

Abstract
BACKGROUND & AIMS
The signaling molecule and transcriptional regulator SMAD6, which inhibits the transforming growth factor beta (TGFB) signaling pathway, is required for infection of hepatocytes by hepatitis C virus (HCV). We investigated the mechanisms by which SMAD6, and another inhibitory SMAD (SMAD7), promote HCV infection in human hepatoma cells and hepatocytes.
METHODS
We infected Huh7 and Huh7.5.1 cells and primary human hepatocytes with JFH1 HCVcc; we measured HCV binding, intracellular levels of HCV RNA, and expression of target genes. HCV entry in HepG2/miR122/CD81 cells, which support entry and replication of HCV, were transfected with small-interfering (si)RNAs and gene expression profiles were analyzed. Uptake of labeled low-density lipoprotein (LDL) and cholesterol were measured. Cell surface proteins were quantified by flow cytometry. We obtained liver biopsy samples from 69 patients with chronic HCV infection and 19 uninfected individuals (controls) and measured levels of syndecan 1 (SDC1), SMAD7, and SMAD6 mRNAs.
RESULTS
siRNA knockdown of SMAD6 blocked the binding and infection of cell lines and primary human hepatocytes by HCV, whereas SMAD6 overexpression increased infection by HCV. We found levels of mRNAs encoding heparan sulfate proteoglycans (HSPGs), particularly SDC1 mRNA, and cell surface levels of heparan sulfate to be reduced in cells following SMAD6 knockdown. SMAD6 knockdown also reduced transcription of gene encoding lipoprotein and cholesterol uptake receptors, including the LDL receptor (LDLR), the very LDLR (VLDLR), and the scavenger receptor class B member 1 (SCARB1 or SR-BI) in hepatocytes; SMAD6 knockdown also reduced cell uptake of cholesterol and lipoprotein. Overexpression of SMAD6 increased expression of these genes. Similar effects were observed with knockdown and overexpression of SMAD7. HCV infection of cells increased expression of SMAD6, which required the activity of nuclear factor-kB (NF-kB), but not TGFB. Liver tissues from patients with chronic HCV infection had significantly higher levels of SMAD6, SMAD7, and HSPG mRNAs than controls.
Conclusions
In studies of hepatoma cell lines and human primary hepatocytes, we found that infection with HCV leads to activation of NF-κB, leading to increased expression of SMAD6 and SMAD7. Upregulation of SMAD6 and SMAD7 lead to increased expression of HSPGs, such as SDC1, as well as LDLR, VLDLR, and SR-BI, which promote HCV entry and propagation, as well as cell uptake of cholesterol and lipoprotein.
Keywords: I-SMAD, viral entry, BMP, CTBP1
Hepatitis C virus (HCV) is an etiologic agent of chronic hepatitis that can progress to end-stage live disease including fibrosis, cirrhosis and hepatocellular carcinoma1. Approximately 3% of the world population are estimated to be infected with HCV, and among them around 170 million people are chronically infected, leading to about 350,000 death each year globally2. Current therapeutic regimens consist of peginterferon, ribavirin, and recently approved direct-acting antivirals (DAAs) that are highly effective for most chronic hepatitis C patients3. HCV extensively interacts with host factors throughout the viral life cycle and thus trigger various pathological processes in hepatocytes4, 5. As such, interrogating these host dependencies of HCV may reveal not only novel targets for development of host-derived antivirals, but also cellular mechanisms underlying HCV-mediated liver disease.
HCV infects hepatocytes through a highly coordinated process that involves complex interactions of its envelope proteins (E1/E2) with cellular co-receptors and a number of other entry factors. The entry pathway consists of multiple sequential steps, starting from viral attachment or binding, to receptor-dependent intake followed by clathrin-mediated endocytosis, and finally fusion in early endosomes6. HCV attachment occurs when apolipoproteins present on lipoviral particles (LVPs) bind to heparan sulfate proteoglycans (HSPGs) expressed on cell surface7. Concurrently, low-density lipoprotein receptor (LDLR)8 and scavenger receptor class B1 (SR-BI)9 also interact with HCV LVPs to initiate HCV entry. SR-BI can also bind to HDL, VLDL and the oxidized forms of LDL to promote cholesterol uptake in hepatocytes10. In addition, SR-BI possesses cholesterol-transfer activity that may help unmask virus particles from their associated lipoproteins during the entry process11.
Through systems biology and integrative functional genomics approaches, we recently identified SMAD6, among a number of other cellular factors, as novel HCV host dependencies12. SMAD6 belongs to the inhibitory SMAD (I-SMAD) family that includes SMAD7, both of which negatively regulate TGF-β signaling pathway through feedback loop effects13. Here we explore the intrinsic roles and mechanisms of these I-SMADs in modulating HCV infection. We showed that both I-SMADs are requisite for HCV entry, acting mainly on the viral attachment step via novel transcriptional mechanisms that regulate both cell surface HSPG expression and cholesterol uptake. HCV infection, on the other hand, co-opts this proviral pathway for its own advantage through up-regulation of I-SMADs and their downstream signaling pathways.
Materials and Methods
HCV Infection
Huh7 or Huh7.5.1 cells were infected with JFH1 HCVcc at a multiplicity of infection (MOI) of 0.5 and passaged every three days after infection. Intracellular HCV RNA and target gene mRNA levels were measured every day until 7 days post-infection. Primary human hepatocytes were infected at a MOI of 1 for 7 days before harvested for various assays.
HCV Entry and Binding Assays
Huh7.5.1 or HepG2/miR122/CD81 cells were transfected with siRNAs for 72h and then infected with HCVpp, VSV-Gpp or MLVpp for 48h. Firefly luciferase activity, reflecting the entry level of the pseudoparticles, was measured using the Luciferase Assay System (Promega) according the manufacture’s protocols. For HCV binding assay, Huh7.5.1 cells were treated with various siRNAs for 72h, and subsequently infected with JFH1 HCVcc at 4°C for 2 h. Afterwards, unbound virus was removed by washing cells extensively with cold PBS, and virus bound to cells were determined by quantifying HCV RNA levels by RT-qPCR.
Flow Cytometry Analysis of Cell Surface HSPGs
Huh7.5.1 cells were seeded onto 12-wells plates and treated with siNT or SMAD6 siRNA as described above. After 72 h, cells were harvested and immunostained with a mouse anti-heparan sulfate delta antibody (3G10 epitope, US biological, 1:100), followed by incubation with a FITC-conjugated goat anti-mouse IgG antibody (Invitrogen, 1:1000). After extensively washing, cells were resuspended in cold PBS and subjected to flow cytometry on a FACS Canto flow cytometer (BD Biosciences) that collects ~40,000 gated events. FlowJo flow cytometry analysis software (Flowjo LLC) was used for analysis of HSPG expression levels under various treatment conditions.
Promoter Reporter Activity Assay
Huh7.5.1 cells were transfected with various siRNAs for 72 h or with Flag-tagged SMAD6 plasmid for 48 h, and then transfected with various indicated promoter constructs that encode a luciferase reporter (SwitchGear Genomics). After 48 h, cells were lysed and luciferase activities were measured with the Lightswitch Luciferase Assay System (SwitchGear Genomics).
Low-density Lipoprotein and Cholesterol Uptake Assay
For LDL uptake assay, Huh7.5.1 were transfected with various siRNAs for 72 h, and subsequently treated with 12.5 μg/mL of BODIPY-labeled LDL (Life Technologies) at 37°C for 12 h. Cells were then fixed with 4% paraformaldehyde before nuclear staining with Hoechst (Invitrogen). Images were acquired using ZEN 2009 software on a Zeiss confocal microscope (Carl Zeiss). For cholesterol uptake assay, cells pre-treated with various indicated siRNAs were incubated with 10 μg/mL of NBD cholesterol (Cayman Chemical Company) for 48 h. Cell based assay buffer was subsequently added and meanwhile cell nuclei were stained with Hoechst. NBD cholesterol uptake in cells was analyzed immediately under a fluorescent microscope (Carl Zeiss).
Microarray analysis
Huh7.5.1 cells were treated with siNT or siSMAD6 for 72 h, and then mock infected or infected with HCV at an MOI of 1. At 48 h post-infection, total cellular RNA was extracted and purified. RNA quality and quantity were analyzed using an Agilent Bioanalyzer. Cellular RNA was then amplified using an Agilent Enzo kit. Amplified complementary RNA was hybridized to an Affymetrix Human 133 Plus 2.0 microarray chip containing 54,675 gene transcripts. For microarray analysis, a >1.5-fold change in expression combining a >95% probability of being differentially expressed (P < 0.05) was considered to be biologically significant. Bioinformatics and statistical analysis of microarray data were performed at the NIDDK Genomics Core Facility.
Patient Samples
Sixty-nine patients were provided by the Liver Clinic at the National Institutes of Health (NIH) Clinical Center from a large cohort of CHC patients (n = 712). To exclude other factors, patients with other causes of liver diseases (including HBV or HIV co-infection), excessive alcohol consumption, decompensated liver disease, active substance abuse or severe systemic disease were not chosen. All tested samples were included in this retrospective analysis based on the availability of adequate stored liver biopsy specimen for analysis. All patients gave written informed consent for participation in clinical research and genetic testing. Normal human liver tissues (frozen) were provided by the NIH-supported Liver tissue Cell Distribution system (LTCDS).
Results
SMAD6 Enhances HCV Entry at the Attachment Stage
SMAD6, an I-SMAD and a versatile transcription factor that mediates multiple cellular signaling pathways, was identified as a putative HCV entry factor through our functional genomics screens conducted in the Huh7.5.1 cells12. Here we first validated the effect of SMAD6 on HCV entry in HepG2/miR122/CD81 cells that support efficient viral entry and replication14. Silencing of SMAD6 by pooled siRNA effectively blocked the infection of HCV pseudoparticle (HCVpp) genotypes 1a and 1b, but not that of VSV-Gpp or MLVpp, two control pseudoviruses (Figure 1A), suggesting that SMAD6 is uniquely exploited by HCV to infect hepatocytes. The function of SMAD6 in mediating HCV infection was further confirmed by the HCVcc assays. Efficient knockdown of SMAD6 by siRNA drastically reduced HCV RNA and core protein expression in Huh7.5.1 cells (Figures 1B–1C). Similarly, in primary human hepatocytes (PHHs), SMAD6 silencing resulted in marked decrease of HCV propagation (Figure 1D). SMAD6 siRNA robustly depleted target gene expression in both Huh7.5.1 cells and PHHs (Figures 1C and 1D). Treatment of cells with four individual SMAD6 siRNAs within the SMARTpool inhibited HCV infection that was proportionate to their knockdown efficiencies (Figure S1A–1B), revealing its physiological relevance in modulating HCV infection in hepatocytes. We also performed a gain-of-function assay by transfecting a Flag-tagged SMAD6 plasmid into Huh7.5.1 cells, and demonstrated that SMAD6 overexpression significantly enhanced HCV infection (Figure 1E). Furthermore, transfecting a siRNA-resistant mutant form of SMAD6 restored HCV infection in cells treated with SMAD6 siRNA (Figure S1C), supporting the phenotype-specific role of SMAD6 in mediating HCV infection.
Figure 1.
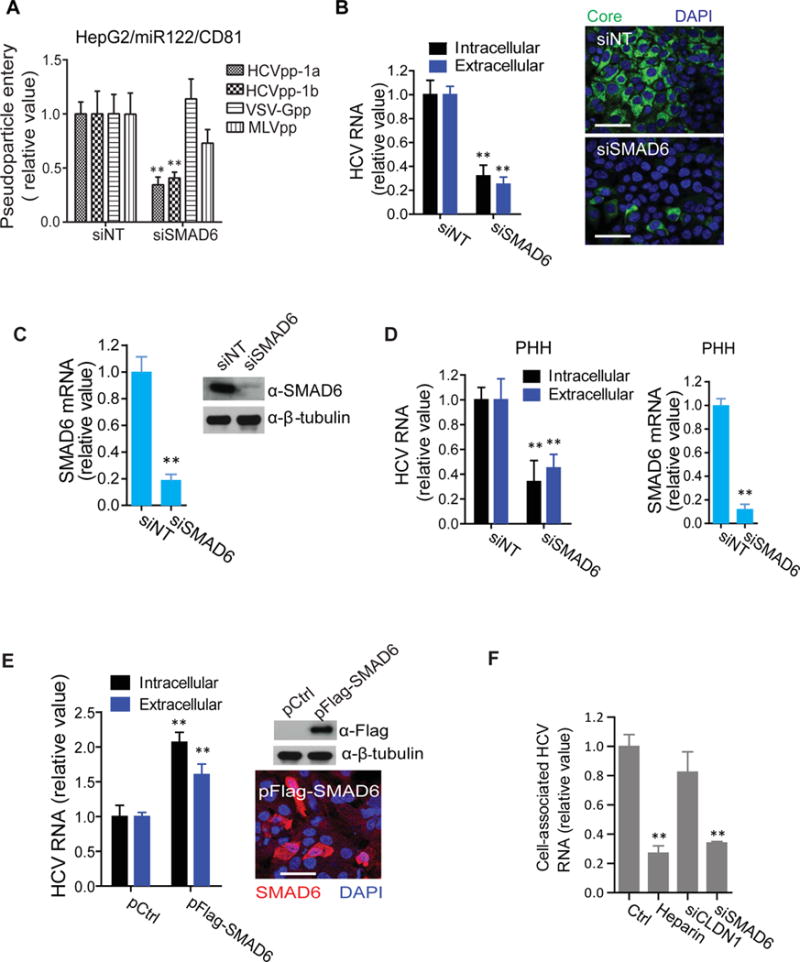
SMAD6 modulates HCV attachment, entry and infection. (A) Effect of SMAD6 knockdown on viral entry in HepG2/miR-122/CD81 cells. Cells were treated with non-targeting control (NT) or SMAD6 siRNA, and then infected with various pseudoviruses. Two days after infection, firefly luciferase activity was measured. (B) Effect of SMAD6 silencing on HCV RNA abundance (right panel) and core protein expression (left panel) in Huh7.5.1 cells. Intracellular and extracellular HCV RNA levels were determined by qPCR, and HCV core protein was immunostained with an anti-core monoclonal antibody. Representative images are shown. Green: core; blue: nuclei. Scale bar, 50 μm. (C) Knockdown efficiency of siSMAD6 in Huh7.5.1 cells. SMAD6 mRNA or protein levels were determined by qPCR or Western blot, respectively. β–tubulin was used as a loading control for Western blot. (D) SMAD6 silencing in PHHs inhibits HCV infection. HCV RNA and SMAD6 mRNA levels were quantified by qPCR. (E) SMAD6 overexpression enhances HCV infection. Huh7.5.1 cells were transfected with control (Ctrl) or Flag-tagged SMAD6 plasmid for 24 h, and then infected with HCV. At 48 h post-infection, HCV RNA levels was measured by qPCR, and SMAD6 protein level was determined by both Western blot and immunofluorescence using an anti-Flag antibody. For Western blot, β–tubulin serves as a loading control. A representative microscopic image is shown in the lower right panel. Red: SMAD6; blue: nuclei. Scale bar, 50 μm. (F) Effect of SMAD6 silencing on HCV binding. Huh7.5.1 cells were treated with SMAD6 or CLDN1 siRNA for 72 h or with heparin (200 g/mL) for 2 h and then incubated with HCV at 4°C for 2 h. Unbound virus was removed by extensive washes with cold PBS. Intracellular HCV RNA levels were determined by qPCR. All values were normalized as relative to siNT or Ctrl (set as 1), and represent the mean ± SD, n = 3 (B–F) or 5 (A). The asterisks indicate statistically significant differences (**p < 0.01).
Next, to examine whether SMAD6 is involved in the initial attachment step or downstream steps in the viral entry process, we performed HCVcc binding assay. Treatment of cells with SMAD6 siRNA inhibited HCV binding at 4°C to an extent comparable to treatment with heparin (Figure 1F) – a homologue of highly sulfated heparan sulfate that has been shown to selectively block HCV envelope glycoprotein binding7. In contrast, knockdown of CLDN1, a post-binding entry factor15, had no effect on HCV binding (Figure 1F). Collectively, these data suggest that SMAD6 is involved in HCV attachment that initiates viral entry.
SMAD6 Modulates HCV Entry Through Transcriptional Regulation of Cell Surface Heparan Sulfate Expression
To identify the mechanism and cellular signaling pathways that mediate the effect of SMAD6 on cell binding of HCV, we performed microarray gene expression analysis and evaluated global transcriptomic changes in hepatocytes null of SMAD6. Intriguingly, the mRNA levels of multiple HSPG core proteins16, particularly those of syndecan-1 (SDC1) and syndecan-2 (SDC2) were abundantly reduced upon SMAD6 silencing, either in the absence or in the presence of HCV infection (Figure 2A). Quantitative RT-PCR (qPCR) validated the SMAD6 depletion-based microarray results (Figure 2B), whereas overexpression of the protein significantly upregulated the HSPGs (Figure 2C), indicating a regulatory role of SMAD6 in HSPG expression in hepatocytes. Furthermore, knockdown of SMAD6 by siRNA in Huh7.5.1 cells substantially reduced cell surface heparan sulfate contents, examined by immunofluorescence and flow cytometry using a heparan sulfate-specific antibody (Figures 2D and 2E). Cell surface HSPGs, in particular SDC1, are critical host factors that mediate HCV attachment during viral entry7, 17, 18. As expected, SDC1 knockdown in Huh7.5.1 cells significantly inhibited HCV binding (Figure S2). To assess whether the transcription factor SMAD6 directly regulates SDC1 promoter activity, we either silenced or overexpressed SMAD6 in Huh7.5.1 cells, and demonstrated that the SDC1 promoter-driven luciferase reporter activity was significantly inhibited or enhanced, respectively (Figures 2F). Taken together, these results revealed a previously undescribed SMAD6-guided pathway that mediates the initial attachment step of HCV entry through transcriptional regulation of cell surface HSPGs.
Figure 2.
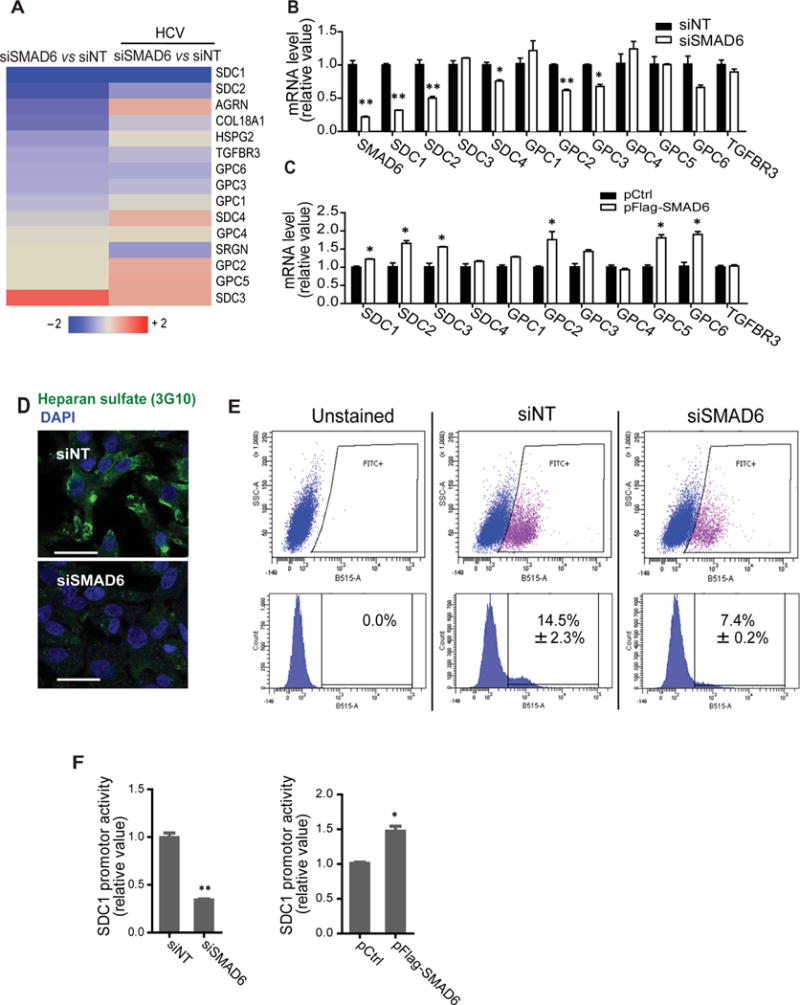
Regulation of cell surface heparan sulfate contents by SMAD6. (A) Heatmap illustrating siSMAD6-mediated regulation of HSPGs either in the absence or in the presence of HCV infection. Fold changes of mRNA levels of all known HSPG core proteins in Huh7.5.1 cells relative to siNT are shown. Values in the heatmap are depicted in a continuum of blue (downregulation) to red (upregulation). (B, C) Effects of SMAD6 knockdown (B) or overexpression (C) on mRNA levels of various HSPG core protein in Huh 7.5.1 cells, determined by qPCR. (D, E) Reduction of cell surface heparan sulfate contents upon SMAD6 silencing. Huh7.5.1 cells were depleted of SMAD6 expression via siRNA treatment for 72 h, and then immunostained with an anti-heparan sulfate delta (3G10 epitope) antibody for either immunofluorescence/confocal microscopy (D) or flow cytometry (E) that analyzes cell surface HSPG levels. Images shown are representatives of triplicated experiments. Scale bars represent 50 μM. (F) Effects of SMAD6 knockdown or overexpression on SDC1 promoter activity. Huh7.5.1 cells were treated with control or SMAD6 siRNA (left panel) or transfected with Flag-tagged SMAD6 (right panel) before transfection with a SDC1 promoter-driven luciferase reporter construct. Luciferase activities were measured after 48 h and are normalized to control cells. (B, C, F) Values were normalized as relative to siNT or Ctrl (set as 1), and represent the mean ± SD, n = 3 (B, C) or 5 (F). The asterisks indicate statistically significant differences (*p < 0.05; **p < 0.01).
Involvement of SMAD6 Co-repressors in HCV Entry
Like SMAD6, SMAD7 is another I-SMAD that acts as an antagonist of the canonical TGF-β pathway and its upstream bone morphogenetic protein (BMP) signaling19–21. Interestingly, we found that SMAD7 possesses similar function as SMAD6 in modulating HCV infection. SMAD7 knockdown by siRNA significantly restricted HCV infection and entry in Huh7.5.1 cells (Figures 3A, 3B and S3A), while overexpression of the protein led to a proviral effect (Figure 3C). Interestingly, silencing of various R-SMADs or Co-SMADs including SMAD2, SMAD3, SMAD4 and SMAD5, which are positive regulators of the TGF-β signaling, did not affect HCV entry or production (Figures 3A, S3A and S3B).
Figure 3.
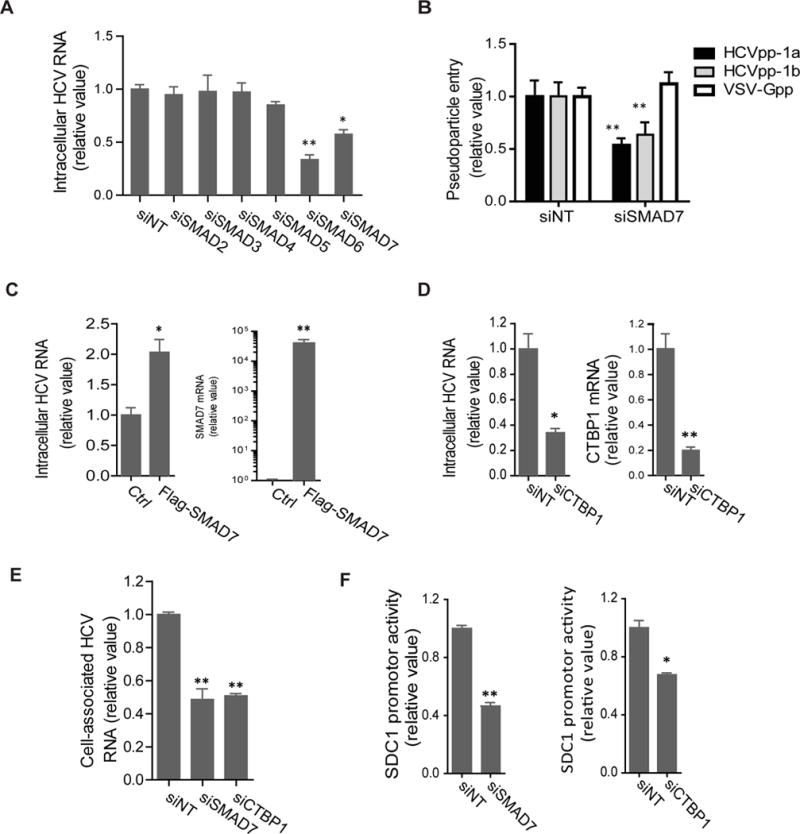
SMAD6 co-repressors regulate HSPG expression and HCV entry. (A) Silencing of various SMAD family members exerts varying effects on HCV infection. Intracellular HCV RNA levels were quantified at 48 h post-infection in Huh7.5.1 cells pre-treated with various indicated siRNAs. (B) Effect of SMAD7 knockdown on HCV genotypes 1a and 1b entry in Huh 7.5.1 cells. (C) SMAD7 overexpression enhances HCV infection. Intracellular HCV RNA and SMAD7 mRNA levels in Ctrl or Flag-SMAD7-transfected cells were determined by qPCR. (D) CTBP1 knockdown restricts HCV infection. Intracellular HCV RNA and CTBP1 mRNA levels in siNT or siCTBP1-treated cells were measured by qPCR. (E) Effect of SMAD7 or CTBP1 silencing on HCV attachment. SMAD7 or CTBP1 were depleted in Huh7.5.1 cells by siRNA treatment, and HCV binding ability at 4°C was evaluated by quantifying levels of cell-associated HCV RNA. (F) Silencing of SMAD7 or CTBP1 by siRNAs decreases SDC1 promoter activity in Huh7.5.1 cells. All values were normalized as relative to siNT or Ctrl (set as 1), and represent the mean ± SD, n = 5 (A, F) or 3 (B–E). The asterisks indicate statistically significant differences (*p < 0.05; **p < 0.01).
Since SMAD6 has been shown to recruit co-repressor C-terminal binding protein (CTBP1) in suppression of BMP-induced transcription22, we examined the potential role of CTBP1 in HCV infection. SiRNA-mediated knockdown of CTBP1 resulted in a significant reduction of HCV RNA level in Huh7.5.1 cells (Figure 3D), suggesting a similar proviral function of CTBP1 as the I-SMADs. CTBP1 or SMAD7 depletion in cells significantly inhibited HCV binding (Figure 3E), indicating that these I-SMAD co-repressors regulate the initial attachment step of HCV entry. Likewise, silencing of SMAD7 or CTBP1, but not SMAD3 or SMAD4, markedly attenuated SDC1 promoter activity (Figures 3F and S3C). In addition, SMAD7 or CTBP1 silencing, like knockdown of SMAD6, substantially reduced the mRNA levels of various HSPG core proteins in hepatocytes (Figure S3D), whereas overexpression of SMAD7 significantly up-regulated these HSPGs genes expression (Figure S3E). These data further validate the intrinsic role of I-SMADs and co-repressors in regulating HSPG expression and thus affecting HCV entry.
BMP6 and BMP7 Induce I-SMAD and HSPG Expression and Promote HCV Entry
I-SMADs are induced by BMP/TGF-β signaling pathway, while they also function as feedback inhibitors of the BMP/TGF-β signaling19–21. HSPGs, shown above to be regulated by I-SMADs and co-repressors (Figures 2 and 3), serve as BMP co-receptors that are associated with TGF-β components to modulate BMP/TGF-β signaling23–25. In order to examine whether the TGF-β superfamily members, including the BMPs and TGF-β1, affect HCV entry, we treated cells with these cytokines and performed viral infection assays. We showed that BMP6 and BMP7 treatment, like overexpression of I-SMADs in cells, significantly increased HCV core protein and RNA production (Figures 4A and 4B). This proviral function seems to be BMP6/7-specific, as treatment of cells with other BMPs, including BMP2 and BMP4, had no effect on HCV infection (Figure 4B). By contrast, TGF-β1 treatment markedly reduced HCV infection (Figures 4A and 4B), supporting the previously identified antiviral function of the TGF-β pathway in HCV infection26. We further showed that BMP6 and BMP7 facilitate HCV attachment in the early entry process, while other BMPs had no effect (Figure 4C). All cytokines tested are not associated with any significant cytotoxicity at the concentrations used (Figure S4A).
Figure 4.

BMP6/7 enhance I-SMAD expression and HCV binding. (A–C) TGF-β or BMP treatment executes varying effects on HCV infection. Huh7.5.1 cells were treated with TGF-β1 (0.1 ng/mL), BMP2 (0.1 g/mL), BMP4 (20 ng/mL), BMP6 (0.1 μg/mL) or BMP7 (0.5 μg/mL) for 24 h, and then infected with HCV at 37°C for 48 h (A, B) or 4°C for 2 h (C). HCV core staining (A), viral RNA quantification (B) and HCV binding assay (C) were subsequently performed. (A) Representative immunofluorescence images are shown. Green: core; blue: nuclei. Scale bar, 200 μm. The right panel shows quantification of core staining results. (D, E) BMP6/7 treatment induces I-SMAD (D) and HSPG (E) expression in Huh7.5.1 cells. All values were normalized as relative to Ctrl (set as 1), and represent the mean ± SD, n = 3. The asterisks indicate statistically significant differences (*p < 0.05; **p < 0.01).
Interestingly, BMP6 or BMP7 treatment drastically induced the expression of I-SMADs in Huh7.5.1 cells (Figure 4D), whereas the effects on the other SMAD family members were none (SMAD3 and SMAD5) or minor (SMAD4) (Figure S4B). In addition, BMP6 and BMP7, but not TGF-β1 or BMP-2/4 treatments, significantly induced the mRNA levels of various HSPG core proteins in hepatocytes (Figures 4E and S4C), likely reflecting their divergent effects on I-SMAD expression (Figure 4D). It is noteworthy that BMP2, while modestly upregulating SMAD6, did not significantly enhance HSPG core mRNA levels (Figure S4C). It is possible that the modest effect of BMP2 on SMAD6 expression was not sufficient to cause a significant increase in HSPG expression. To summarize, these data demonstrated a BMP6/7-triggered, I-SMAD-mediated, and canonical TGF-β pathway-independent transcriptional mechanism that preferentially acts on the initial attachment step of HCV infection via upregulation of HSPG expression on cell surface.
SMAD6 Regulates Hepatocellular Lipoprotein and Cholesterol Uptake
HCV, circulating in the form of LVPs that highly resemble very low-density lipoprotein (VLDL), exploits cell surface receptors that are involved in cholesterol and lipid uptake to gain entry into hepatocytes27. Intriguingly, in our microarray analysis as mentioned above, multiple lipoprotein and cholesterol uptake receptors, including LDLR, SR-BI and VLDLR were markedly downregulated at the mRNA levels as a result of SMAD6 knockdown (Figure 5A). Confirmatory qPCR assays validated the SMAD6 siRNA-mediated effects (Figure 5B), while over-expression of SMAD6 up-regulated these genes (Figure 5C). These data indicate that SMAD6 may also transcriptionally regulate hepatic cholesterol and lipid uptake through its effect on the expression of various lipo-receptors. Indeed, silencing of SMAD6 by siRNA in Huh7.5.1 cells markedly reduced cellular uptake of NBD cholesterol and BODIPY-labeled LDL as measured by fluorescent microscopy and subsequent quantitative analysis (Figure 5D and 5E). Interestingly, cell surface HSPGs have also been attributed to hepatic lipoprotein uptake28, 29. As expected, depletion of SDC1 in hepatocytes reduced cellular uptake of cholesterol and LDL which is similar to the effects of SR-BI, LDLR or VLDLR silencing (Figure 5D and 5E). These data collectively suggest that SMAD6 directs a transcriptional mechanism in regulating hepatic lipoprotein and cholesterol uptake that is important in HCV entry. As such, we conclude that SMAD6 mediates HCV attachment and entry via two concurrent pathways that both involve its transcriptional activities.
Figure 5.

SMAD6 regulates cholesterol and lipid uptake in hepatocytes. (A) Heatmap eliciting the suppression of various lipoprotein and cholesterol uptake receptors by SMAD6 silencing in Huh7.5.1 cells. Reduction of gene expression in the heatmap is illustrated as blue comparing with siNT. (B) Validation of the microarray results on siSMAD6-medaited reduction of VLDLR, LDLR and SR-BI expression by qPCR. (C) Effect of SMAD6 overexpression on VLDLR, LDLR and SR-BI expression. (B, C) Values were normalized as relative to siNT (set as 1), and represent the mean ± SD, n = 3. The asterisks indicate statistically significant differences (*p < 0.05; **p < 0.01). (D, E) Effects of SMAD6, SDC1, SR-BI, LDLR or VLDLR silencing on uptake of NBD cholesterol (D) or BODIPY-labeled LDL (E) into Huh7.5.1 cells, examined by fluorescent microscopy. Representative immunofluorescence images are shown. Green: cholesterol (D) or LDL (E); blue: nuclei. Scale bar, 200 μm. Quantitative analyses of cholesterol and LDL up taken from above shown images were performed using Image J by counting various florescent spots, and the results are shown in the bottom panels. Values were normalized as relative to siNT (set as 1), and the asterisks indicate statistically significant differences (**p < 0.01; NS, not significant).
HCV Infection Induces I-SMAD and HSPG Expression
Viruses like HCV employ various strategies, such as inducing or activating host proviral machineries or subverting host antiviral responses to achieve productive infection. Here we examined whether HCV infection manipulates the I-SMAD–HSPG pathway for its own benefit. First, we infected Huh7 cells with the JFH1 strain of HCV, and evaluated viral replication and relevant expression levels of I-SMADs and HSPGs (Figure S5A). We found that I-SMAD mRNA levels were substantially elevated from day 5 of the infection, and increased by over 3.5-fold on day 7, the longest duration of HCV infection we can keep in vitro without cell death (Figure 6A). HCV infection also markedly increased SMAD6 protein level in Huh7 cells (Figure 6B). Similarly, in PHHs infected with HCV, I-SMAD mRNA levels were increased by over 2.5-fold on day 7 (Figure 6C).
Figure 6.
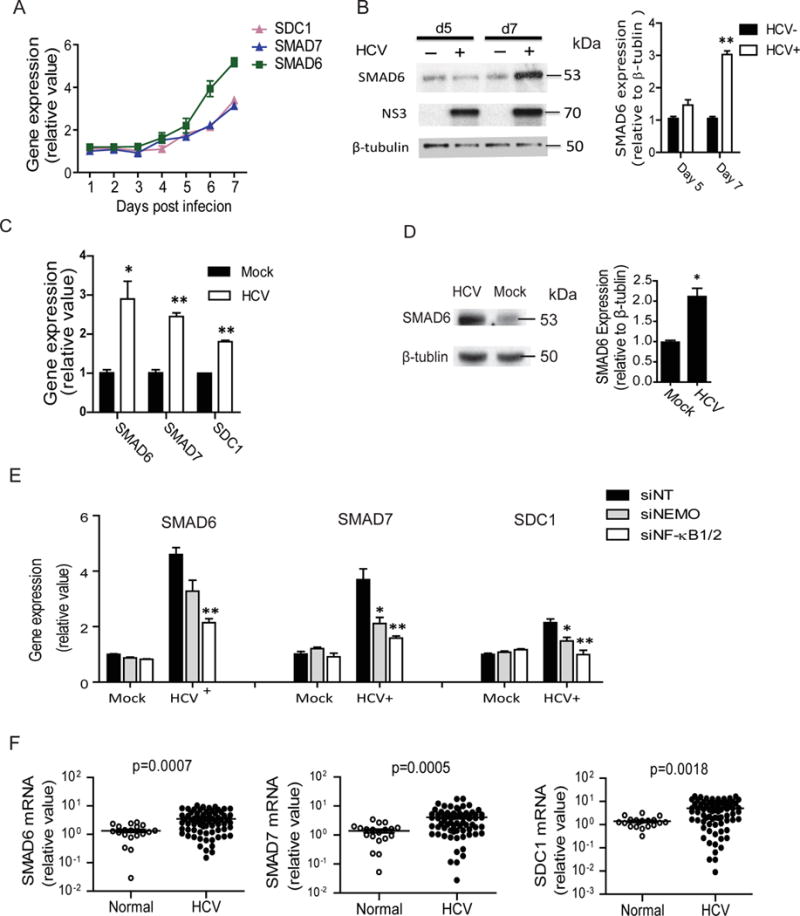
HCV infection upregulates I-SMAD and HSPG expression. (A) I-SMAD and SDC1 mRNA levels increased over time upon HCV infection in Huh7 cells. HCV infection was maintained for 7 days. (B) SMAD6 protein level also increased at 5 and 7 days post-infection in Huh7 cells. (C) The expression of I-SMAD and SDC1 was induced by HCV in PHHs. Various indicated mRNA levels were quantified at 7 days post-infection. (D) HCV infection increases SMAD6 protein level in PHHs as determined by Western blot. β–tubulin was used as a loading control. Representative Western blot gels and quantification of band intensities are shown. (E) HCV infection induces I-SMAD and SDC1 expression via the NF-κB pathway. Huh7 cells were infected with HCV JFH1 strain for 24 h, and then treated with siRNAs targeting NF-κB1/2 or NEMO. The expression of I-SMADs and SDC1 were subsequently assessed by RT-qPCR. (C–E) Values were normalized as relative to mock infection (set as 1), and represent the mean ± SD, n = 3. The asterisks indicate statistically significant differences (*p < 0.05; **p < 0.01). (F) Enhanced I-SMADs and SDC1 expression in the liver biopsy samples of chronic hepatitis C patients (n=69), comparing with those of HCV-naïve patients (n=19).
In addition, SMAD6 protein level was increased in HCV-infected PHHs examined by Western blot (Figure 6D). Likewise, the expression levels of SDC1 and various other HSPG core proteins were significantly elevated during HCV infection in both Huh7 cells and PHHs (Figures 6A, 6C and S5A, S5B), indicating that the I-SMAD–HSPG pathway is induced upon HCV infection.
We then investigated the mediator of I-SMAD upregulation in the setting of HCV infection. Expression of I-SMADs can be regulated by multiple extracellular signals, including EGF/MAPK, TNF-α/NF-κB, IFN-γ/JAK1/STAT1, and TGF-β/BMP30. Genes in these pathways (MAPK1, NF-κB1, NF-κB2, STAT1, TGF-β1, TGF-β2, TGF-β3, BMP6 and BMP7) were studied in HCV-infected cells (Figure S5C). Among them, TGF-β and NF-κB components were shown to be upregulated by HCV infection. Indeed, both pathways have previously been shown to be activated by HCV4, 31–33. To explore whether these signaling pathways are involved in the HCV-triggered induction of I-SMADs, we first treated Huh7 cells with SB-431542, a potent and specific inhibitor of TGF-β signaling. We showed that SB-431542 treatment did not diminish the induction of SMAD6 expression by HCV infection (Figure S5D), implying that the TGF-β pathway is not involved. We then studied the NF-κB pathway. Cells were depleted of NEMO – a crucial NF-κB-activating kinase, or NF-κB1/2 by siRNA treatment to silence the NF-κB signaling (Figure S5E). We also treated cells with a NF-κB specific inhibitor – caffeic acid phenethyl este (CAPE) (Figure S5F). Interestingly, HCV-triggered induction of I-SMAD and HSPG expression was substantially abrogated in the NF-κB-deficient cells (Figures 6E, S5F and S5G), suggesting that NF-κB activation is required for HCV-mediated upregulation of I-SMAD and HSPG expression in hepatocytes.
Finally, we asked whether the upregulation of I-SMADs and HSPGs is clinically relevant to HCV infection. We examined the expression levels of these genes in liver biopsy samples of HCV-naïve (n = 19) or chronic hepatitis C (n = 69) patients. Significantly higher mRNA levels of I-SMADs and SDC1 were observed in the liver tissues infected with HCV (Figure 6F). These results support that chronic HCV infection leads to the induction of a specific proviral host pathway involving the I-SMADs and HSPGs, which may in turn contribute to viral persistence.
Discussion
HCV enters host cells through a coordinated cascade of pathways engaging a number of entry factors, including CD8134, tight junction proteins CLDN115 and OCLN35, the receptor tyrosine kinases epidermal growth factor receptor (EGFR) and ephrin receptor A2 (EphA2)36, Niemann-Pick C1-like 1 (NPC1L1)37 and iron uptake receptor transferrin receptor 1 (TRF1)38. Indeed, how this complexed chain of cell signals and virus-host interactions is orchestrated and regulated to the advantage of the virus is not yet clear. Applying integrative functional genomics and systems biology approaches, we recently dissected global HCV–host interactions and uncovered SMAD6 as a novel host proviral factor for HCV39. In the present study, we interrogated the precise functions and mechanisms of action of SMAD6 in modulating HCV infection. We found that SMAD6 and the other I-SMAD, SMAD7, are both required for HCV entry. Present as important transcription factors in cells, these I-SMADs were shown to regulate the expression of HSPGs (particularly syndecans) on cell surface, thereby affecting the initial attachment step of the viral entry process. I-SMADs were also shown to manipulate cholesterol uptake via multiple pathways (mediated by SR-BI, VLDLR or LDLR) that are closely associated with HCV entry. Intriguingly, HCV infection, through enhancing I-SMAD and HSPG expression in hepatocytes, co-opts this intrinsic proviral pathway for its own advantage. A working model of these novel interactions is illustrated in Figure 7.
Figure 7.
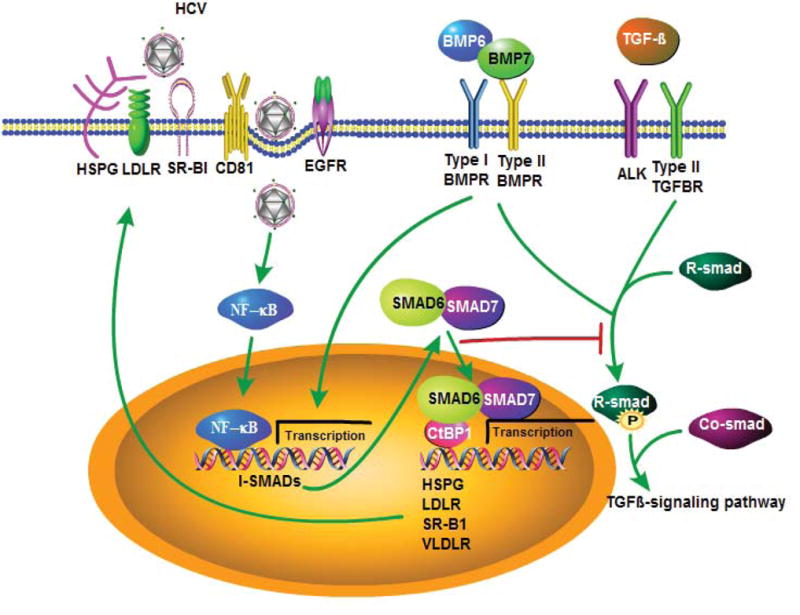
A proposed model for I-SMAD-regulated signaling that enhances HSPG expression, cholesterol uptake and HCV entry. HCV entry is mediated by viral binding to HSPGs, LDLR and SR-BI (and other entry factors, not shown) on the host cell surface, thereby triggering a cassette of signaling to facilitate the internalization of HCV virions. HCV infection also induces the expression of SMAD6 and SMAD7, two I-SMADs of the TGF-β signaling pathway via NF-κB regulation. The I-SMADs translocate to the nucleus and transcriptionally activate the expression HSPG core protein and other cholesterol uptake receptors. Increased expression level of heparin sulfate on the cell surface in turn enhances HCV binding. Additionally, the I-SMADs and HSPGs can be induced by BMP6 and BMP7, the ligands in the BMP/TGF-β pathway.
Here we identified I-SMADs as novel HCV entry factors acting at the early viral attachment stage. The I-SMADs belong to the SMAD family of proteins that are important signal transducers and key regulators of proliferation, differentiation, migration, apoptosis and immune response40. The SMAD family consists of eight members, and among them, five (SMAD1, 2, 3, 5 and 8) are R-SMADs, all containing a phosphorylation domain that can be activated by type I receptor of the TGF-β receptor family. Specifically, SMAD2 and 3 mediate the signaling through TGF-β, whereas SMAD1, 5 and 8 mediate the BMP signaling. The R-SMADs, once phosphorylated through receptor binding, complex with SMAD4, which plays a role in recruiting the downstream co-regulators for transcriptional activation of various cellular processes. SMAD6 and 7 are both I-SMADs and are induced by the TGF-β and BMP signaling while also act as TGF-β/BMP feedback inhibitors. The main function of the I-SMADs is to prevent ligand-induced activation of SMAD proteins in the TGF-β signaling pathway19–21. In fact, these diverse properties and functions of SMAD family members may account for their divergent effects on HCV entry and infection observed in this study.
Accumulating evidence has demonstrated that I-SMADs may act as transcriptional co-activators that are independent of the R-SMAD signaling41, 42. The C-terminus of the I-SMADs has a MH2 domain that is indeed conserved among all SMAD proteins. Domain mapping studies and relative structure-based data suggested that MH2 domains, in response to TGF-β or BMP stimulation, form a complex with transcription activation potential43. Surprisingly, our data indicate that only the I-SMADs but not the R-SMADs or SMAD4 possess transcription regulatory activities on the HSPG expression in hepatocytes that is essential for HCV attachment and entry, implicating a previously unidentified and unique function of I-SMADs that is distinct from the other SMAD proteins.
In our study, two secreted signaling molecules and TGF-β superfamily members, BMP6 and BMP7, were shown to enhance HCV attachment and entry by inducing I-SMAD and HSPG expression. Interestingly, BMP6 was found to facilitate SMAD phosphorylation, a process that can be inhibited by adding exogenous heparan sulfate. Therefore, BMP6 may functionally interact with HSPGs44. Indeed, HSPGs can act as BMP co-receptors, bind to TGF-β components and play an important role in regulating BMP/TGF-β signaling23, 24. We showed that BMP6/7 but not BMP2/4 affected HCV binding and I-SMAD and HSPG expression. Mechanistically, BMP6/7 interact with type II receptors Act RII and Act RIIB on the cell surface, and recruit type I receptors Alk2, Alk3 and Alk6, while the receptor-binding affinity is reversed for BMP2/4, which preferentially bind to the type I receptors Alk3 and Alk6 and recruit type II receptors to the heteromeric signaling complexes45. The difference in receptor binding activities between two BMP sets may explain the BMP6/7-specific function in modulating I-SMADs. It is interesting that BMP6, which regulates iron uptake via hepcidin46, appears to have a previously unrecognized function in regulating cellular cholesterol uptake via induction of I-SMADs, linking iron homeostasis and lipid metabolism47, 48.
It is known that I-SMADs are regulated by TGF-β/BMP and other signaling, including EGF/MAPK, IFN-γ/STAT and TNF-α/NF-κB pathways30. We found that only NF-κB signaling but not TGF-β/BMP. STAT1 or MAPK1 is responsible for the I-SMAD induction upon HCV infection. Indeed, the NF-κB pathway has been implicated in multiple viral infections49. The correlation between NF-κB and I-SMAD-mediated pathway identified in this study suggests that HCV, by activating the innate NF-κB pathway - probably via engaging pattern recognition receptors (RIG-I for example), upregulates I-SMADs to facilitate viral propagation.
Heparan sulfate is expressed on most mammalian cell surface as proteoglycan and consists of a core and highly sulfated glycosaminoglycan (GAG) side chains. HSPGs bind to various extracellular proteins, adhesion molecules and growth factors and have been shown to mediate the binding and entry of a variety of viruses including HCV7,50. The role of cell surface HSPGs in HCV binding appears to be mediated by their specific interaction with ApoE present on viral LVPs18, 28. HSPG core proteins consist of membrane-bound syndecans (SDC1-4) and glypicans (GPC1-6), serglycin expressed in secretory vesicles, and perlecan (also known as HSPG2)50. The majority of these HSPG core proteins were shown to be regulated by I-SMADs in our study. A previous report demonstrated that multiple SDCs mediate HCV attachment to hepatocytes4, further supporting our notion that I-SMADs modulate HCV binding and entry through up-regulation of HSPGs.
Collectively, our study, for the first time, reveals a transcription regulatory mechanism that mediates viral entry. The pathways underlying the regulation of HCV entry process involve I-SMADs, cell surface HSPGs and cholesterol uptake receptors. In addition, this specific I-SMAD–HSPG axis is induced by HCV infection, likely via the activation of NF-κB, either in cell culture models or in liver biopsies of chronic hepatitis C patients. Apart from the two I-SMADs, other members of SMAD proteins, including SMAD2/3/4/5 – positive regulators of the TGF-β signaling pathway – are dispensable for HCV entry and HSPG regulation, indicating that the functions of I-SMADs observed in this study are unique and independent of the canonical TGF-β pathway. Our study not only provides crucial insight into the complex mechanism of HCV entry but also reveals previously undescribed biological pathways in lipid and cholesterol metabolism that may link inflammation, immune regulation and metabolic dysfunction.
Supplementary Material
Acknowledgments
We thank Brianna Lowey for technical assistance and members of the Liver Diseases Branch for helpful discussion. We also thank Chithra Keembiyehetty and Weiping Chen of the NIDDK Genomics Core for assistance in microarray experiments and data analysis. Normal human liver tissues were obtained through the Liver Tissue Cell Distribution System, Minneapolis, Minnesota, which was funded by NIH Contract # HHSN276201200017C.
Grant Support: This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health.
Abbreviations used
- BMP
bone morphogenetic protein
- CD81
cluster of differentiation 81
- CLDN1
claudin-1
- CTBP1
co-repressor C-terminal binding protein
- HCVpp
HCV pseudoparticle
- LDLR
low density lipoprotein receptor
- VLDLR
very low density lipoprotein receptor
- NEMO
NF-kappa-B essential modulator
- NF-κB
nuclear factor-kappa B
- PHHs
primary human hepatocytes
- SDC
syndecan
- SR-BI
scavenger receptor class B member 1
- TGF-β
Transforming growth factor beta
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: The authors disclose no conflicts.
Transcript Profiling: The accession number of repository for expression microarray data is …
Author Contributions: Study concept and design: QL, TJL; Acquisition of data: FZ, CS, HC, QL; Analysis and interpretation of data: FZ, QL, TJL; Writing of the manuscript: FZ, QL, TJL.
Author names in bold designate shared co-first authors.
References
- 1.Liang TJ, Heller T. Pathogenesis of hepatitis C-associated hepatocellular carcinoma. Gastroenterology. 2004;127:S62–71. doi: 10.1053/j.gastro.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 2.Mohd Hanafiah K, Groeger J, Flaxman AD, et al. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology. 2013;57:1333–42. doi: 10.1002/hep.26141. [DOI] [PubMed] [Google Scholar]
- 3.Liang TJ, Ghany MG. Current and future therapies for hepatitis C virus infection. N Engl J Med. 2013;368:1907–17. doi: 10.1056/NEJMra1213651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Q, Pene V, Krishnamurthy S, et al. Hepatitis C virus infection activates an innate pathway involving IKK-alpha in lipogenesis and viral assembly. Nat Med. 2013;19:722–9. doi: 10.1038/nm.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scheel TK, Rice CM. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med. 2013;19:837–49. doi: 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ploss A, Evans MJ. Hepatitis C virus host cell entry. Curr Opin Virol. 2012;2:14–9. doi: 10.1016/j.coviro.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barth H, Schafer C, Adah MI, et al. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J Biol Chem. 2003;278:41003–12. doi: 10.1074/jbc.M302267200. [DOI] [PubMed] [Google Scholar]
- 8.Agnello V, Abel G, Elfahal M, et al. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc Natl Acad Sci U S A. 1999;96:12766–71. doi: 10.1073/pnas.96.22.12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barth H, Cerino R, Arcuri M, et al. Scavenger receptor class B type I and hepatitis C virus infection of primary tupaia hepatocytes. J Virol. 2005;79:5774–85. doi: 10.1128/JVI.79.9.5774-5785.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Acton S, Rigotti A, Landschulz KT, et al. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271:518–20. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 11.Bankwitz D, Steinmann E, Bitzegeio J, et al. Hepatitis C virus hypervariable region 1 modulates receptor interactions, conceals the CD81 binding site, and protects conserved neutralizing epitopes. J Virol. 2010;84:5751–63. doi: 10.1128/JVI.02200-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Q, Zhang YY, Chiu S, et al. Integrative functional genomics of hepatitis C virus infection identifies host dependencies in complete viral replication cycle. PLoS Pathog. 2014;10:e1004163. doi: 10.1371/journal.ppat.1004163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benchabane H, Wrana JL. GATA- and Smad1-dependent enhancers in the Smad7 gene differentially interpret bone morphogenetic protein concentrations. Mol Cell Biol. 2003;23:6646–61. doi: 10.1128/MCB.23.18.6646-6661.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Narbus CM, Israelow B, Sourisseau M, et al. HepG2 cells expressing microRNA miR-122 support the entire hepatitis C virus life cycle. J Virol. 2011;85:12087–92. doi: 10.1128/JVI.05843-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Evans MJ, von Hahn T, Tscherne DM, et al. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature. 2007;446:801–5. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 16.Sarrazin S, Lamanna WC, Esko JD. Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi Q, Jiang J, Luo G. Syndecan-1 serves as the major receptor for attachment of hepatitis C virus to the surfaces of hepatocytes. J Virol. 2013;87:6866–75. doi: 10.1128/JVI.03475-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu Y, Martinez P, Seron K, et al. Characterization of hepatitis C virus interaction with heparan sulfate proteoglycans. J Virol. 2015;89:3846–58. doi: 10.1128/JVI.03647-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hayashi H, Abdollah S, Qiu Y, et al. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell. 1997;89:1165–73. doi: 10.1016/s0092-8674(00)80303-7. [DOI] [PubMed] [Google Scholar]
- 20.Imamura T, Takase M, Nishihara A, et al. Smad6 inhibits signalling by the TGF-beta superfamily. Nature. 1997;389:622–6. doi: 10.1038/39355. [DOI] [PubMed] [Google Scholar]
- 21.Murakami G, Watabe T, Takaoka K, et al. Cooperative inhibition of bone morphogenetic protein signaling by Smurf1 and inhibitory Smads. Mol Biol Cell. 2003;14:2809–17. doi: 10.1091/mbc.E02-07-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin X, Liang YY, Sun B, et al. Smad6 recruits transcription corepressor CtBP to repress bone morphogenetic protein-induced transcription. Mol Cell Biol. 2003;23:9081–93. doi: 10.1128/MCB.23.24.9081-9093.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Irie A, Habuchi H, Kimata K, et al. Heparan sulfate is required for bone morphogenetic protein-7 signaling. Biochem Biophys Res Commun. 2003;308:858–65. doi: 10.1016/s0006-291x(03)01500-6. [DOI] [PubMed] [Google Scholar]
- 24.Kuo WJ, Digman MA, Lander AD. Heparan sulfate acts as a bone morphogenetic protein coreceptor by facilitating ligand-induced receptor hetero-oligomerization. Mol Biol Cell. 2010;21:4028–41. doi: 10.1091/mbc.E10-04-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Santander C, Brandan E. Betaglycan induces TGF-beta signaling in a ligand-independent manner, through activation of the p38 pathway. Cell Signal. 2006;18:1482–91. doi: 10.1016/j.cellsig.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 26.Murata T, Ohshima T, Yamaji M, et al. Suppression of hepatitis C virus replicon by TGF-beta. Virology. 2005;331:407–17. doi: 10.1016/j.virol.2004.10.036. [DOI] [PubMed] [Google Scholar]
- 27.Schaefer EA, Chung RT. HCV and host lipids: an intimate connection. Semin Liver Dis. 2013;33:358–68. doi: 10.1055/s-0033-1358524. [DOI] [PubMed] [Google Scholar]
- 28.Jiang J, Cun W, Wu X, et al. Hepatitis C virus attachment mediated by apolipoprotein E binding to cell surface heparan sulfate. J Virol. 2012;86:7256–67. doi: 10.1128/JVI.07222-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lefevre M, Felmlee DJ, Parnot M, et al. Syndecan 4 is involved in mediating HCV entry through interaction with lipoviral particle-associated apolipoprotein E. PLoS One. 2014;9:e95550. doi: 10.1371/journal.pone.0095550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 31.Lee J, Tian Y, Chan ST, et al. TNF-alpha Induced by Hepatitis C Virus via TLR7 and TLR8 in Hepatocytes Supports Interferon Signaling via an Autocrine Mechanism. PLoS Pathog. 2015;11:e1004937. doi: 10.1371/journal.ppat.1004937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin W, Tsai WL, Shao RX, et al. Hepatitis C virus regulates transforming growth factor beta1 production through the generation of reactive oxygen species in a nuclear factor kappaB-dependent manner. Gastroenterology. 2010;138:2509–18. 2518 e1. doi: 10.1053/j.gastro.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Q, Sodroski C, Lowey B, et al. Hepatitis C virus depends on E-cadherin as an entry factor and regulates its expression in epithelial to mesenchymal transition. PNAS. 2016 doi: 10.1073/pnas.1602701113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pileri P, Uematsu Y, Campagnoli S, et al. Binding of hepatitis C virus to CD81. Science. 1998;282:938–41. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- 35.Ploss A, Evans MJ, Gaysinskaya VA, et al. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature. 2009;457:882–6. doi: 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lupberger J, Zeisel MB, Xiao F, et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med. 2011;17:589–95. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sainz B, Jr, Barretto N, Martin DN, et al. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat Med. 2012;18:281–5. doi: 10.1038/nm.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin DN, Uprichard SL. Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc Natl Acad Sci U S A. 2013;110:10777–82. doi: 10.1073/pnas.1301764110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Q, Brass AL, Ng A, et al. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc Natl Acad Sci U S A. 2009;106:16410–5. doi: 10.1073/pnas.0907439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Massague J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–91. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 41.Glesne D, Huberman E. Smad6 is a protein kinase X phosphorylation substrate and is required for HL-60 cell differentiation. Oncogene. 2006;25:4086–98. doi: 10.1038/sj.onc.1209436. [DOI] [PubMed] [Google Scholar]
- 42.Hong S, Kim HY, Kim J, et al. Smad7 protein induces interferon regulatory factor 1-dependent transcriptional activation of caspase 8 to restore tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis. J Biol Chem. 2013;288:3560–70. doi: 10.1074/jbc.M112.400408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, Feng X, We R, et al. Receptor-associated Mad homologues synergize as effectors of the TGF-beta response. Nature. 1996;383:168–72. doi: 10.1038/383168a0. [DOI] [PubMed] [Google Scholar]
- 44.Brkljacic J, Pauk M, Erjavec I, et al. Exogenous heparin binds and inhibits bone morphogenetic protein 6 biological activity. Int Orthop. 2013;37:529–41. doi: 10.1007/s00264-012-1714-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wiley DM, Jin SW. Bone Morphogenetic Protein functions as a context-dependent angiogenic cue in vertebrates. Seminars in Cell & Developmental Biology. 2011;22:1012–1018. doi: 10.1016/j.semcdb.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parrow NL, Fleming RE. Bone morphogenetic proteins as regulators of iron metabolism. Annu Rev Nutr. 2014;34:77–94. doi: 10.1146/annurev-nutr-071813-105646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fernandez-Real JM, Manco M. Effects of iron overload on chronic metabolic diseases. Lancet Diabetes Endocrinol. 2014;2:513–26. doi: 10.1016/S2213-8587(13)70174-8. [DOI] [PubMed] [Google Scholar]
- 48.Dongiovanni P, Fracanzani AL, Fargion S, et al. Iron in fatty liver and in the metabolic syndrome: a promising therapeutic target. J Hepatol. 2011;55:920–32. doi: 10.1016/j.jhep.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 49.Marianneau P, Cardona A, Edelman L, et al. Dengue virus replication in human hepatoma cells activates NF-kappaB which in turn induces apoptotic cell death. J Virol. 1997;71:3244–9. doi: 10.1128/jvi.71.4.3244-3249.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–7. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.