1995 | 3679 | 230 | VanA type vancomycin resistance operon genes, which can synthesize peptidoglycan with modified C-terminal d-Ala-d-Ala to d-alanine-d-lactate | Teicoplanin; vancomycin | + | + |
| 2661 | 1992 | 3682 | 222 | Resistance-nodulation-cell division transporter system. Multidrug resistance efflux pump. Macrolide-specific efflux system | Macrolide | + | + |
| 2663 | 1990 | 3684 | 224 | VanE type vancomycin resistance operon genes, which can synthesize peptidoglycan with modified C-terminal d-Ala-d-Ala to d-alanine-d-serine | Vancomycin | + | + |
| 2831 | 1853 | 0043 | 304 | ABC transporter system, bacitracin efflux pump | Bacitracin | + | + |
| 2922 | 2024 | 0104 | 192 | Virginiamycin A acetyltransferase, which can inactivate the target drug | Streptogramin_a | − | + |
| 3194 | 2245 | 0335 | 227 | Resistance-nodulation-cell division transporter system. Multidrug resistance efflux pump. Macrolide-specific efflux system | Macrolide | + | + |
| 3599 | 2639 | 0706 | 274 | Undecaprenyl pyrophosphate phosphatase, which consists in the sequestration of Undecaprenyl pyrophosphate | Bacitracin | + | + |
| 3614 | 2654 | 0729 | 235 | ABC transporter system, bacitracin efflux pump | Bacitracin | + | + |
| 3781 | 2789 | 0865 | 225 | Resistance-nodulation-cell division transporter system. Multidrug resistance efflux pump. Macrolide-specific efflux system | Macrolide | + | + |
| 3846 | 2855 | 0930 | 224 | Resistance-nodulation-cell division transporter system. Multidrug resistance efflux pump. Macrolide-specific efflux system | Macrolide | + | + |
| − | − | 1192 | 71 | Resistance-nodulation-cell division transporter system. Multidrug resistance efflux pump. Macrolide-specific efflux system | Macrolide | − | − |
| − | − | 3284 | 174 | VanG type vancomycin resistance operon genes, which can synthesize peptidoglycan with modified C-terminal d-Ala-d-Ala to d-alanine–d-serine | Vancomycin | − | − |
“+” indicates corresponding genes present; “−” indicates no corresponding genes
Prophage identification
Based on the prediction by PHAST, the ST201 genome sequences contained seven to eight prophages (Table 3). Strain LC693 contained three intact, three incomplete and one questionable phages. Among those prophages, three prophages (the 19.7-kb, the 71.7-kb and the 67.2-kb one) were also present in the other two ST201 strains, but they were missing in the ST1 strain and the ST11 strain. Another 27.1-kb prophage was not only shared by the other two ST201 isolates but also shared by the ST1 and ST11 strains. Moreover, the homologous region (97–98% identity; 82–99% coverage) of this putative phage was also found in genomes of C. difficile strains of other clades such as strains 630 and 08ACD0030 (clade 1), strains M68 and CF5 (clade 4).
Table 3.
Prophages predicted in the three ST201 genomes
| Phage region | Region length (kb) | Completeness | #CDS | Possible phage | GC (%) |
|---|---|---|---|---|---|
| Strain LC693 | |||||
| 1 | 28.3 | Incomplete | 24 | PHAGE_Clostr_phiCD505_NC_028764 (6) | 27.38 |
| 2 | 19.7 | Incomplete | 47 | PHAGE_Clostr_CDMH1_NC_024144 (21) | 28.15 |
| 3 | 71.7 | Intact | 102 | PHAGE_Clostr_phiCDHM19_NC_028996 (35) | 29.33 |
| 4 | 27.1 | Incomplete | 31 | PHAGE_Clostr_phiCDHM19_NC_028996 (11) | 28.04 |
| 5 | 7.4 | Questionable | 6 | PHAGE_Paenib_Xenia_NC_028837 (2) | 30.32 |
| 6 | 67.2 | Intact | 88 | PHAGE_Clostr_phiMMP02_NC_019421 (30) | 28.69 |
| 7 | 24.1 | Intact | 45 | PHAGE_Clostr_c_st_NC_007581 (6) | 33.26 |
| Strain VL-0104 | |||||
| 1 | 22.4 | Incomplete | 19 | PHAGE_Clostr_c_st_NC_007581 (3) | 28.77 |
| 2 | 48.3 | Intact | 58 | PHAGE_Clostr_CDMH1_NC_024144 (22) | 28.44 |
| 3 | 27.1 | Incomplete | 31 | PHAGE_Clostr_phiCDHM19_NC_028996 (11) | 28.04 |
| 4 | 48.4 | Intact | 69 | PHAGE_Clostr_phiCD505_NC_028764 (24) | 28.27 |
| 5 | 7.4 | Questionable | 6 | PHAGE_Paenib_Xenia_NC_028837 (2) | 30.32 |
| 6 | 15.6 | Incomplete | 27 | PHAGE_Clostr_phiCD27_NC_011398 (14) | 29.98 |
| 7 | 11.2 | Questionable | 18 | PHAGE_Clostr_c_st_NC_007581 (4) | 31.68 |
| Strain VL-0391 | |||||
| 1 | 22.6 | Incomplete | 18 | PHAGE_Clostr_c_st_NC_007581 (3) | 28.79 |
| 2 | 33.3 | Intact | 39 | PHAGE_Clostr_phiMMP03_NC_028959 (8) | 27.45 |
| 3 | 27.1 | Incomplete | 31 | PHAGE_Clostr_phiCDHM19_NC_028996 (11) | 28.04 |
| 4 | 51.4 | Intact | 69 | PHAGE_Clostr_phiCD505_NC_028764 (23) | 28.06 |
| 5 | 7.4 | Questionable | 6 | PHAGE_Paenib_Xenia_NC_028837 (2) | 30.32 |
| 6 | 34.9 | Questionable | 37 | PHAGE_Clostr_CDMH1_NC_024144 (15) | 31.25 |
| 7 | 25.3 | Incomplete | 34 | PHAGE_Clostr_phiCD27_NC_011398 (12) | 31.52 |
| 8 | 11.1 | Intact | 23 | PHAGE_Clostr_c_st_NC_007581 (3) | 32.47 |
Single nucleotide polymorphisms
Single nucleotide polymorphisms analysis showed that the ST201 genomes harbored approximately 53,288 SNPs (52,447–54,837) and 107,774 SNPs (107,694–107,889) compared to the ST1 genome and the ST11 genome, respectively (Table 4). Among them, approximately 40,224 (39,065–41,696) and 82,383 (81,424–82,872) SNPs were found in the coding sequence regions across the ST201 genomes, and 14,662 (14,127–15,172) and 25,649 (25,046–26,258) of those SNPs caused non-synonymous changes, respectively. The average ratio of nonsynonymous versus synonymous substitution rate (dN/dS) of the SNPs identified the ST201 genomes against the ST1 genome was 0.57, and 0.45 against the ST11 genome.
Table 4.
SNPs identified in the ST201 genomes against the ST1 and the ST11 genomes
| Nos. of SNPs | Non-synonymous | Synonymous | DN/dS | |
|---|---|---|---|---|
| LC693 vs. R20291 | 54,837 (41,696a) | 15,172 | 26,524 | 0.57 |
| VL-0104 vs. R20291 | 52,580 (39,912a) | 14,687 | 25,225 | 0.58 |
| VL-0319 vs. R20291 | 52,447 (39,065a) | 14,127 | 24,938 | 0.57 |
| Subtotal | 159,864 (120,673a) | 43,986 | 76,687 | 0.57 |
| LC693 vs. M120 | 107,738 (82,855a) | 25,643 | 57,212 | 0.45 |
| VL-0104 vs. M120 | 107,889 (82,872a) | 26,258 | 56,614 | 0.46 |
| VL-0319 vs. M120 | 107,694 (81,424a) | 25,046 | 56,378 | 0.44 |
| Subtotal | 323,321 (247,151a) | 76,947 | 170,204 | 0.45 |
aIndicates the number of SNPs in CDSs
Sequence analysis of PaLoc
Our previous study has determined that the ST201 strain LC693 was TcdA- and TcdB-positive [14], and the two large clotridial toxins TcdA and TcdB are reported to be encoded on the 19.6-kb PaLoc between two conserved genes designed cdd1 and cdu1 [5, 10, 11]. However, the PaLoc region carried by the three ST201 strains discussed here was found to be located in a 28.8-kb region, with a specific fragment of approximately 9-kb in length inserted between the putative tcdE gene and the tcdA gene that was missing in the 19.6-kb PaLoc contained by the epidemic ST1 strain R20291 and ST11 strain M120 (Fig. 2). Interestingly, this 9-kb insertion was also found in the ST54 strain ZJCDC-S82, and it contained approximately 10 predicted genes. Nucleotide sequence comparison using BlastN against the NCBI nucleotide collection database found that this this 9-kb insertion was highly homologous (99% nucleotide sequence identity) to the novel mobile genetic element Tn6218 identified in the PaLocs of clade 3 strains [33]. Correspondingly, orthologs of the four common genes (int, xis, rep, and xre) and five accessory genes (a transcription regulator gene merR; a gene encoding the oxidoreductase; the flavodoxin coding gene; an orf encoding a hypothetical protein containing the cupin domain; and the RNA polymerase σ70 coding gene) carried by Tn6218 determined before [33] were expectedly found in the 9-kb insertion contained by the ST201 strains.
Fig. 2.
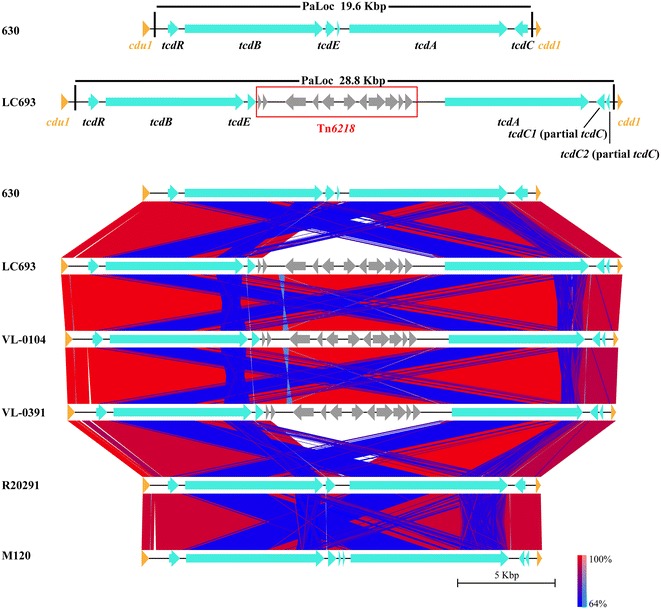
Comparative analysis of PaLoc Clostridium difficile strains discussed in this study. Color code stands for BLASTn identity of those regions between genomes Arrows in the same colors represent putative CDSs with similar roles in different genomes
The TcdA and TcdB encoding genes tcdA and tcdB harbored by the ST201 strains were highly homologous to that carried by the ST1 strain or the ST11 strain. Moreover, those two genes were more conserved among the strains in the same clade other than among those in different clades (Table 5). In addition, the SNPs identified with either the tcdA gene and/or the tcdB gene between the ST201 strains and the ST11 strain were much less than those between the ST201 strains and the ST1 strain (Table 5).
Table 5.
SNPs harbored by the PaLoc comprising genes of the ST201 strains compared with isolates of ST1 and ST11
| Strains | tcdR | tcdB | tcdE | Tn6218 | tcdA | tcdC | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| tnt | xis | rep | xre | merR | Oxidoreductase | Flavodoxin | orf | σ70 | ||||||
| Against R20291 (ST1/027, clade 2) | ||||||||||||||
| LC693 | 13 | 459 | 3 (72 del) | – | – | – | – | – | – | – | – | – | 100 | 5 (144 del) |
| VL-0104 | 13 | 459 | 3 (72 del) | – | – | – | – | – | – | – | – | – | 100 | 5 (144 del) |
| VL-0391 | 13 | 459 | 3 (72 del) | – | – | – | – | – | – | – | – | – | 100 | 5 (144 del) |
| Against M120 (ST11/078, clade 5) | ||||||||||||||
| LC693 | 18 | 175 | 2 (72 del) | – | – | – | – | – | – | – | – | – | 90 | 3 (264 del + 20 in) |
| VL-0104 | 18 | 175 | 2 (72 del) | – | – | – | – | – | – | – | – | – | 90 | 3 (264 del + 20 in) |
| VL-0391 | 18 | 175 | 2 (72 del) | – | – | – | – | – | – | – | – | – | 90 | 3 (264 del + 20 in) |
Among the toxin-expression regulating genes, tcdR was also conserved, as only 13 SNPs (between ST201 and ST1) and 18 SNPs (between ST201 and ST11) were identified between different clade strains. However, more variations were observed within the tcdE gene and the tcdC gene among the strains in different clades. The tcdE gene carried by the three ST201 strains in clade 3 had a 72-bp deletion at the N-terminal of the gene compared to the ST1 strain in clade 2 and/or the ST11 strain in clade 5 (Fig. 2; Table 5). However, for the tcdC gene, it was very interesting that there were two potential genes in the putative tcdC region of the ST201 genomes as well as in M120 compared to strain R20291 (Fig. 2). Further analysis using the putative tcdC region of strain LC693 comparing with the typical tcdC nucleotide sequence of strain 630 found a nucleotide change occurred at position 185 (C → T) which caused the formation of a stop codon here and led to an early termination of translation and the disruption of the gene (Fig. 4). These mutations resulted in a truncated TcdC protein in the ST201 strains. In addition, an 18-bp deletion was found at positions 330–347 in the putative tcdC region of strain LC693 compared to 630 (Fig. 3). Those changed patterns were also found in the other two ST201 strains VL-0104 and VL-0391 (Fig. 3). More interestingly, the tcdC harbored by strain R20291 had 120-bp deletion compared to the typical tcdC carried by strain 630, and the 18-bp deletion identified in the ST201 genomes at positions 330–347 compared to strain 630 was also found in R20291 (Fig. 3).
Fig. 4.
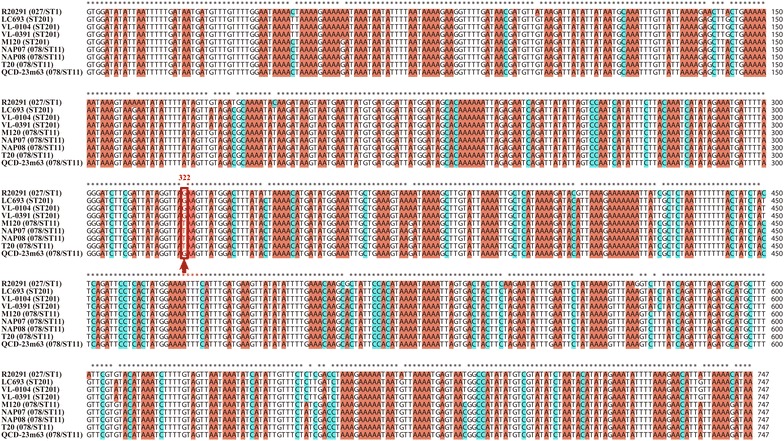
Sequence comparisons of cdtR among Clostridium difficile strains discussed in this study
Fig. 3.
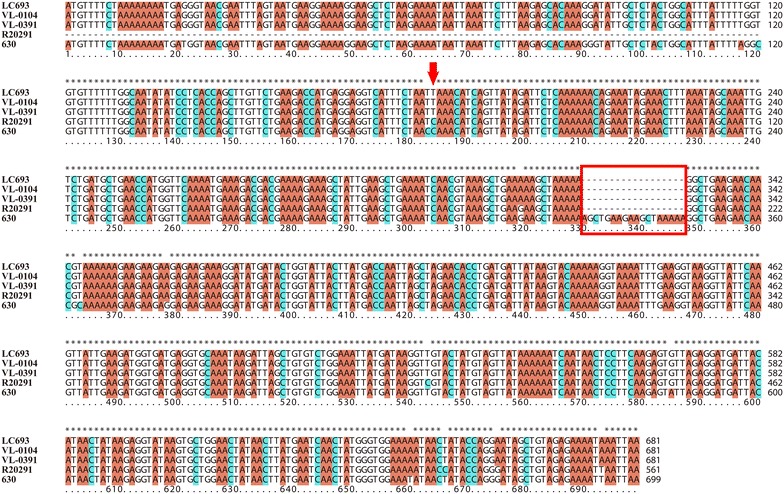
Sequence comparisons of tcdC among Clostridium difficile strains discussed in this study
Sequence analysis of CdtLoc
In addition to TcdA and TcdB, the ST201 strain LC693 is also determined as binary-toxin-positive [14]. Sequence comparisons using the nucleotide sequence of the putative CdtLoc locus against the whole genome sequences the other two ST201 strains VL-0104 and VL-0391 as well as the epidemic ST1/027 strain R20291 and the ST11/078 strain M120 demonstrated that the other two ST201 strains also contained the CdtLoc region. Unlike the PaLoc harbored by the clade 3 strains, there were no insertions of mobile genetic elements in the CdtLoc region. Among the three genes carried by CdtLoc, cdtA and ctdB were highly conserved between the ST201 strains and the ST1/ST11 strains. However, the ctdR was found to be conserved among the strains excluding the ST11/078 strain M120. The cdtR gene of strain M120 was found to have a nucleotide change occurred at position 322 (G → T) compared to the ctdR carried by either the strain R20291 or the three ST201 strains, and this change caused the formation of a stop codon and therefore resulted in a truncated CdtR in M120. Interestingly, this changed pattern was also found in most 078/ST11 strains (Fig. 4).
Whole genome sequence comparison
Whole genome sequences comparison showed that the ST201 genomes and the ST1 and the ST11 genomes were highly matched and homologous (Fig. 5a). Comparative analysis identified a shared set of 2585 core genes and a pan genome of more than 1404 genes as well as 31 genes unique to strain VL-0104; 109 unique to VL-0391; 129 unique to LC693; 377 unique to the epidemic ST1/027 strain R20291; and 458 unique to the ST11/078 strain M120 (Fig. 5b). Functional comparison of the core genes and the strain-specific genes against the COG database showed that the core genes mainly participated in carbohydrate transport and metabolism, amino acid transport and metabolism, energy production and conversion, cell membrane biogenesis, inorganic ion transport and metabolism, signal transduction mechanisms, transcription, replication, recombination and repair, coenzyme transport and metabolism, translation, ribosomal structure and biogenesis, nucleotide transport and metabolism, lipid transport and metabolism, posttranslational modification, protein turnover, chaperones, and hypothetical proteins. For the 129 strain-specific genes for LC693, approximately 85 were phage-related genes, and 19, 6, 43, 15, and 2 of them were clustered in the 28.3-, 19.7-, 71.7-, 67.2-, and 24.1-kb prophage that identified in the strain, respectively (Table 3); the rest of them encoded hypothetical proteins, phage-related proteins outside the predicted prophage regions, and proteins in amino acid transport and metabolism, ribosomal structure and biogenesis, transcription, cell membrane biogenesis, inorganic ion transport and metabolism, and defense. The 31 strain-specific genes for VL-0104 encoded proteins mainly participating in cell cycle control, carbohydrate transport and metabolism, transcription, replication, recombination and repair, cell membrane biogenesis, mobilization, and hypothetical proteins. For strain VL-0391, the 109 unique genes encoded proteins associated with energy production and conversion, cell cycle control, amino acid transport and metabolism, carbohydrate transport and metabolism, coenzyme transport and metabolism, lipid transport and metabolism, translation, ribosomal structure and biogenesis, transcription, replication, recombination and repair, cell membrane biogenesis, cell motility, posttranslational modification, inorganic ion transport and metabolism, secondary metabolites biosynthesis, transport and catabolism, signal transduction, intracellular trafficking, secretion, and vesicular transport, and bacterial defense mechanisms.
Fig. 5.
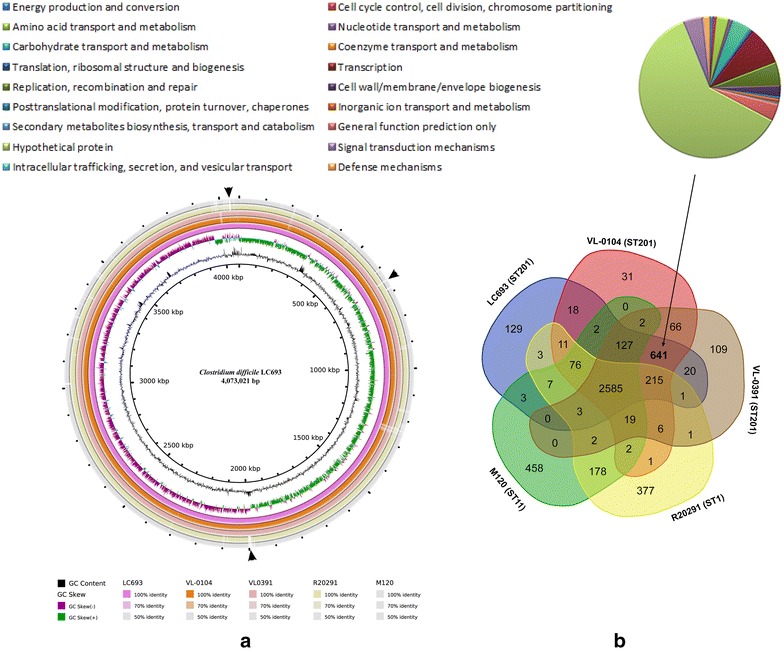
Comparative genomic analysis of Clostridium difficile ST201 strains with the epidemic 027/ST1 strain R20291 and 078/ST11 strain M120. a Whole genome sequences comparison of the strains. Circles from inside to outside indicate GC content of strain LC693, GC skew of strain LC693, C. difficile strains LC693, VL-0104, VL-0391, R20291 and M120. Different DNA BLAST identities are shown using different colors. b Venn diagram shows shared genes and unique gene among the strains. Pie chart displays COG functional catalogues of the 641 predicted genes specific for the ST201 strains
The three ST201 strains contained 641 genes which were absent in both the ST1 and ST11 strains (Fig. 5b; Additional file 1: Table S1). Those ST201 strains-specific genes contained those predicted as phage-related genes that were carried by either the ST201 strains-shared 19.7-kb, the 71.7-kb or the 67.2-kb prophage. Those ST201 strains-specific genes also included those forming the 9-kb insertion Tn6218 which was generally found in the clade 3 PaLoc but was absent in other clade strains. In particular, the ST201 strains-specific genes also covered many genes involved in the bacterial fitness and pathogenesis. For example, the type I restriction–modification system was found to have a potential role in the virulence of some bacterial pathogens such as Haemophilus [34] and Salmonella enterica serovar Enteritidis [35]. The ferric iron ABC transporter and the iron compound ABC uptake transporter ATP-binding protein was helpful to uptake iron, which is not only an essential element for bacterial survival, but also acts an environmental signal that regulates the expression of many virulence factors [36]. The histidine kinase and response regulator forms the bacterial two-component system, which is undoubtedly important for bacterial survival and virulence regulation [37]. The antitoxin protein HigA was favorable for bacteria to escape the toxin and was feasible to survival the infection loci [38].
Discussion
Clostridium difficile infection is widely accepted as one of the most common healthcare and economy problems throughout the world especially in North America and Europe [4, 39–41]. More worrisome, the emergence and prevalence of the 027/ST1 has significantly increased the morbidity and mortality of CDI [7, 42, 43]. Besides, the emergent 078/ST11 strains are reported to share the same genetic virulence characteristics as 027/ST1 and cause severe disease at a similar rate [8]. However, both of those two types of strains are rarely reported in China. The 027 has not been detected in China before 2013, and cases of C. difficile 078 have not been reported yet [14]. Instead, a number of severe diarrhea-associated C. difficile toxigenic strains belonging to clades distinct from the 027/ST1 and 078/ST11 strains have been reported in China [16, 44]. This might indicate that the dominant genotypes of C. difficile spreading in China are different from those circulating in North America and Europe. Consistently, phylogenetic analysis showed that the novel binary toxin-positive C. difficile associated with severe diarrhea isolated in China discussed here belonged to clade 3, while all epidemic 027/ST1 and 078/ST11 strains were concentrated in clade 2 and clade 5, respectively (Fig. 1). Those results are in accordance with our previously reported phylogenetic tree generated using whole-genomic comparison [14]. What is more, the clade 3 branch also included another China—sourced toxigenic C. difficile strain ZJCDC-S82 which is also reported as a severe diarrhea-associated strain [44]. In addition, the other three recently-reported binary toxin-positive C. difficile (strains 103, 133, and 106) recovered from three ICU patients in China are also clade 3 strains [16]. These findings suggest that C. difficile clade 3 strains might contribute to the occurrence of CDI in China. In the phylogenetic tree, the same evolutionary branch includes C. difficile strains isolated from different places (Fig. 1), suggesting that there were no correlations between the bacterial genetic diversity and its geographic location. Meanwhile, even though all 027 or 078 strains were concentrated on the same clade, there were still strains sharing the same ribotype/sequence type being clustered in different clades (Fig. 1), suggesting that there was little correlation between the bacterial genetic diversity and its sequence type/ribotype. The phylogenetic analysis also showed that the clade 3 strains had a closer evolutionary relationship with the 027 strains that with the 078 strains (Fig. 1). Consistence with this, much less SNPs were identified between the ST201 strains in clade 3 against the 027 strain R20291 in clade than against the 078 strain M120 in clade 5 (Table 4). Besides, the average dN/dS of the ST201 strains against both the 027 strain and the 078 strain were significantly smaller than 1, suggesting a strong purifying selection during the evolutionary process [6].
The genomes of the binary toxin-positive ST201 strains as well as the epidemic 027 and 078 strains contained more than 40 antibiotic-resistance-related genes which confer the strains resistance to multiple antibiotics (Table 2). It has been proposed that the use of antibiotics is the most important risk factor for CDI [4], because C. difficile is resistant to multiple antibiotics that are commonly used for treating bacterial infections in clinical settings [2, 45]. Therefore, so many antibiotic resistance-related genes harbored in the ST201 strains may contribute to the bacterial pathogenesis. What is more, a large proportion (37.5%) of those antibiotic resistance genes were predicted to be associated with resistance to vancomycin, a kind of antibiotic commonly used for CDI treatment in clinic [46, 47]. Our result from antimicrobial susceptibility test demonstrated that strain LC693 is resistant to vancomycin, suggesting that those genes confer resistance of vancomycin to the strain. This might explain that enteral vancomycin is useless for treating the patient who is infected by strain LC693 [14].
Toxin expression is considered to be a key factor for the development of CDI [4], and PaLoc is responsible for encoding the clostridial toxins and regulating their expression [10]. Like the PaLoc reported in other clade 3 strains before [16, 33, 44], the PaLoc carried by the three ST201 strains discussed in this study as well as another clade 3 strain ZJCDC-S82 contained a mobile genetic element designated Tn6218 inserted between tcdE and tcdA (Fig. 2). It is suggested that the insertion of Tn6218 in PaLoc is clade-specific [16]. Consistence with this, this insertion element was not found in the PaLoc of R20291 in clade 2 and M120 in clade 5. In addition, the Tn6218 in the three ST201 genomes were found to be flanked by two AT rich sequences. Previous studies suggested that those two AT rich sequences might have inserted into clade 3 PaLoc prior to the insertion of Tn6218 and provide the insertion site of Tn6218 [16]. For the other components of the PaLoc, it is worth to mention that although phylogenetic analysis using either MLST or whole genome comparison demonstrated a closer evolutionary relationship between the ST201 strains and the 027 strain R20291 (Fig. 1), the ST201 strains and the 078 strain M120 shared a more homologous tcdA and/or tcdB (Table 5). Further analysis needs to be performed to determine whether the toxin-yielding profile of the ST201 strain is closer to the 027 strain or to the 078 strain. For the toxin expression regulating genes, the tcdC gene is proposed to be a negative regulator for the toxin production, and the mutations within tcdC is observed to contribute to the toxin-production of some 027 strains [48, 49]. In our study, two main kinds of mutations were found within the tcdC gene ST201 carried by the ST201 strains compared to strain 630. The first one was an 18-bp deletion at positions 330–347 in the tcdC, and this mutation pattern was also found in the epidemic 027 strain R20291 (Fig. 3). However, this 18-bp in frame mutation has been found to have no effect on toxin production [50]. Instead, previous study reported that a deletion at position 117 in tcdC of the 027/ST1 strains compared to strain 630 resulted in the formation of a stop codon and truncation of the protein, and then caused increased toxin production further [51]. Even though this kind of mutation was not observed in the ST201 tcdC compared to the 630 tcdC, a nucleotide change occurred at position 185 (C → T) of the ST201 tcdC, which caused the formation of a stop codon here and therefore led to an early termination of translation as well as the disruption of the gene, may have a similar contribution to the toxin production of the ST201 strains.
Another factor contributing to the pathogenesis of the ST201 strains was the presence of the CdtLoc responsible for encoding the binary toxin in bacterial genomes. Previous data have found that the patients infected with C. difficile producing CDT had higher fatality rate (approximately 60%) than those infected with CDT-deficient strains [13]. A more recent study found that the binary toxin enhanced two PCR-ribotype 027 strains (R20291 and M7404) in mice by suppressing protective colonic eosinophilia [52]. Sequence comparisons demonstrated that the CdtLoc harbored in the ST201 strains was highly homologous to that of strain R20291, and the three genes cdtA, ctdB and ctdR carried by the ST201 CdtLoc were intact and also highly homologous to their corresponding genes harbored by R20291, respectively. These data suggest that the CdtLoc in the ST201 strain is active and the binary toxin encoded by it contributes to the pathogenesis of the ST201 strain. In particular, previous studies found that CdtR increased production of TcdA, TcdB and CDT in two epidemic 027 strains including R20291, but this regulation was not found in the 078 strain [53]. A R20291-cdtR-higly homologous cdtR identified in the ST201 strains may also have a similar role in positively regulating the production of the C. difficile toxins, and a truncated CdtR identified in most 078/ST11 strains may explain why the CdtR-mediated toxin regulation does not occur in the 078/ST11 strains [53]. In addition, whole genome sequence comparison identified a series of virulence-associated genes shared by the three ST201 genomes but not shared by both the R20291 genome and the M120 genome, the presence of these genes may also have a contribution to the bacterial pathogenesis.
Conclusions
We summarized the genomic characterization of three binary toxin-positive ST201 strains in clade 3 in this study. While the presence of multiple fitness and virulence associated genes might form the pathogenesis basis of the binary toxin-positive ST201 strain, two main contents are likely to play the main role. (1) The presence of a number of antibiotic resistance associated genes in the strain especially the vancomycin resistant genes might increase the treatment difficulty of the bacterial infection; (2) the toxin producing required genes of the ST201 strain were highly homologous to the epidemic 027/ST1 strain; these genes might increase the virulence of the bacterium. Our work reveals the pathogenesis-basis of the ST201 binary toxin-positive strains in part. To our knowledge, this is the first time that the genomic characterization of the ST201 strains in clade 3 was discussed. As studies on clade 3 strains especially C. difficle ST201 are limited, the present study would have a contribution to understanding the pathogenesis basis of C. difficle ST201.
Authors’ contributions
ZP, AW and CL conceived and designed the project; JD and CF contributed to the bacterial isolation; SL and XM performed the bacterial DNA isolation and genome sequencing; ZP, WL, BT and BW performed the genome data organization, submission and analysis; ZP and YW wrote the paper, and ZP, ZX, BW, AW, and CL revised and re-edit the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We sincerely thank Beijing Novogene Bioinformatics Technology Co., Ltd (Beijing, China) for its help in bacterial genome sequencing.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
This Whole Genome Shotgun project of Clostridium difficile LC693 has been deposited at DDBJ/ENA/GenBank under the Accession NCXL00000000.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China [Grant Number 81601803], Young Scientists Fund of Xiangya Hospital [Grant Number 2014Q05] and Xiangya Sinobioway Health Research Fund [Grant Number xywm2015I11].
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- CDI
Clostridium difficile infection
- CDS
coding DNA sequences
- CDT
binary toxin
- CdtLoc
binary toxin encoding locus
- MLST
multilocus sequence typing
- ORF
open reading frame
- PaLoc
pathogenicity locus
- SNP
single nucleotide polymorphism
Additional file
Additional file 1: Table S1. Genes identified to be specific for Clostridium difficile ST201 strains but absent in both 027/ST1 strain R20291 and 078/ST11 strain M120.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s13099-017-0191-z) contains supplementary material, which is available to authorized users.
Contributor Information
Zhong Peng, Email: pengzhong525@163.com.
Sidi Liu, Email: 807401745@qq.com.
Xiujuan Meng, Email: mxj0324@163.com.
Wan Liang, Email: liangwan521521@163.com.
Zhuofei Xu, Email: zhuofei.xu@gmail.com.
Biao Tang, Email: tb_411@163.com.
Yuanguo Wang, Email: wygcy2012@gmail.com.
Juping Duan, Email: Conan_1@126.com.
Chenchao Fu, Email: fuchenchao112@163.com.
Bin Wu, Email: wub@mail.hzau.edu.cn.
Anhua Wu, Email: xywuanhua@csu.edu.cn.
Chunhui Li, Email: lichunhui@csu.edu.cn.
References
- 1.Burke KE, Lamont JT. Clostridium difficile infection: a worldwide disease. Gut Liver. 2014;8(1):1–6. doi: 10.5009/gnl.2014.8.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peng Z, Jin D, Kim HB, Stratton CW, Wu B, Tang YW, Sun X. Update on antimicrobial resistance in Clostridium difficile: resistance mechanisms and antimicrobial susceptibility testing. J Clin Microbiol. 2017;55:1998–2008. doi: 10.1128/JCM.02250-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ofosu A. Clostridium difficile infection: a review of current and emerging therapies. Ann Gastroenterol Q Publ Hell Soc Gastroenterol. 2016;29(2):147. doi: 10.20524/aog.2016.0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leffler DA, Lamont JT. Clostridium difficile infection. N Engl J Med. 2015;372(16):1539–1548. doi: 10.1056/NEJMra1403772. [DOI] [PubMed] [Google Scholar]
- 5.Chowdhury PR, DeMaere M, Chapman T, Worden P, Charles IG, Darling AE, Djordjevic SP. Comparative genomic analysis of toxin-negative strains of Clostridium difficile from humans and animals with symptoms of gastrointestinal disease. BMC Microbiol. 2016;16(1):41. doi: 10.1186/s12866-016-0653-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He M, Sebaihia M, Lawley TD, Stabler RA, Dawson LF, Martin MJ, Holt KE, Seth-Smith HM, Quail MA, Rance R. Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc Natl Acad Sci. 2010;107(16):7527–7532. doi: 10.1073/pnas.0914322107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lim SK, Stuart RL, Mackin K, Carter G, Kotsanas D, Francis M, Easton M, Dimovski K, Elliot B, Riley TV. Emergence of a ribotype 244 strain of Clostridium difficile associated with severe disease and related to the epidemic ribotype 027 strain. Clin Infect Dis. 2014;58(12):1723–1730. doi: 10.1093/cid/ciu203. [DOI] [PubMed] [Google Scholar]
- 8.Goorhuis A, Bakker D, Corver J, Debast SB, Harmanus C, Notermans DW, Bergwerff AA, Dekker FW, Kuijper EJ. Emergence of Clostridium difficile infection due to a new hypervirulent strain, polymerase chain reaction ribotype 078. Clin Infect Dis. 2008;47(9):1162–1170. doi: 10.1086/592257. [DOI] [PubMed] [Google Scholar]
- 9.Voth DE, Ballard JD. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev. 2005;18(2):247–263. doi: 10.1128/CMR.18.2.247-263.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hammond GA, Johnson JL. The toxigenic element of Clostridium difficile strain VPI 10463. Microb Pathog. 1995;19(4):203–213. doi: 10.1016/S0882-4010(95)90263-5. [DOI] [PubMed] [Google Scholar]
- 11.Cohen SH, Tang YJ, Silva J. Analysis of the pathogenicity locus in Clostridium difficile strains. J Infect Dis. 2000;181(2):659–663. doi: 10.1086/315248. [DOI] [PubMed] [Google Scholar]
- 12.Eckert C, Emirian A, Le Monnier A, Cathala L, De Montclos H, Goret J, Berger P, Petit A, De Chevigny A, Jean-Pierre H. Prevalence and pathogenicity of binary toxin-positive Clostridium difficile strains that do not produce toxins A and B. New Microbes New Infect. 2015;3:12–17. doi: 10.1016/j.nmni.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bacci S. Binary toxin and death after clostridium difficile infection. Emerg Infect Dis. 2011;17(6):976–982. doi: 10.3201/eid/1706.101483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li C, Liu S, Zhou P, Duan J, Dou Q, Zhang R, Chen H, Cheng Y, Wu A. Emergence of a novel binary toxin-positive strain of Clostridium difficile associated with severe diarrhea that was not ribotype 027 and 078 in China. Infect Control Hosp Epidemiol. 2015;36(09):1112–1114. doi: 10.1017/ice.2015.120. [DOI] [PubMed] [Google Scholar]
- 15.Pituch H, Kreft D, Obuch-Woszczatyński P, Wultańska D, Meisel-Mikołajczyk F, Łuczak M, van Belkum A. Clonal spread of a Clostridium difficile strain with a complete set of toxin A, toxin B, and binary toxin genes among polish patients with Clostridium difficile-associated diarrhea. J Clin Microbiol. 2005;43(1):472–475. doi: 10.1128/JCM.43.1.472-475.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen R, Feng Y, Wang X, Yang J, Zhang X, Lü X, Zong Z. Whole genome sequences of three Clade 3 Clostridium difficile strains carrying binary toxin genes in China. Sci Rep. 2017;7:43555. [DOI] [PMC free article] [PubMed]
- 17.Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20(2):265–272. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24(5):713–714. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- 19.Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14(7):1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edwards DJ, Holt KE. Beginner’s guide to comparative bacterial genome analysis using next-generation sequence data. Microb Inform Exp. 2013;3(1):2. doi: 10.1186/2042-5783-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc Natl Acad Sci USA. 2005;102(39):13950–13955. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu Z, Chen X, Li L, Li T, Wang S, Chen H, Zhou R. Comparative genomic characterization of Actinobacillus pleuropneumoniae. J Bacteriol. 2010;192(21):5625–5636. doi: 10.1128/JB.00535-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M. The RAST Server: rapid annotations using subsystems technology. BMC Genom. 2008;9(1):75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28(1):33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39:W347–W352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu B, Pop M. ARDB—antibiotic resistance genes database. Nucleic Acids Res. 2009;37(suppl 1):D443–D447. doi: 10.1093/nar/gkn656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen L, Yang J, Yu J, Yao Z, Sun L, Shen Y, Jin Q. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 2005;33(suppl 1):D325–D328. doi: 10.1093/nar/gki008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alikhan N-F, Petty NK, Zakour NLB, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genom. 2011;12(1):402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE. 2010;5(6):e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sullivan MJ, Petty NK, Beatson SA. Easyfig: a genome comparison visualizer. Bioinformatics. 2011;27(7):1009–1010. doi: 10.1093/bioinformatics/btr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng Z, Liang W, Liu W, Wu B, Tang B, Tan C, Zhou R, Chen H. Genomic characterization of Pasteurella multocida HB01, a serotype A bovine isolate from China. Gene. 2016;581(1):85–93. doi: 10.1016/j.gene.2016.01.041. [DOI] [PubMed] [Google Scholar]
- 32.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dingle KE, Elliott B, Robinson E, Griffiths D, Eyre DW, Stoesser N, Vaughan A, Golubchik T, Fawley WN, Wilcox MH. Evolutionary history of the Clostridium difficile pathogenicity locus. Genome Biol Evol. 2014;6(1):36–52. doi: 10.1093/gbe/evt204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, Xu X, Zhang S, Guo F, Cai X, Chen H. Identification and analysis of potential virulence-associated genes in Haemophilus parasuis based on genomic subtraction. Microb Pathog. 2011;51(4):291–296. doi: 10.1016/j.micpath.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 35.Silva CA, Blondel CJ, Quezada CP, Porwollik S, Andrews-Polymenis HL, Toro CS, Zaldívar M, Contreras I, McClelland M, Santiviago CA. Infection of mice by Salmonella enterica serovar Enteritidis involves additional genes that are absent in the genome of serovar Typhimurium. Infect Immun. 2012;80(2):839–849. doi: 10.1128/IAI.05497-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacques M. Surface polysaccharides and iron-uptake systems of Actinobacillus pleuropneumoniae. Can J Vet Res. 2004;68(2):81. [PMC free article] [PubMed] [Google Scholar]
- 37.Stock AM, Robinson VL, Goudreau PN. Two-component signal transduction. Annu Rev Biochem. 2000;69(1):183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 38.Schureck MA, Maehigashi T, Miles SJ, Marquez J, Cho SE, Erdman R, Dunham CM. Structure of the Proteus vulgaris HigB-(HigA) 2-HigB toxin-antitoxin complex. J Biol Chem. 2014;289(2):1060–1070. doi: 10.1074/jbc.M113.512095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’donoghue C, Kyne L. Update on Clostridium difficile infection. Current Opinion Gastroenterol. 2011;27(1):38–47. doi: 10.1097/MOG.0b013e3283411634. [DOI] [PubMed] [Google Scholar]
- 40.Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, Farley MM, Holzbauer SM, Meek JI, Phipps EC. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372(9):825–834. doi: 10.1056/NEJMoa1408913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li C, Duan J, Liu S, Meng X, Fu C, Zeng C, Wu A. Assessing the risk and disease burden of Clostridium difficile infection among patients with hospital-acquired pneumonia at a University Hospital in Central China. Infection. 2017. doi:10.1007/s15010-017-1024-1. [DOI] [PMC free article] [PubMed]
- 42.Napolitano LM, Edmiston CE. Clostridium difficile disease: diagnosis, pathogenesis, and treatment update. Surgery. 2015;162(2):325–348. doi: 10.1016/j.surg.2017.01.018. [DOI] [PubMed] [Google Scholar]
- 43.Chen W, Liu WE, Li YM, Luo S, Zhong YM. Preparation and preliminary application of monoclonal antibodies to the receptor binding region of Clostridium difficile toxin B. Mol Med Rep. 2015;12(5):7712–7720. doi: 10.3892/mmr.2015.4369. [DOI] [PubMed] [Google Scholar]
- 44.Luo Y, Huang C, Ye J, Fang W, Gu W, Chen Z, Li H, Wang X, Jin D. Genome sequence and analysis of Peptoclostridium difficile strain ZJCDC-S82. Evolut Bioinform Online. 2016;12:41. doi: 10.4137/EBO.S32476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johanesen PA, Mackin KE, Hutton ML, Awad MM, Larcombe S, Amy JM, Lyras D. Disruption of the gut microbiome: clostridium difficile infection and the threat of antibiotic resistance. Genes. 2015;6(4):1347–1360. doi: 10.3390/genes6041347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cohen SH, Gerding DN, Johnson S, Kelly CP, Loo VG, McDonald LC, Pepin J, Wilcox MH. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the society for healthcare epidemiology of America (SHEA) and the infectious diseases society of America (IDSA) Infect Control Hosp Epidemiol. 2010;31(05):431–455. doi: 10.1086/651706. [DOI] [PubMed] [Google Scholar]
- 47.Bauer MP, Kuijper E, Van Dissel JT. European society of clinical microbiology and infectious diseases (ESCMID): treatment guidance document for Clostridium difficile infection (CDI) Clin Microbiol Infect. 2009;15(12):1067–1079. doi: 10.1111/j.1469-0691.2009.03099.x. [DOI] [PubMed] [Google Scholar]
- 48.Warny M, Pepin J, Fang A, Killgore G, Thompson A, Brazier J, Frost E, McDonald LC. Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet. 2005;366(9491):1079–1084. doi: 10.1016/S0140-6736(05)67420-X. [DOI] [PubMed] [Google Scholar]
- 49.Carter GP, Douce GR, Govind R, Howarth PM, Mackin KE, Spencer J, Buckley AM, Antunes A, Kotsanas D, Jenkin GA. The anti-sigma factor TcdC modulates hypervirulence in an epidemic BI/NAP1/027 clinical isolate of Clostridium difficile. PLoS Pathog. 2011;7(10):e1002317. doi: 10.1371/journal.ppat.1002317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dupuy B, Govind R, Antunes A, Matamouros S. Clostridium difficile toxin synthesis is negatively regulated by TcdC. J Med Microbiol. 2008;57(6):685–689. doi: 10.1099/jmm.0.47775-0. [DOI] [PubMed] [Google Scholar]
- 51.Stabler RA, He M, Dawson L, Martin M, Valiente E, Corton C, Lawley TD, Sebaihia M, Quail MA, Rose G. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol. 2009;10(9):R102. doi: 10.1186/gb-2009-10-9-r102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cowardin CA, Buonomo EL, Saleh MM, Wilson MG, Burgess SL, Kuehne SA, Schwan C, Eichhoff AM, Koch-Nolte F, Lyras D. The binary toxin CDT enhances Clostridium difficile virulence by suppressing protective colonic eosinophilia. Nat Microbiol. 2016;1:16108. doi: 10.1038/nmicrobiol.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lyon SA, Hutton ML, Rood JI, Cheung JK, Lyras D. CdtR regulates TcdA and TcdB production in Clostridium difficile. PLoS Pathog. 2016;12(7):e1005758. doi: 10.1371/journal.ppat.1005758. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This Whole Genome Shotgun project of Clostridium difficile LC693 has been deposited at DDBJ/ENA/GenBank under the Accession NCXL00000000.