

Abstract
Cadmium (Cd) is a carcinogenic heavy metal which is implicated in breast cancer development. While the mechanisms of Cd-induced breast cancer initiation and promotion have been studied, the molecular processes involved in breast cancer progression remain to be investigated. The purpose of the present study was to evaluate the influence of Cd on metastasis-associated phenotypes, such as cell adhesion, migration, and invasion in triple-negative breast cancer cells. Treatment of MDA-MB-231 cells with 1 μM Cd increased cell spreading and cell migration. This was associated with the activation of integrin β1, FAK, Src, and Rac1. Treatment with Cd also inhibited GSK3β activity and induced T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription, indicating the involvement of β-catenin signaling. Furthermore, treatment with 3 μM Cd for 4 weeks increased the expression of β-catenin and enhanced TCF/LEF-mediated transcription. Furthermore, enhanced expressions of integrins α5 and β1, paxillin, and vimentin indicated that prolonged Cd treatment reorganized the cytoskeleton, which aided malignancy, as evidenced by enhanced matrix metalloprotease 2/9 (MMP2/9) secretion and cell invasion. Prolonged Cd treatment also caused an increase in cell growth. Together, these results indicate that Cd alters key signaling processes involved in the regulation of cytoskeleton to enhance cancer cell migration, invasion, adhesion, and proliferation.
Keywords: Cadmium, Breast cancer, Cell migration, Integrin, β-catenin
Introduction
Cadmium (Cd) is a toxic heavy metal and exposure to this metal has been associated with a variety of cancers, including breast cancer (Stayner et al., 1992; Il’yasova and Schwartz, 2005; Sahmoun et al., 2005; Julin et al., 2012a, b). Studies in mice and rats have confirmed that Cd treatment results in the development of lung, breast, and bladder cancer (Takenaka et al., 1983; Shiraishi et al., 1994; Johnson et al., 2003; Schrauzer, 2008; Davis et al., 2013). The International Agency for Research on Cancer has categorized Cd as a Group 1B carcinogen.
Carcinogenesis is a continuous process comprised of initiation, promotion, and progression. Several studies indicate that Cd promotes breast cancer cell growth, with some suggesting that it also promotes metastasis (Yang et al., 2004; Benbrahim-Tallaa et al., 2009; Siewit et al., 2010). Promotion of cell growth by Cd is described as a xenoestrogenic effect (Siewit et al., 2010; Ponce et al., 2013; Song et al., 2015) involving membrane receptors (Yu et al., 2010; Wei et al., 2015). Waalkes et al. (2000) reported that chronic Cd treatment increased metastatic potential of sc-injected sarcomas in rats. Metastasis is a multi-step process that includes dissemination, migration, intravasation, extravasation, and colonization to form secondary tumors. Specific mechanisms by which Cd may promote these steps in breast cancer cells are poorly understood. Ponce et al. (2015) reported that Cd interferes with cell-cell interaction by disrupting E-cadherin, enhancing the ability of breast cancer cells to disseminate from the primary tumor.
After disseminating from primary tumors, metastatic breast cancer cells enter a prolonged period of latency before forming secondary tumors. The rate-limiting steps seem to be extravasation and cell proliferation (Luzzi et al., 1998). During extravasation, metastatic cancer cells migrate through endothelial barriers and settle in new microenvironments via cell adhesion molecules (Felding-Habermann et al., 2001). An important feature in metastatic cell movement is alteration of cellular microfilament dynamics and extracellular matrix (ECM) remodeling. Integrins adhere to ECM proteins and alter cytoskeletal dynamics. FAK-Src complex transduces integrin signaling to downstream Rac/Rho, which initiates assembly of microfilaments, leading to cell adhesion and migration (Mitra and Schlaepfer, 2006). Crosstalk with PI3K and MAPK also promotes cell survival and proliferation (Giancotti and Ruoslahti, 1999). In lung epithelial cells, 0.5–1 μM Cd stimulated actin filament formation (Go et al., 2013). Also, Baroni et al. (2015) reported that 10 μM Cd increased integrin β1 and β5 gene expression and directly affected the production of ECM proteins in human bronchial epithelial cells. Accordingly, it seems reasonable to hypothesize that Cd may affect metastasized breast cancer cell adhesion through alterations in integrin activation.
Colonization at metastasized sites requires cells to proliferate to counterbalance cell death. Activation of the Wnt/β-catenin pathway, that normally regulates cell proliferation during embryonic development, has been implicated in tumorigenesis in colon cancer, leukemia, and breast cancer (Reya and Clevers, 2005). The Wnt pathway is initiated after Wnt ligands bind to Frizzled and Lrp 5/6 receptors. Receptor occupancy leads to accumulation of an important downstream signaling protein, β-catenin. GSK3β-mediated phosphorylation translocates β-catenin into the nucleus. There, it forms co-transcriptional activators with TCF/LEF, driving transcription of many oncogenes (Eastman and Grosschedl, 1999). Chakaraborty et al. (2010b) reported that chronic treatment of mice resulted in increased release of Wnt ligand and transcription of Wnt receptors. The same group (Chakraborty et al., 2010a) also reported that Cd enhanced nuclear translocation of β-catenin in human renal epithelial cells, which triggered TCF/LEF-mediated transcription. In addition, in rat renal epithelial cells, Cd increased free β-catenin accumulation through disruption of cadherins (Edwards et al., 2013). Based on these studies, it seems that Cd may stimulate secondary tumor growth through activation of Wnt/β-catenin-dependent pathways.
Cell migration and metastasis involve increased ECM interaction, activation of adhesion-related signaling, crosstalk with β-catenin and mitogenic signaling. Such effects have recently been reported in triple-negative MDA-MB-231 breast cancer cells exposed to ionizing radiation which is known to paradoxically stimulate metastatic cancer growth (Bouchard et al., 2016; Galarza et al., 2016). Promotion of MDA-MB-231 cell migration also occurs following treatment with TNF-α (Wolczyk, et al., 2016). The purpose of the present study was to test the hypothesis that Cd exerts stimulatory effects on migration and invasion in breast cancer cells. Results presented here were derived mainly from the triple-negative MDA-MB-231 cells.
Materials and Methods
Materials
DMEM and trypsin-versene were purchased from Lonza (Walkersville, MD). Fetal bovine serum (FBS) was purchased from Atlanta Biologicals (Flowery Branch, GA). CdCl2, PMSF and protease inhibitor were purchased from Sigma-Aldrich (Dallas, TX). Bis-acrylamide powder, BCA kit, protein ladder, and extracellular matrix components were purchased from Thermo-Fisher Scientific (Pittsburgh, PA). Phosphatase inhibitor cocktail tablets were purchased from Roche (Indianapolis, IN). Gelatin-zymogram gel, Laemmli buffer and blotting buffers were obtained from Bio-Rad (Hercules, CA). Phorbol 12-myristate 13-acetate (PMA) and diphenyleneiodonium chloride (DPI) were purchased from Cayman Chemical (Ann Arbor, Michigan). Active-integrin β1, GAPDH antibodies, integrins- linked kinase (ILK) inhibitor Cpd 22, Amicon centrifugal filter units (NMWL 10), slide chambers and Transwell inserts were purchased from EMD Millipore (Billerica, MA). Phosphor Rac1, α-actinin, phosphor-ERK 1/2 (Thr 202/Tyr 204), ERK, phosphor-Src (Tyr 416), Src, phosphor-FAK (Tyr 397) and FAK primary antibodies were from Cell Signaling Technology (Danvers, MA). CF5.5 goat anti-rabbit IgG was purchased from Santa Cruz Biotechnology (Dallas, TX). IRDye 680 goat anti-rabbit and goat anti-mouse IgG antibodies were purchased from Licor (Lincoln, NE). TOPFlash and pRL-TK plasmids were purchased from Addgene (Cambridge, MA). Genjet DNA transfection reagent was purchased from SignaGen Laboratories (Gaithersburg, MD). β-catenin siRNA (siGENOME Human CTNNB1 1499 – SMARTpool, catalog #M-003482-00-0005), nonsense siRNA (catalog #D-001206-01-05), transfection reagent, and RNase were from GE Healthcare Dharmacon (Chicago, IL). Nuclear Extract Kit was purchased from Active Motif (Carlsbad, CA). Cell Counting Kit-8 was purchased from Dojindo Molecular Technologies (Rockville, MD). Cultrex 3-D spheroid cell invasion assay kit was obtained from Trevigen (Gaithersburg, MD). E-PlateView 16-well plates were purchased from ACEA Biosciences (San Diego, CA). Dual-luciferase assay kit was from Promega (Madison, WI).
Cell lines and cell culture
Breast cancer derived triple-negative MDA-MB-231 cells and hormone-responsive MCF7 cells were obtained from the American Type Culture Collection (Manassas, VA). The cells were cultured in DMEM supplemented with 10% FBS and penicillin (100 U/mL)/streptomycin (100 U/mL). Cells were maintained in a humidified incubator (37°C) with 5% CO2.
Wound healing assay
The wound healing assay was performed in a 12-well plate. MDA-MB-231 cells were seeded in three replicates and cultured in DMEM with 10 % FBS. Wounds were generated by scratching the 90% confluent cell monolayer with a 1–20 μL pipet tip. The unattached cells were washed away with PBS and attached cells were treated with 0.5–1 μM Cd in 10 % FBS-supplemented DMEM. Images of the wounds were acquired every 24 h under phase contrast microscopy. The scratch area was measured using MRI Wound Healing Tool in ImageJ software and relative scratch closure was calculated based on the change in the scratch area at time zero.
Electrical impedance assay
MDA-MB-231 cells were seeded at a density of 8,000 cells/well in E-PlateView 16 plates and cultured in DMEM with 10% FBS. The bottom of the plate was covered with gold microelectrode sensors that generated an electric field when a low voltage (less than 20 mV) was applied between electrodes. Cells in the logarithmic growth phase were treated with DMEM containing 1 – 10 μM Cd, or 10 μM Cd and 0.5 μM DPI or 1 μM Cpd 22. Cell adhesion was monitored by using xCELLigence RTCA system at a sweeping interval of 15 s. Results were profiled and plotted by using the RTCA software.
Immunofluorescent staining
MDA-MB-231 cells were seeded in slide chambers and grown in DMEM with 10 % FBS. After 24 h, the cells were treated with 1 or 3 μM Cd in DMEM for 3 h. For specimen preparation, cells were fixed by treatment with 4% paraformaldehyde in PBS for 15 min followed by permeation with 0.3% Triton X-100 in 1% BSA-supplemented PBS. Cells were then blocked in 5% BSA-supplemented PBS. Finally, the cells were incubated with primary antibody for α-actinin overnight. After washing with PBS for 5 min, three times, the cells were stained using secondary antibody, CF 5.5 goat anti-rabbit IgG. Nuclei were stained with DAPI. Images of the stained cells were captured with a Nikon NE2000 fluorescence microscope.
Western blot analysis
Whole cell lysates were prepared in RIPA buffer. Nuclear and cytoplasmic fraction samples were prepared using an Active Motif nuclear extraction kit. Protein concentration in lysates was determined using the BCA kit. Proteins were denatured by heating the lysate to 95°C in Laemmli buffer containing 5% β-mercaptoethanol. Samples requiring non-reduced proteins were prepared in Laemmli buffer without β-mercaptoethanol. Proteins in the samples were then separated by SDS-PAGE electrophoresis and transferred to nitrocellulose membranes. The membranes were incubated with 5% non-fat milk for 1 h followed by incubation with the primary antibody overnight. The membranes were rinsed for 5 min three times in PBS containing 0.5% Tween 20, and incubated with secondary antibody for 1 h, followed by three additional washings in PBS containing 0.5% Tween 20, before imaging. The membranes were scanned by an Odyssey Infrared Imager. The optical density of the bands was quantified by the Odyssey image analysis software.
Luciferase reporter assay
MDA-MB-231 cells were seeded in 96-well plates and cultured in DMEM with 10% FBS. After 24 h, the cells were transfected with pRL-TK (2 ng/well) and TOPFlash (50 ng/well) plasmids for 48 h. The cells were serum starved by incubating in blank medium overnight, prior to treatment with Cd for 8 h. Firefly and renilla luciferase activities were generated by using Promega Dual-Luciferase assay and measured in a Glomax luminometer. Relative luciferase activity was quantified as the ratio of firefly to renilla luciferase activities.
Transwell assay
Transwell compartments (8 μm pore size) were prepared and inserted in a 24-well plate. The membranes were coated with 2 μg/mL collagen, 2 μg/mL fibronectin, 8 μg/mL vitronectin, and 8 μg/mL laminin. Cells were seeded into upper chambers in DMEM supplemented with 10% FBS. After incubation for 48 h, cells in the upper chamber were carefully removed with a cotton swab. The cells remaining on the underside of the membranes were fixed in 70% ethanol for 10 min and then stained with 0.5% crystal violet solution. Membranes with stained cells were imaged using an EVOS microscope at scan mode. To quantify the number of cells that migrated to the lower chamber, crystal violet stained cells were de-stained in 20% methanol. Migrated cell numbers in control and Cd-treated groups were determined by the absorbance of de-staining solutions at 450 nm in a Spectromax microplate reader. The relative migration tendency of Cd-treated cells was quantified as the ratio of absorbance reading by the treated cells over that of the control cells.
Gel zymography analysis
Precast gelatin-zymogram SDS-polyacrylamide gels were co-polymerized with gelatin. Media from cultured cells were concentrated 10-fold by centrifuging in Amicon filter units at 4,000 g for 30 min before adding to non-denaturing loading buffer solution. Protein concentration of all samples was determined and normalized to the same level before loading the samples onto gels. Electrophoresis of gelatin-zymogram gels was performed at 100V for 1 h. Gels were washed and incubated in renaturing buffer for 40 min and incubated in developing buffer overnight at 37°C. Then gels were stained with 0.1% Coomassie R-250 brilliant blue for 1 h and washed for 30 min until clear bands developed. Gelatinolytic activity (relative intensity of clear bands) of secreted MMP 9 and MMP 2 was quantified by densitometry analysis using the Odyssey image analysis software.
Cell proliferation assay
Untreated and 4-week Cd-treated MDA-MB-231 cells were harvested and seeded in a 96-well plate at a density of 10,000 cells/well in DMEM supplemented with 10 % FBS. After 4 days, cell growth was measured by Cell-Counting Kit 8. Absorbance was read at 450 nm and relative cell growth of Cd-treated cells was compared to untreated control cells.
3D cell culture
A spheroid invasion assay was used to estimate cell invasiveness. The experiment was performed by using a Cultrex 3-D spheroid cell invasion assay kit. Control and 4-week Cd-treated cells were harvested and seeded at a density of 5,000 cells/well in low-adhesive round-bottom 96-well plates. The cells were cultured in DMEM supplemented with 10 % FBS and basal membrane extract. After 4 days, the cells assembled into a compact spheroid. At that point, Matrigel was added to each well to provide a solidified invasion matrix. The growth of spheroids was observed microscopically and imaged daily.
Transfection with siRNA
MDA-MB-231 cells were cultured in DMEM containing 10% FBS and transfected with 2 μL β-catenin siRNA (20 μM) and 2 μL transfection reagent for 48 h. Cells transfected withnonsense siRNA were used as negative control.
Data analysis
All experiments were repeated at least three times. Western blot and zymography data were analyzed and quantified by Odyssey imaging analysis software. One-way ANOVA was performed by Prism with Tukey’s post hoc test on all data and p<0.05 was considered statistically significant.
Results
Changes in cytoskeletal dynamics
The wound healing assay was employed to study cell migration. Scratches were created on cell monolayers and the cells were treated with 0.5 and 1 μM Cd to determine if Cd would cause the cells in the surrounding area to migrate and fill the gap created by the scratch. As shown in Fig. 1A, scratches in the Cd-treated cells appeared to heal quicker than in the untreated control cells. Quantitively, at both concentrations of Cd 75% of the scratched area healed in 48 h as compared to only 42% healing in the control cells. These results indicated that non-toxic concentration of Cd facilitated cell migration in the triple-negative MDA-MB-231 breast cancer cells.
Fig. 1.


Cell spreading and adhesion in Cd-treated cells. (A) Wound healing assay. Before treatment with Cd, cross-scratches were made evenly on MDA-MB-231 cell monolayers. The cells were then treated with 0.5 or 1 μM Cd for 2 days. Representative images of cells migrating into the scratched area are shown. Bar graphs were generated using ImageJ software to measure scratch closure over 2 days, relative to the original scratch. Results are plotted as mean±SEM (n=3. *Sigificantly higher than the untreated control cells (p < 0.05). (B) Real-time electrical impedance assay. MDA-MB-231 cells in the logarithmic growth phase were treated with 1–10 μM Cd alone or with 10 μM Cd and 1 μM Cpd 22 or 0.5 μM DPI. Electrical impedance was measured every 15 s. Increased attachment between cell and electrode coated-plates was quantified as elevations in cell index (n=3). (C) Immuno- fluorescence staining for α-actinin. Attached MDA-MB-231 cells were treated with 1 or 3 μM Cd for 3 h. Then cells were fixed and stained with α-actinin antibody. DAPI was added to the slide before microscopic imaging. Upper images were taken under 10 X objective lens. Lower images were taken under oil lens at 100 X. Arrows point to α-actinin in filopodia.
Next, a real-time electrical impedance assay was employed to track cell spreading. Adding 1 or 3 μM Cd to attached cells caused a moderate increase in the cell spreading index that peaked between 6–8 h (Fig. 1B top). Higher concentration of Cd caused an even greater increase (1.6-fold) in cell spreading as compared to the control cells. However, this concentration was cytotoxic and cells began to lift off with the passage of time. Addition of 0.5 μM ROS inhibitor, DPI delayed this effect (Fig. 1B bottom). In contrast, the presence of 1 μM ILK inhibitor Cpd 22 completely abolished the Cd-induced cell spreading.
Cell migration and spreading in response to Cd treatment were also assessed by analyzing the level of a dynamic cytoskeletal construction indicator, α-actinin, which cross-links actin filaments (Otey et al., 1990). Cells treated with Cd for 3 h exhibited longitudinal filamentous α-actinin staining (Fig. 1C). Its spatial distribution indicated that Cd stimulated the buildup of cytoskeletal proteins for cell spreading and subsequently cell migration.
Activation of integrin and downstream signaling
Upon stimulation, integrin transduces adhesion signal to focal adhesion kinases, which is essential in assembly of actin filaments (Mitra and Schlaepfer, 2006). The effect of Cd treatment on integrin activation and downstream kinases was examined by Western blot analyses in not only the triple negative MDA-MB-231 cells, but also in hormone-responsive MCF7 cells. As shown in Fig. 2A, treatment with 1 or 3 μM Cd or 100 nM PMA (positive control) for 15 min to 1 h caused about 2-fold increase in active integrin β1 expression in both the MDA-MB 231 cells and the MCF7 cells. Cd-treated MDA-MB-231 cells also displayed similar increases in phosphorylation of Src (Tyr 416) and FAK (Tyr 397), which indicated activation of kinases at focal adhesion sites. Rac1 is a downstream target of focal adhesion kinase, and is involved with regulation of actin polymerization (Vial et al., 2003). Here, 1 and 3 μM Cd significantly elevated Rac1 phosphorylation, which was associated with the appearance of filamentous α-actinin (Fig. 1C). Additionally, ERK 1/2 signaling was activated by Cd treatment, presumably because of integrin pathway crosstalk with MAPK signaling (Vial et al., 2003). The activation of integrin itself and downstream signal transduction indicated that integrin was involved in Cd-induced cell adhesion.
Fig. 2.


Activation of integrin β1 and downstream signaling pathway upon Cd treatment. The cells were treated with 1 or 3 μM Cd for 15 min or 1 h. Cells treated with 100 nM PMA for 1 h were positive controls. (A) Expression of active integrin β1 in MDA-MB-231 and MCF7 cells. (B) Expression of integrin downstream kinases in MDA-MB-231 cells. Representative Western blot images of active integrin β1, pSrc (Tyr 416), pFAK (Tyr 397), pERK1/2 (Thr 202/Tyr 204), and pRac1 are shown. Densities relative to the untreated control cells are plotted as mean±SEM (n =3). *Significantly higher than the untreated control cells (p < 0.05).
β-catenin signaling and expression
To examine activation of the Wnt/β-catenin pathway in Cd-treated breast cancer cells, the MDA-MB-231 cells were transfected with TOPFlash plasmids which contained repeated TCF/LEF sites upstream of the luciferase reporter. As depicted in Fig. 3A, treatment with 3 μM Cd for 1 h increased luciferase activity 1.8-fold in the transfected cells, suggesting increased TCF/LEF-mediated transcription. This was further confirmed by the observation that Cd treatment enhanced phosphorylation of GSK3β (Ser 9) 1.5-fold, thus resulting in a decrease in the activity of GSK3β (Fig. 3B). To further examine that Cd stimulated the Wnt/β-catenin pathway, the ILK inhibitor Cpd 22 was utilized in the TOPFlash luciferase reporter assay. As shown in Fig. 3C, Cpd 22 significantly curtailed the Cd-induced increase in luciferase activity. It is evident that the Wnt/β-catenin pathway was activated in response to Cd treatment. The results also suggest that integrin signaling is involved in Cd-induced TCF/LEF transcription.
Fig. 3.

Effect of Cd treatment on the Wnt/β-catenin pathway. (A) Luciferase reporter assay. The cells were transfected with TOPFlash and pRL-TK plasmids for 2 days. Transfected cells were serum-starved overnight and treated with 1 or 3 μM Cd for 8 h. Firefly and renilla luciferase activities were then measured. The ratio of firefly to renilla luciferase activities was normalized to control and relative luciferase activity is plotted as mean±SEM. (B) Inhibition of GSK3β by Cd. Serum-starved cells were treated with 1 and 3 μM Cd for 15 min or 1 h. Representative Western blot images of phosphorylated-GSK 3β (Ser 9) and GSK 3β are shown. Densities relative to the untreated control group are plotted as mean±SEM (n =3). *Significantly higher than the control (p < 0.05). (C) Effect of ILK inhibition by Cpd 22. The cells were transfected with TOPFlash and pRL-TK plasmids for 2 days. Transfected cells were serum-starved overnight and treated with either 1 μM Cpd 22 alone, or along with 3 μM Cd, for 8 h. The ratio of firefly to renilla luciferase activities normalized to control is plotted as mean±SEM. *Significantly higher than the vehicle control (p < 0.05). #Significantly different at p <0.05.
Next, the levels of β-catenin were determined in whole cell lysate, cytosol and nuclear extract of MDA-MB-231 cells treated with 3 μM Cd for 4 week. Western blot analyses showed that prolonged Cd treatment led to a 2.9-fold increase in β-catenin level in the cell lysate, a 1.9-fold increase in both the cytosolic and nuclear fractions (Fig. 4A). Accumulation of β-catenin generated a larger pool of co-activator for TCF/LEF-mediated transcription. As shown in Fig. 4B, luciferase activity in the 4-week Cd-treated cells was significantly higher than in the untreated cells, indicating that the basal TCF/LEF-mediated transcriptional activity was elevated after prolonged Cd treatment. When these cells were treated with 1 μM Cd for 8 h, the TCF/LEF-mediated transcription activity was even greater (2.8-fold) in the Cd-treated cells as compared to the non-treated cells. Knockdown of β-catenin by siRNA eliminated TCF/LEF-mediated transcriptional activity in both the untreated and 4-week Cd-treated cells. Thus, the activation of TCF/LEF-mediated transcription upon prolonged Cd-treatment was dependent on β-catenin expression.
Fig. 4.

Effect of prolonged Cd treatment on β-catenin levels. The cells were cultured in DMEM supplemented with 3 μM Cd for 4 weeks. Control cells were cultured in medium alone. (A) Expression of β-catenin in cellular fractions. Representative Western blot images of β-catenin expression are shown. Lamin A/C and GAPDH were loading controls for the nuclear and cytosol fractions, respectively. Densities relative to the respective fractions of 4-week untreated cells are plotted as mean±SEM (n=3). (B) Luciferase reporter assay. Untreated and 4-week Cd-treated cells were transfected with TOPFlash and pRL-TK plasmids, serum-starved overnight, and treated with 1 μM Cd for 8 h. Firefly and renilla luciferase activities were measured. The ratio of firefly and renilla luciferase activities was normalized to untreated control cells and plotted as mean±SEM. *Significantly higher than the 4-week untreated control cells. #Significantly different at p < 0.05.
Cytoskeleton reorganization upon prolonged Cd treatment
To study how prolonged Cd treatment affects the cytoskeleton, the MDA-MB-231 cells were treated with 3 μM Cd for 4 weeks and stained with vimentin. These cells exhibited different spatial distribution of intermediate filaments when compared to the control cells (Fig. 5A). Further exploration revealed that Cd treatment increased the expression of not only vimentin but also of integrins α5 and β1, and paxillin by 5.3-, 2.1-, 6.3-, and 3-fold, respectively (Fig. 5B). The increases in intermediate filaments, integrins, and scaffold proteins indicated that prolonged Cd treatment reorganized the cytoskeleton.
Fig. 5.

Effect of prolonged Cd treatment on the cytoskeleton. The cells were cultured in DMEM containing 3 μM Cd for 4 weeks. Control cells were cultured in medium alone. (A) Immuno- fluorescence staining for vimentin. Cells were fixed and stained with the vimentin antibody. DAPI was added to the slides before microscopic imaging. (B) Western blots of integrin α5 and β1, paxillin, and vimentin. Untreated control cells and Cd-treated cells were seeded in 6-well plates. After 48 h, attached cells were harvested and analyzed for expression of integrin α5 and β1, paxillin, and vimentin and representative images are shown. Densities relative to the control cells are plotted as mean±SEM (n =3). *Significantly higher than the control cells (p < 0.05).
Increase in cell migration, invasion and proliferation upon prolonged Cd treatment
Transwell assay and MMP secretion were used to assess cell migration and invasion. To evaluate the effect of Cd on cell proliferation, cell growth was compared between the Cd-treated and control cells. Furthermore, the spheroid invasion assay was used to comprehensively examine cell invasion and proliferation.
Microscopic pictures of the underside of Transwell membrane inserts showed that a greater number of Cd-treated cells migrated through the membrane toward the lower chamber than the untreated cells (Fig. 6A, top). Quantified result indicated that 4 weeks of Cd treatment increased cell migration by 1.6-fold (Fig. 6A). Activities of MMP 2 and 9 in cell culture medium were detected by in-gel zymography. As shown in Fig. 6B, the clear bands in the gel image indicated gelatinolytic activity of MMPs. Determined by molecular weight, the intensities of clear bands at 58 and 82 KDa represented activities of MMP 2 and 9, respectively. Together, the secretion of MMP 2 and 9 in Cd-treated cells was 3.4-fold higher than in the control cells.
Fig. 6.

Effect of prolonged Cd treatment on cell migration. The cells were cultured in DMEM containing 3 μM Cd for 4 weeks. The control cells were cultured in medium alone. (A) Transwell assay. Control and Cd-treated cells were seeded in Transwell inserts. After 48 h, migrated cells were stained by crystal violet. Quantification of migrated cells was conducted by measuring the absorbance of de-staining solution. (B) Gelatin zymograph of MMPs. Equal number of control and Cd-treated cells were cultured in serum-free medium for 2 days. The cell culture medium was collected and the supernatant after centrifugation was concentrated 10-fold prior to separation on gelatin-zymogram gels. MMP2/9 levels were visualized as clear bands at 58 and 82 KDa. Representative gel image is shown. Densities relative to those of the control cells are plotted as mean±SEM (n=3). *Significantly higher than the control cells (p < 0.05).
To investigate whether 4 weeks of treatment with 3 μM Cd altered growth rate of MDA-MB-231 cells, the cells were harvested and cultured in the absence of Cd for 4 days. As shown in Fig. 7, the pretreated cells exhibited 30% higher growth than the control cells.
Fig. 7.
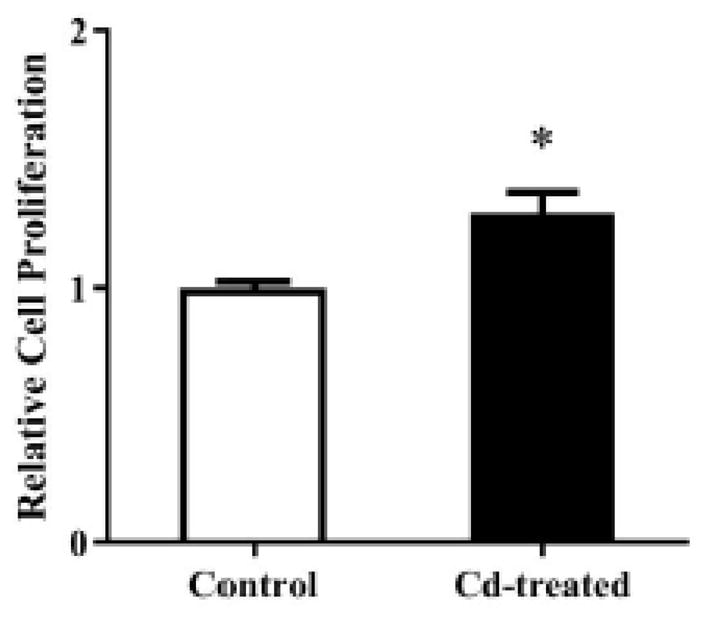
Effect of prolonged Cd treatment on cell proliferation. The cells were cultured in 10% FBS-supplemented DMEM containing either no Cd or 3 μM Cd. After 4 weeks, the cells were harvested and seeded at equal densities in DMEM supplemented with 10% FBS, but no Cd, for 4 days. Cell proliferation was quantified using Cell Counting Kit-8. Absorbance at 450 nM relative to the control cells was plotted as mean±SEM (n=3). *Significantly higher than the control cells (p < 0.05).
Finally, the invasiveness of prolonged Cd-treated cells was evaluated in a 3D spheroid invasion assay. The Cd-treated cells exhibited a shady outer ring around the spheroid core appeared as early as on day-6 and became very pronounced on day-12 (arrow) (Fig. 8). This ring was indicative of cell invasion into the surrounding Matrigel. In comparison, in control cells this ring became evident on day-12. Prolonged Cd treatment thus enhanced the migratory and invasive properties of breast cancer cells.
Fig. 8.

Effect of prolonged Cd treatment on cell invasion. The cells were cultured in DMEM supplemented with 3 μM Cd for 4 weeks. Control cells were cultured in medium alone. The cells were harvested and seeded in a round bottom 96-well plate at an equal density and grown in DMEM supplemented with basal membrane extract. Matrigel was added into each well after cells aggregated into spheroids. Cellular invasion of the spheroid was assessed by the appearance of a shady outer ring which was composed of cells invading into the Matrigel.
Discussion
Metastasis is a consecutive process which includes invasion into surrounding tissue, intravasation, extravasation, and colonization into secondary tumors (Nguyen et al., 2009). Cell movement during this process is conducted through sequential activation of cell adhesion receptors which dynamically regulate cytoskeletal construction, leading to cell spreading and migration (Felding-Habermann et al., 2001).
The results presented here suggest that Cd enhances metastasized triple-negative MDA-MB-231 breast cancer cell adhesion through integrin activation. Prolonged Cd treatment appears to stimulate malignancy, as evidenced by cytoskeleton reorganization and enhanced cell migration and invasion. Enhanced cell proliferation further facilitates metastasized cell growth. These are key indicators of metastatic changes consisting of cell migration to a new environment and growth into a secondary tumor. Moreover, cellular responses shortly after Cd treatment correlated with alterations in cells treated with Cd for 4 weeks. First, short-term Cd treatment led to integrin β1 activation, which preceded cell adhesion and cell migration. Prolonged Cd treatment also elevated integrin α5 and β1 expression and displayed markedly higher migration and invasion characteristics. Second, short-term Cd treatment activated β-catenin signaling. Prolonged Cd treatment also elevated expression of β-catenin, which rendered Cd-treated cells more responsive to the activation of Wnt/β-catenin signaling that contributes to cancer progression.
Cd-induced cytoskeletal perturbation has been well studied in animal cells. Exposure to Cd leads to disassembly of microtubules in mouse 3T3 cells (Perrino and Chou, 1986). In Cd-treated mouse mesangial cells, actin filaments become irregular and are lost because of disturbances in focal adhesion (Choong et al., 2013). In contrast to these findings of cytoskeleton disruption upon Cd treatment, the results of the present study show that Cd treatment increases α-actinin, a binder of actin filaments polymerization, indicating active cytoskeleton buildup in the MDA-MB-231 cells (Fig. 1C). Real-time tracking of cell attachment indicated that Cd treatment stimulated cell spreading. These results led to examination of the effects of Cd on receptors that regulate cell adhesion (Huttenlocher et al., 1996). The present study is the first to report that Cd treatment induces cell adhesion through integrin activation (Fig. 2). In this study, it was found that Cd, instead of disrupting focal adhesion as reported by Choong et al. (2013), induced phosphorylation of focal adhesion kinases in breast cancer cells. This is a significant finding, because metastatic colonization at distant sites requires activation of integrin- mediated signaling (Reticker-Flynn et al., 2012). Furthermore, increased expressions of intermediate filament (vimentin) and scaffold protein (paxillin) upon Cd treatment for 4 weeks (Fig. 5) provided evidence of cytoskeleton reorganization (Fife et al., 2014). The elevation of vimentin increases focal contact size, which is critical for cancer cell movement (Tsuruta and Jones, 2003).
Integrin plays an important role in tumor cell invasion and migration (Hood and Cheresh, 2002). Disseminated tumor cells use integrin and protease to develop a mesenchymal-type migration pattern (Friedl and Wolf, 2003). Several studies reported that integrin-associated MMPs became localized to cell-ECM sites when integrin was bound to the ECM (Mueller et al., 1999; Dumin et al., 2001). The present study observed in the Transwell assay an increase in cell migration in cells treated with Cd for 4 weeks (Fig. 6A). Apparently, migrated cells had to degrade ECM on the membrane to enter the lower chamber because ECM blocks the membrane pores. The enhancement of MMP2/9 secretion (Fig. 6B) was parallel to integrin expression in Cd-treated breast cancer cells (Fig. 5B). Increases in integrin and MMP levels are important for local tumor invasion because their interactions likely facilitate ECM degradation. Moreover, enhanced MMP secretion promotes the release of growth factors, including transforming growth factor β (TGFβ) and vascular endothelial growth factor (VEGF) (Bergers et al., 2000; Yu and Stamenkovic, 2000; Mu et al., 2002). The released growth factors bind to membrane receptors which further stimulate cell proliferation and migration (Schenk et al., 2003).
Canonical Wnt/β-catenin activation may facilitate carcinogenesis (Reya and Clevers, 2005) since it is marked by translocation of β-catenin to the nucleus with subsequent triggering of TCF/LEF-mediated transcriptional machinery. In the present study, Cd activated TOPFlash luciferase reporter in the breast cancer cells (Fig. 3A), similar to what has been reported in Cd-treated human kidney cells (Chakraborty et al., 2010a; Edwards et al., 2013). In the kidney cells, the former investigators reported that Cd caused release of membrane-bound β-catenin to cytosol and nuclei due to disruption of E-cadherin-catenin at the cadherins junction complex. However, since the MDA-MB-231 cells do not express E- and N-cadherins, the related molecular events summarized in Fig. 9 are independent of the E-cadherin disruption.
Fig. 9.

Schematic diagram summarizing the effects of Cd on integrin and β-catenin signaling. (A) Cd activates integrins on the cell membrane. Focal adhesion kinase (FAK) then transduces integrin signaling to downstream Rac1 to regulate actin assembly. (B) Cd inhibits GSK3β, resulting in β-catenin’s escape from degradation. Free β-catenin translocates into the nucleus and stimulates TCF/LEF-mediated oncogene transcription, which contributes to carcinogenesis.
β-catenin is an important co-activator of TCF/LEF-mediated transcription. It is caged by GSK3β for degradation. The result presented in Fig. 3B show that Cd inhibited GSK3β activity, resulting in β-catenin’s escape from degradation. The free β-catenin translocated into the nucleus and increased TCF/LEF-mediated transcription (Fig. 3A). Furthermore, prolonged Cd treatment elevated the expression of β-catenin (Fig. 4A). Although the mechanism responsible for β-catenin accumulation remains unclear, some insight might be provided by the observation that upregulation of β-catenin rendered cells more responsive to Wnt/β-catenin pathway stimulation and knockdown of β-catenin eliminated TCF/LEF-mediated luciferase activity in the TOPFlash assay (Fig. 4B).
In triple-negative breast cancer cells, the integrin-dependent metastasis-associated phenotypes are accompanied by the upregulation of Wnt/β-catenin pathway (De et al., 2016). The present study examined whether Cd could activate Wnt/β-catenin pathway without engaging integrins in the MDA-MB-231 cells. It was found that TCF/LEF-mediated transcription was diminished if cells were treated with Cd in low-adhesive plates (data not shown). This suggested that the involvement of integrin was important in Wnt/β-catenin activation. Furthermore, ILK inhibitor Cpd 22 suppressed Cd-induced activation of TCF/LEF-mediated transcription (Fig. 3C). Since integrin signaling crosstalks with the Wnt/β-catenin pathway at the GSK3β node (Burkhalter et al., 2011), Cd likely activates ILK, which in turn inhibits GSK3β, resulting in β-catenin-initiated TCF/LEF-mediated transcription. It was recently reported that integrin signaling affects Wnt/β-catenin pathway through Rac1 and Cdc42 activation (De et al., 2017). It is plausible that Cd, by causing Rac1 activation, further facilitates β-catenin translocation into the nucleus.
Breast tumor cells that migrate to distant organs frequently enter a long latency period before developing into life-threatening metastasized breast cancer. During this process, environmental factors may affect tumor malignancy. Therefore, it is important to understand how environmental risks contribute to breast cancer progression. In this regard, the results reported here may provide some insight into this process. This study reports for the first time that an integrin-related mechanism is involved in the enhancement of breast cancer cell adhesion and migration by the environmental contaminant Cd. Furthermore, it shows that prolonged Cd treatment results in β-catenin accumulation, which renders breast cancer cells more sensitive to the activation of Wnt pathway. Based on the in vitro observation of Cd-induced integrin activation, it could be extrapolated that the metastasized cancer cells might be more likely to lodge at distant organs after Cd exposure in vivo. There is growing evidence that tumor spheroids represent tumor conditions in vivo. Spheroids exhibit several traits which 2D monolayer cultures lack, such as characteristic tumor morphology, cell-cell contact, and hypoxic centers. The results from this in vitro assay indicated that prolonged Cd treatment increases the migration and invasion ability of metastasized breast cancer cell, which may shorten the latency of breast cancer relapse.
In summary, Cd treatment activates integrin in triple-negative breast cancer cells, substantially increases their adhesion and migration. A schematic diagram of how Cd’s effects on integrin, GSK3β, and β-catenin result in the activation of TCF/LEF-mediated transcription is presented in Fig. 9. The results of this study contribute to a better understanding of how Cd stimulates triple-negative breast cancer cell progression and may be of value in designing better strategies for cancer interventions in the future.
Highlights.
Cd enhances cell adhesion in triple-negative breast cancer cells through integrin activation.
Inhibition of GSK3β by Cd leads to activation of β-catenin pathway.
Accumulation of β-catenin facilitates TCF/LEF-mediated-transcription.
Prolonged Cd treatment stimulates cell migration, invasion, and proliferation.
Acknowledgments
This research was made possible by use of the RI-INBRE Centralized Research Core Facility support by grant # 2P20GM103430 from NIGMS, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baroni T, Lilli C, Bellucci C, Luca G, Mancuso F, Fallarino F, Falabella G, Arato I, Calvitti M, Marinucci L, Muzi G, Dell’Omo M, Gambelunghe A, Bodo M. In vitro cadmium effects on ECM gene expression in human bronchial epithelial cells. Cytokine. 2015;72:9–16. doi: 10.1016/j.cyto.2014.12.002. [DOI] [PubMed] [Google Scholar]
- Benbrahim-Tallaa L, Tokar EJ, Diwan BA, Dill AL, Coppin JF, Waalkes MP. Cadmium malignantly transforms normal human breast epithelial cells into a basal-like phenotype. Environ Health Perspect. 2009;117:1847–1852. doi: 10.1289/ehp.0900999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Hanahan D. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nature Cell Biol. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard G, Therriault H, Geha S, Bérubé-Lauzière Y, Bujold R, Saucier C, Paquette B. Stimulation of triple negative breast cancer cell migration and metastases formation is prevented by chloroquine in a pre-irradiated mouse model. BMC Cancer. 2016;16:361. doi: 10.1186/s12885-016-2393-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhalter RJ, Symowicz J, Hudson LG, Gottardi CJ, Stack MS. Integrin regulation of beta-catenin signaling in ovarian carcinoma. J Biol Chem. 2011;286:23467–23475. doi: 10.1074/jbc.M110.199539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty PK, Lee WK, Molitor M, Wolff NA, Thevenod F. Cadmium induces Wnt signaling to upregulate proliferation and survival genes in sub-confluent kidney proximal tubule cells. Mol Cancer. 2010a;9:102. doi: 10.1186/1476-4598-9-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty PK, Scharner B, Jurasovic J, Messner B, Bernhard D, Thevenod F. Chronic cadmium exposure induces transcriptional activation of the Wnt pathway and upregulation of epithelial-to-mesenchymal transition markers in mouse kidney. Toxicol Lett. 2010b;198:69–76. doi: 10.1016/j.toxlet.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Choong G, Liu Y, Templeton DM. Cadmium affects focal adhesion kinase (FAK) in mesangial cells: involvement of CaMK-II and the actin cytoskeleton. J Cell Biochem. 2013;114:1832–1842. doi: 10.1002/jcb.24529. [DOI] [PubMed] [Google Scholar]
- Davis J, Khan G, Martin MB, Hilakivi-Clarke L. Effects of maternal dietary exposure to cadmium during pregnancy on mammary cancer risk among female offspring. J Carcinog. 2013;12:11. doi: 10.4103/1477-3163.114219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De P, Carlson JH, Wu H, Marcus A, Leyland-Jones B, Dey N. Wnt-beta-catenin pathway signals metastasis-associated tumor cell phenotypes in triple negative breast cancers. Oncotarget. 2016;7:43124–43149. doi: 10.18632/oncotarget.8988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De P, Carlson JH, Jepperson T, Willis S, Leyland-Jones B, Dey N. RAC1 GTP-ase signals wnt-beta-catenin pathway mediated integrin-directed metastasis-associated tumor cell phenotypes in triple negative breast cancers. Oncotarget. 2017;7:3072–3103. doi: 10.18632/oncotarget.13618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumin JA, Dickeson SK, Stricker TP, Bhattacharyya-Pakrasi M, Roby JD, Santoro SA, Parks WC. Pro-collagenase-1 (matrix metalloproteinase-1) binds the alpha(2)beta(1) integrin upon release from keratinocytes migrating on type I collagen. J Biol Chem. 2001;276:29368–29374. doi: 10.1074/jbc.M104179200. [DOI] [PubMed] [Google Scholar]
- Eastman Q, Grosschedl R. Regulation of LEF-1/TCF transcription factors by Wnt and other signals. Current Opin Cell Biol. 1999;11:233–240. doi: 10.1016/s0955-0674(99)80031-3. [DOI] [PubMed] [Google Scholar]
- Edwards JR, Kolman K, Lamar PC, Chandar N, Fay MJ, Prozialeck WC. Effects of cadmium on the sub-cellular localization of beta-catenin and beta-catenin-regulated gene expression in NRK-52E cells. Biometals. 2013;26:33–42. doi: 10.1007/s10534-012-9592-0. [DOI] [PubMed] [Google Scholar]
- Felding-Habermann B, O’Toole TE, Smith JW, Fransvea E, Ruggeri ZM, Ginsberg MH, Hughes PE, Pampori N, Shattil SJ, Saven A, Mueller BM. Integrin activation controls metastasis in human breast cancer. Proc Nat Acad Sci. 2001;98:1853–1858. doi: 10.1073/pnas.98.4.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fife CM, McCarroll JA, Kavallaris M. Movers and shakers: cell cytoskeleton in cancer metastasis. Brit J Pharmacol. 2014;171:5507–5523. doi: 10.1111/bph.12704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nature Rev Cancer. 2003;3:362–374. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- Galarza TE, Mohamad NA, Táquez Delgado MA, Vedoya GM, Crescenti EJ, Bergoc RM, Gabriela A, Martín GA, Cricco GP. Histamine prevents radiation- induced mesenchymal changes in breast cancer cells. Pharmacol Res. 2016;11:731–739. doi: 10.1016/j.phrs.2016.07.039. [DOI] [PubMed] [Google Scholar]
- Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- Go YM, Orr M, Jones DP. Actin cytoskeleton redox proteome oxidation by cadmium. American journal of physiology Lung Cell Mol Physiol. 2013;305:L831–843. doi: 10.1152/ajplung.00203.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nature reviews. Cancer. 2002;2:91–100. doi: 10.1038/nrc727. [DOI] [PubMed] [Google Scholar]
- Huttenlocher A, Ginsberg MH, Horwitz AF. Modulation of cell migration by integrin- mediated cytoskeletal linkages and ligand-binding affinity. J Cell Biol. 1996;134:1551–1562. doi: 10.1083/jcb.134.6.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Il’yasova D, Schwartz GG. Cadmium and renal cancer. Toxicol Appl Pharmacol. 2005;207:179–186. doi: 10.1016/j.taap.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Johnson MD, Kenney N, Stoica A, Hilakivi-Clarke L, Singh B, Chepko G, Clarke R, Sholler PF, Lirio AA, Foss C, Reiter R, Trock B, Paik S, Martin MB. Cadmium mimics the in vivo effects of estrogen in the uterus and mammary gland. Nat Med. 2003;9:1081–1084. doi: 10.1038/nm902. [DOI] [PubMed] [Google Scholar]
- Julin B, Wolk A, Bergkvist L, Bottai M, Akesson A. Dietary cadmium exposure and risk of postmenopausal breast cancer: a population-based prospective cohort study. Cancer Res. 2012a;72:1459–1466. doi: 10.1158/0008-5472.CAN-11-0735. [DOI] [PubMed] [Google Scholar]
- Julin B, Wolk A, Johansson JE, Andersson SO, Andren O, Akesson A. Dietary cadmium exposure and prostate cancer incidence: a population-based prospective cohort study. Brit J Cancer. 2012b;107:895–900. doi: 10.1038/bjc.2012.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, Groom AC. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol. 1998;153:865–873. doi: 10.1016/S0002-9440(10)65628-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Current Opin Cell Biol. 2006;18:516–523. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, Nishimura SL. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J Cell Biol. 2002;157:493–507. doi: 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SC, Ghersi G, Akiyama SK, Sang QX, Howard L, Pineiro-Sanchez M, Nakahara H, Yeh Y, Chen WT. A novel protease-docking function of integrin at invadopodia. J Biol Chem. 1999;274:24947–24952. doi: 10.1074/jbc.274.35.24947. [DOI] [PubMed] [Google Scholar]
- Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nature reviews. Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- Otey CA, Pavalko FM, Burridge K. An interaction between alpha-actinin and the beta 1 integrin subunit in vitro. J Cell Biol. 1990;111:721–729. doi: 10.1083/jcb.111.2.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrino BA, Chou IN. Role of calmodulin in cadmium-induced microtubule disassembly. Cell Bol Internat Reports. 1986;10:565–573. doi: 10.1016/0309-1651(86)90031-7. [DOI] [PubMed] [Google Scholar]
- Ponce E, Aquino NB, Louie MC. Chronic cadmium exposure stimulates SDF-1 expression in an ER alpha dependent manner. PLoS One. 2013;8:e72639. doi: 10.1371/journal.pone.0072639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponce E, Louie MC, Sevigny MB. Acute and chronic cadmium exposure promotes E-cadherin degradation in MCF7 breast cancer cells. Mol Carcinog. 2015;54:1014–1025. doi: 10.1002/mc.22170. [DOI] [PubMed] [Google Scholar]
- Reticker-Flynn NE, Malta DF, Winslow MM, Lamar JM, Xu MJ, Underhill GH, Hynes RO, Jacks TE, Bhatia SN. A combinatorial extracellular matrix platform identifies cell-extracellular matrix interactions that correlate with metastasis. Nature Commun. 2012;3:1122. doi: 10.1038/ncomms2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya T, Clevers H. Wnt signaling in stem cells and cancer. Nature. 2005;434:843–850. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- Sahmoun AE, Case LD, Jackson SA, Schwartz GG. Cadmium and prostate cancer: a critical epidemiologic analysis. Cancer Invest. 2005;23:256–263. doi: 10.1081/cnv-200055968. [DOI] [PubMed] [Google Scholar]
- Schenk S, Hintermann E, Bilban M, Koshikawa N, Hojilla C, Khokha R, Quaranta V. Binding to EGF receptor of a laminin-5 EGF-like fragment liberated during MMP-dependent mammary gland involution. J Cell Biol. 2003;161:197–209. doi: 10.1083/jcb.200208145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrauzer GN. Interactive effects of selenium and cadmium on mammary tumor development and growth in MMTV-infected female mice. A model study on the roles of cadmium and selenium in human breast cancer. Biol Trace Elem Res. 2008;123:27–34. doi: 10.1007/s12011-008-8091-1. [DOI] [PubMed] [Google Scholar]
- Shiraishi N, Rehm S, Waalkes MP. Effect of chlorpromazine pretreatment on cadmium toxicity in the male Wistar (WF/NCr) rat. J Toxicol Environ Health. 1994;42:193–208. doi: 10.1080/15287399409531873. [DOI] [PubMed] [Google Scholar]
- Siewit CL, Gengler B, Vegas E, Puckett R, Louie MC. Cadmium promotes breast cancer cell proliferation by potentiating the interaction between ERalpha and c-Jun. Mol Endocrinol. 2010;24:981–992. doi: 10.1210/me.2009-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Wei Z, Shaikh ZA. Requirement of ERalpha and basal activities of EGFR and Src kinase in Cd-induced activation of MAPK/ERK pathway in human breast cancer MCF-7 cells. Toxicol Appl Pharmacol. 2015;287:26–34. doi: 10.1016/j.taap.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stayner L, Smith R, Thun M, Schnorr T, Lemen R. A quantitative assessment of lung cancer risk and occupational cadmium exposure. IARC Scientific Publications. 1992:447–455. [PubMed] [Google Scholar]
- Takenaka S, Oldiges H, Konig H, Hochrainer D, Oberdorster G. Carcinogenicity of cadmium chloride aerosols in W rats. J Nat Can Inst. 1983;70:367–373. [PubMed] [Google Scholar]
- Tsuruta D, Jones JC. The vimentin cytoskeleton regulates focal contact size and adhesion of endothelial cells subjected to shear stress. J Cell Sci. 2003;116:4977–4984. doi: 10.1242/jcs.00823. [DOI] [PubMed] [Google Scholar]
- Vial E, Sahai E, Marshall CJ. ERK-MAPK signaling coordinately regulates activity of Rac1 and RhoA for tumor cell motility. Cancer Cell. 2003;4:67–79. doi: 10.1016/s1535-6108(03)00162-4. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Rehm S, Cherian MG. Repeated cadmium exposures enhance the malignant progression of ensuing tumors in rats. Toxicol Sci. 2000;54:110–120. doi: 10.1093/toxsci/54.1.110. [DOI] [PubMed] [Google Scholar]
- Wei Z, Song X, Shaikh ZA. Cadmium promotes the proliferation of triple-negative breast cancer cells through EGFR-mediated cell cycle regulation. Toxicol Appl Pharmacol. 2015;289:98–108. doi: 10.1016/j.taap.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolczyk D, Zaremba-Czogalla M, Hryniewicz-Jankowska A, Tabola R, Grabowski K, Sikorski AF, Augoff K. TNF-α promotes breast cancer cell migration and enhances the concentration of membrane-associated proteases in lipid rafts. Cell Oncol (Dordr) 2016;39:353–363. doi: 10.1007/s13402-016-0280-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang PM, Chiu SJ, Lin KA, Lin LY. Effect of cadmium on cell cycle progression in Chinese hamster ovary cells. Chem Biol Interact. 2004;149:125–136. doi: 10.1016/j.cbi.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Develop. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- Yu X, Filardo EJ, Shaikh ZA. The membrane estrogen receptor GPR30 mediates cadmium-induced proliferation of breast cancer cells. Toxicol Appl Pharmacol. 2010;245:83–90. doi: 10.1016/j.taap.2010.02.005. [DOI] [PubMed] [Google Scholar]