

Abstract
O -Phosphoserine (Sep), the most abundant phosphoamino acid in the eukaryotic phosphoproteome, is not encoded in the genetic code, but synthesized posttranslationally. Here, we present an engineered system for specific cotranslational Sep incorporation (directed by UAG) into any desired position in a protein by an Escherichia coli strain that harbors a Sep-accepting transfer RNA (tRNASep), its cognate Sep–tRNA synthetase (SepRS), and an engineered EF-Tu (EF-Sep). Expanding the genetic code rested on reengineering EF-Tu to relax its quality-control function and permit Sep-tRNASep binding. To test our system, we synthesized the activated form of human mitogen-activated ERK activating kinase 1 (MEK1) with either one or two Sep residues cotranslationally inserted in their canonical positions (Sep218, Sep222). This system has general utility in protein engineering, molecular biology, and disease research.
O-Phosphoserine (Sep) was identified 80 years ago as a constituent of phosphoproteins from egg yolk (1). Since then, the extent and importance of the eukaryotic phosphoproteome has been realized and has provided insight into large interconnected networks of kinases and phosphatases (2). Protein kinases represent one of the largest eukaryotic gene families, composing nearly 2% of all human genes (3). Sep is the most abundant phosphoamino acid; based on an analysis of >2000 HeLa cell phosphoproteins, the relative abundance of Sep is 7.3 and 48 times higher than that of phosphothreonine and phosphotyrosine, respectively (2). A major research limitation is the inability to biosynthesize these phosphoproteins for detailed studies of their enzyme or substrate properties.
The discovery of Sep–tRNA synthetase (SepRS), a unique aminoacyl–tRNA synthetase devoted to Sep-tRNACys formation in methanogenic archaea, provided an opportunity to develop our Sep-insertion strategy. The natural role of SepRS is the formation of Sep-tRNACys, which is then converted to Cys-tRNACys by the enzyme SepCysS in the presence of a sulfur donor (4) (Fig. 1A). Given the high specificity of SepRS for Sep and for tRNACys and our knowledge of the identity elements in this tRNA (5), and based on our understanding of the structure of this enzyme and its catalytic site (6, 7), we devised a system to incorporate Sep into proteins directed by the UAG (amber) codon. For this, we chose Methanocaldococcus jannaschii (Mj) tRNACys and the mesophilic Methanococcus maripaludis (Mmp) SepRS as the orthogonal pair [reviewed in (8)] for the synthesis of phosphoserylated amber suppressor tRNA.
Fig. 1.

Design of tRNASep and its aminoacylation by M. maripaludis SepRS. (A) Pathway of Cys-tRNACys formation in M. maripaludis. ATP, adenosine 5′-triphosphate; AMP, adenosine 5′-monophosphate; PPi, inorganic pyrophosphate; PLP, pyridoxal phosphate. (B) Cloverleaf structure of tRNASep. Arrows indicate the three nucleotide changes compared to M. jannaschii tRNACys. (C) Acylation with Sep of M. jannaschii tRNACys and tRNASep catalyzed by M. maripaludis SepRS. Total E. coli tRNA (▼), or tRNA from E. coli strains expressing the M. jannaschii tRNACys (●) or the tRNASep (○) gene was acylated by M. maripaludis SepRS with [14C]Sep (0.1 mM) in the presence of ATP (10 mM).
We first designed tRNASep (Fig. 1B), an amber suppressor tRNA derived from Mj tRNACys by two mutations in the anticodon, and an additional C20U change that improves aminoacylation by SepRS (5). In vitro aminoacylation by Mmp SepRS showed (Fig. 1C) that the anticodon change lowered (to about 40%) the ability of tRNASep to be aminoacylated when compared to tRNACys. In agreement with earlier data (5), total Escherichia coli tRNA could not be acylated with Sep (Fig. 1C). On the basis of these in vitro data, Mj tRNASep and Mmp SepRS appear to be an orthogonal pair.
Efficient and selective addition of Sep to the E. coli genetic repertoire requires exclusive interaction of SepRS with tRNASep for Sep-tRNASep formation without interfering in the host translation system, as well as a sufficient intracellular concentration of Sep. Because E. coli has a Sep-compatible transporter (9), Sep (2 mM) was added to the LB growth medium, and the endogenous serB gene encoding phosphoserine phosphatase was deleted in the E. coli test strain without affecting growth. To assess whether the Mj tRNASep–Mmp SepRS pair is functional and orthogonal in E. coli in vivo, we performed a suppression assay that used a gene encoding chloramphenicol acetyltransferase (CAT) with a UAG stop codon at the permissive position 112 (wild-type amino acid: Asp) to produce the CAT enzyme. Cell survival was measured in the presence of Sep and varying amounts of chloramphenicol (Cm) where the different half-maximal inhibitory concentration (IC50) values and the tRNASep-dependent CATsynthesis correlate with suppression efficiency (Fig. 2). When only tRNASep was expressed (Fig. 2, column B), Cm resistance increased about 3.3-fold over background (Fig. 2, column A). Thus, tRNASep can be aminoacylated to a certain degree by an unknown E. coli aminoacyl–tRNA synthetase (we later found that Gln is being incorporated at the amber stop codon). In contrast, simultaneous expression of tRNASep and SepRS did not provide Cm resistance (Fig. 2, column C). This may indicate that SepRS can outcompete any endogenous aminoacyl–tRNA synthetase and form Sep-tRNASep; however, this aminoacyl-tRNA is neither delivered to the ribosome nor accommodated on it. Providing additional EF-Tu did not improve the result (Fig. 2, column D). Coexpression of tRNASep, SepRS, and SepCysS should result in formation of Sep-tRNASep and subsequent SepCysS-mediated conversion to Cys-tRNASep (4). Indeed, a 2.3-fold increase in Cm resistance was observed (Fig. 2, column E). This further supports the notion that although Sep-tRNASep is synthesized, it cannot be used properly by the E. coli protein biosynthesis machinery. By contrast, coexpression of tRNASep and Mmp CysRS generated a 12.3-fold increase in Cm resistance (Fig 2, column F), demonstrating that Cys-tRNASep can be readily used for amber codon suppression in the CAT gene.
Fig. 2.
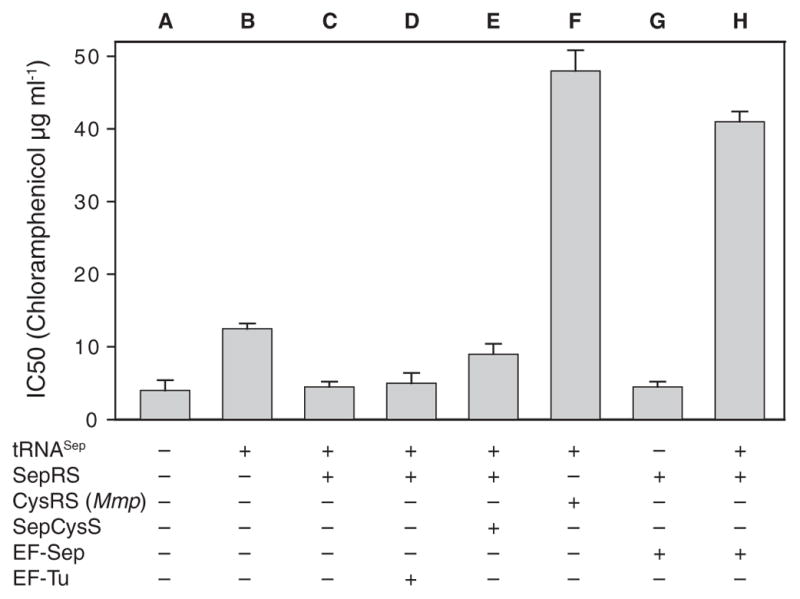
In vivo synthesis of chloramphenicol acetyl-transferase (measured by IC50 value) by tRNASep-dependent read-through of an amber mutation in the CAT gene. tRNASep was co-expressed in the E. coli Top10ΔserB strain with the proteins indicated in the figure (SepRS, M. maripaludis CysRS, SepCysS, EF-Sep, EF-Tu). Selection was carried out on LB agar plates containing 2 mM Sep and various concentrations of chloramphenicol. Error bars indicate SEM.
Given that EF-Tu is a component of quality control in protein synthesis (10), it is plausible that Sep-tRNASep may be rejected by EF-Tu. Chemically synthesized Sep-tRNAGln was a poor substrate for in vitro protein synthesis (11). tRNAs carrying negatively charged amino acids are bound poorly by EF-Tu (12), and molecular dynamics simulations suggested that Sep-tRNACys may not be bound by EF-Tu (13). We tested this assumption in EF-Tu–mediated Sep-tRNA hydrolysis protection experiments (14). Although EF-Tu protected [35S]Cys-tRNACys from deacylation at pH 8.2 (fig. S1), [14C]Sep-tRNACys was significantly deacylated irrespective of the presence of EF-Tu (Fig. 3B and fig. S1). Thus, insufficient binding of Sep-tRNASep to EF-Tu may explain the lack of Sep insertion into protein.
Fig. 3.

Design of EF-Sep. (A) Model of the amino acid binding pocket of E. coli EF-Tu bound to Phe-tRNA (based on Protein Data Bank structure 1OB2). To accommodate Sep-tRNA, an E. coli tufB library was constructed that would allow the six highlighted amino acid residues to change to any of the 20 canonical amino acids. The six mutations in our final EF-Sep are indicated by an arrow. (B) EF-Sep protects Sep-tRNACys from deacylation. M. jannaschii [14C]Sep-tRNACys was incubated at pH 8.2 and at room temperature in the presence or absence of wild-type EF-Tu or EF-Sep. Deacylation of Sep-tRNACys was measured by acid precipitability. Error bars indicate SEM. Abbreviations for the amino acid residues are as follows: D, Asp; E, Glu; F, Phe; G, Gly; H, His; N, Asn; R, Arg; S, Ser; T, Thr; W, Trp; and Y, Tyr.
This observation required the generation of EF-Tu variants able to productively bind Sep-tRNA. We were encouraged by reports that EF-Tu variants allow binding of tRNAs charged with certain unusual amino acids (15, 16). Guided by the structure of the E. coli EF-Tu:Phe-tRNAPhe complex (17), we selected six residues (His67, Asp216, Glu217, Phe219, Thr229, and Asn274) in the amino acid binding pocket of EF-Tu (Fig. 3A) for complete randomization in order to generate EF-Tu variants that bind Sep-tRNA. Variants that permitted SepRS and tRNASep-dependent Sep incorporation were selected in vivo (see SOM). One clone, designated EF-Sep (amino acid variants shown in Fig. 3A), led to a 10-fold increase in Cm resistance (Fig. 2, column H), whereas the combination of SepRS and EF-Sep without tRNASep was not active in the CAT suppression assay (Fig. 2, column G). Thus, it appeared that EF-Sep did bind Sep-tRNASep, a conclusion that was confirmed in the hydrolysis protection assay (Fig. 3B). This assay also shows that EF-Sep still retained some ability to bind Cys-tRNA (fig. S1).
To prove that the observed suppression is due to Sep incorporation, we expressed myoglobin with an amber codon in the Asp127 position (fig. S2A). The expected full-length protein was synthesized only when EF-Sep, SepRS, and tRNASep were coexpressed (fig. S2A). Mass spectrometry–time-of-flight (MS-TOF) and MS/MS analysis showed that Sep is present at the position specified by UAG in both the intact and trypsin-digested proteins (Fig. S2, B and C).
Final validation of our strategy was the synthesis of a Sep-containing human protein MEK1 (mitogen-activated ERK activating kinase 1). This key eukaryotic enzyme of the mitogen-activated signaling cascade is crucial for cell proliferation, development, differentiation, cell cycle progression, and oncogenesis (18). Activation of MEK1 requires posttranslational phosphorylation of Ser218 and Ser222 by MEK activating kinases (e.g., Raf-1, MEKK, or MOS). Substitution of both Ser residues with Glu yields a constitutively active enzyme, albeit with lower activity (19). We generated a clone encoding a MEK1 fusion protein [with the maltose binding protein (MBP) at the N terminus and a His6 tag at the C terminus] in which Ser222 was changed to Glu, and the Ser218 codon was replaced by UAG to encode Sep. After expression in the presence of SepRS, tRNASep, and EF-Sep, 25 μg of full-length MBP-MEK1(Sep218, Glu222) was isolated from 1 liter of culture. The presence of Sep in MBP-MEK1(Sep218,Glu222) protein was demonstrated by its ability to phosphorylate ERK2, which then phosphorylates myelin basic protein. MBP-MEK1(Sep218,Glu222) had a 2500-fold higher specific activity than MBP-MEK1(Ser218,Ser222) and a 70-fold higher specific activity than the constitutively active MBP-MEK1(Glu218,Glu222) (Fig. 4A). MS/MS analysis confirmed the incorporation of Sep at position 218 (Fig. 4B). To determine if our E. coli expression system would allow the simultaneous insertion of two Sep residues into the protein, we changed the Ser codons in positions 218 and 222 to UAG. As expected, the expression efficiency of MBP-MEK1(Sep218,Sep222) was markedly reduced compared to that of wild-type MBP-MEK1 (only about 1 μg of full-length protein was obtained from 1 liter of culture). The presence of Sep at both active-site positions of MEK1 was tested by Western blot analysis with a monoclonal antibody specific to phosphorylation at these two residues (Fig 4C). Only MBP-MEK1(Sep218,Sep222), and to a weaker extent MBP-MEK1(Sep218,Ser222), were detected in this experiment, whereas neither MBP-MEK1(Ser218,Ser222), MBP-MEK(Sep218,Glu222), or MBP-MEK(Glu218,Glu222) was recognized by the antibody. This demonstrates that the addition of SepRS, tRNASep, and EF-Sep endows E. coli with the ability to read UAG as a phosphoserine codon.
Fig. 4.

Properties of E. coli–produced Sep-containing human MEK1. (A) Kinase activity assay. MEK1 was produced in E. coli as a fusion protein with an N-terminal maltose-binding protein (MBP) tag and a C-terminal His6 tag. Residues Ser218 and Ser222, which are targets to phosphorylation by MEK1 activators, were mutated to either Glu218/Glu222 or Sep218/Glu222 to produce active MEK1 variants. Various amounts of MBP-MEK1-His6 with active-site residues Ser218/Ser222 (▼), Glu218/Glu222 (●), or Sep218/Glu222 (○) were used to phosphorylate inactive ERK2 in vitro. ERK2 activity was then measured in a radiometric assay using γ-[32P]ATP and myelin basic protein (MyBP) as substrates. Error bars indicate SEM. (B) MS/MS spectrum confirming the presence of Sep218 in MBP-MEK1(Sep218/Ser222). m/z, mass-to-charge ratio. (C) MBP-MEK1-His6 variants with genetically encoded active-site residues Sep218/Sep222 (lane 1), Ser218/Ser222 (lane 2), Sep218/Ser222 (lane 3), Sep218/Glu222 (lane 4), and Glu218/Glu222 (lane 5) were produced in E. coli and partially purified by Ni2+ affinity chromatography. Proteins were separated by SDS–polyacrylamide gel electrophoresis and either stained with Coomassie (top) or transferred to a nylon membrane and detected with monoclonal antibodies specific for the phosphorylated active site of human MEK (center) or the MBP tag (bottom). Purchased activated glutathione S-transferase (GST)–MEK was used as a control (lane 6). Dark and light arrowheads indicate the positions of MBP-MEK1-His6 and GST-MEK1, respectively. The strong bands (~70 kD size) are probable truncation products caused by termination at UAG.
Our work underscores the key role of EF-Tu in quality control of protein synthesis by ensuring facile delivery of the correct cognate aminoacyl-tRNA to the ribosome (10, 20). Generating an orthogonal aminoacyl–tRNA synthetase:tRNA pair was insufficient to genetically encode Sep. Expansion of the genetic code to include Sep depended critically on reengineering of EF-Tu to bind Sep-tRNASep. This situation is precisely paralleled in the naturally evolved genetic encoding system for selenocysteine, which requires a specialized elongation factor [SelB in prokaryotes, and EFSec in eukaryotes; reviewed in (21)]. Inspired careful manipulation of components of the protein-synthesizing system will allow further expansion of the genetic code without sacrificing organismal fitness.
Orthogonal aminoacyl–tRNA synthetase:tRNA pairs (in our case, SepRS:tRNASep) are critical elements for genetic code expansion, whether produced by evolutionary processes and found in nature [e.g., pyrrolysyl–tRNA synthetase:tRNAPyl (22)] or designed in the laboratory by synthetic biologists [reviewed in (8)]. The tRNA of choice is typically an amber suppressor tRNA; however, a general limitation in product yield is associated with recoding the new amino acid by a stop codon (e.g., UAG), because peptide chain elongation by the designed aminoacylated suppressor tRNA competes with chain termination by the release factor (e.g., RF1). Yet this impediment may soon be removed by the advent of E. coli expression strains with genome-wide UAG codon reassignments and an RF1 deletion (23, 24).
The ability to generate physiologically relevant active kinases and stoichiometrically phosphorylated protein domains could reveal new types of kinase inhibitors for drug development, allow systematic dissection of phosphorylation-dependent protein-protein interactions, and expose new structure-function relationships (18, 25, 26).
Supplementary Material
Acknowledgments
We thank P. Dennis, P. O’Donoghue, and J. Ling for enthusiastic discussions. M.J.H. was a Feodor Lynen Postdoctoral Fellow of the Alexander von Humboldt Foundation (Bonn, Germany). H.-S.P. held a postdoctoral fellowship of the Korean Science Foundation. This work was supported by grants from NSF (MCB-0645283 and MCB-0950474) (to D.S.), National Institute of General Medical Sciences (GM 22854) (to D.S.), National Research Foundation of Korea (ABC-20100029737) (to H.-S.P.), and National Institute of Diabetes and Digestive and Kidney Diseases (K01DK089006) (to J.R.). Yale University holds the U.S. Patent 7,723,069 B2: “Site Specific Incorporation of Phosphoserine into Polypeptides Using Phosphoseryl-tRNA Synthetase” by D. Soll and J. Rinehart. Yale University has applied for a patent that covers the engineered EF-Tu described in this manuscript. Reagents are available under a Yale Material Transfer Agreement.
Footnotes
References and Notes
- 1.Lipmann F. Biochem Z. 1933;262:3. [Google Scholar]
- 2.Olsen JV, et al. Cell. 2006;127:635. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 3.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. Science. 2002;298:1912. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 4.Sauerwald A, et al. Science. 2005;307:1969. doi: 10.1126/science.1108329. [DOI] [PubMed] [Google Scholar]
- 5.Hohn MJ, Park HS, O’Donoghue P, Schnitzbauer M, Söll D. Proc Natl Acad Sci USA. 2006;103:18095. doi: 10.1073/pnas.0608762103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamtekar S, et al. Proc Natl Acad Sci USA. 2007;104:2620. doi: 10.1073/pnas.0611504104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fukunaga R, Yokoyama S. Nat Struct Mol Biol. 2007;14:272. doi: 10.1038/nsmb1219. [DOI] [PubMed] [Google Scholar]
- 8.Liu CC, Schultz PG. Annu Rev Biochem. 2010;79:413. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- 9.Wanner BL, Metcalf WW. FEMS Microbiol Lett. 1992;79:133. doi: 10.1111/j.1574-6968.1992.tb14031.x. [DOI] [PubMed] [Google Scholar]
- 10.LaRiviere FJ, Wolfson AD, Uhlenbeck OC. Science. 2001;294:165. doi: 10.1126/science.1064242. [DOI] [PubMed] [Google Scholar]
- 11.Rothman DM, et al. J Am Chem Soc. 2005;127:846. doi: 10.1021/ja043875c. [DOI] [PubMed] [Google Scholar]
- 12.Dale T, Sanderson LE, Uhlenbeck OC. Biochemistry. 2004;43:6159. doi: 10.1021/bi036290o. [DOI] [PubMed] [Google Scholar]
- 13.Eargle J, Black AA, Sethi A, Trabuco LG, Luthey-Schulten Z. J Mol Biol. 2008;377:1382. doi: 10.1016/j.jmb.2008.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ling J, et al. Proc Natl Acad Sci USA. 2007;104:15299. [Google Scholar]
- 15.Doi Y, Ohtsuki T, Shimizu Y, Ueda T, Sisido M. J Am Chem Soc. 2007;129:14458. doi: 10.1021/ja075557u. [DOI] [PubMed] [Google Scholar]
- 16.Ohtsuki T, Yamamoto H, Doi Y, Sisido M. J Biochem. 2010;148:239. doi: 10.1093/jb/mvq053. [DOI] [PubMed] [Google Scholar]
- 17.Nissen P, et al. Science. 1995;270:1464. doi: 10.1126/science.270.5241.1464. [DOI] [PubMed] [Google Scholar]
- 18.Sebolt-Leopold JS, Herrera R. Nat Rev Cancer. 2004;4:937. doi: 10.1038/nrc1503. [DOI] [PubMed] [Google Scholar]
- 19.Alessi DR, et al. EMBO J. 1994;13:1610. doi: 10.1002/j.1460-2075.1994.tb06424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schrader JM, Chapman SJ, Uhlenbeck OC. Proc Natl Acad Sci USA. 2011;108:5215. doi: 10.1073/pnas.1102128108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoshizawa S, Böck A. Biochim Biophys Acta. 2009;1790:1404. doi: 10.1016/j.bbagen.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 22.Nozawa K, et al. Nature. 2009;457:1163. doi: 10.1038/nature07611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mukai T, et al. Nucleic Acids Res. 2010;38:8188. doi: 10.1093/nar/gkq707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Isaacs FJ, et al. Science. 2011;333:348. doi: 10.1126/science.1205822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scott JD, Pawson T. Science. 2009;326:1220. doi: 10.1126/science.1175668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yaffe MB, et al. Cell. 1997;91:961. doi: 10.1016/s0092-8674(00)80487-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.