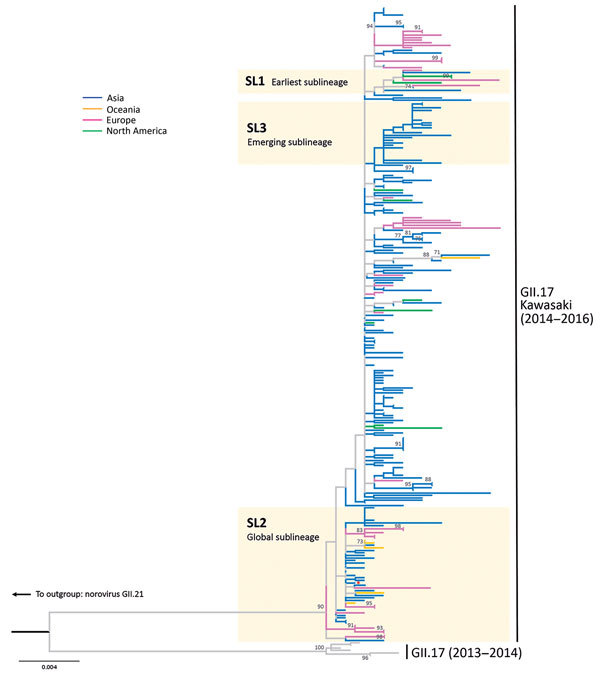
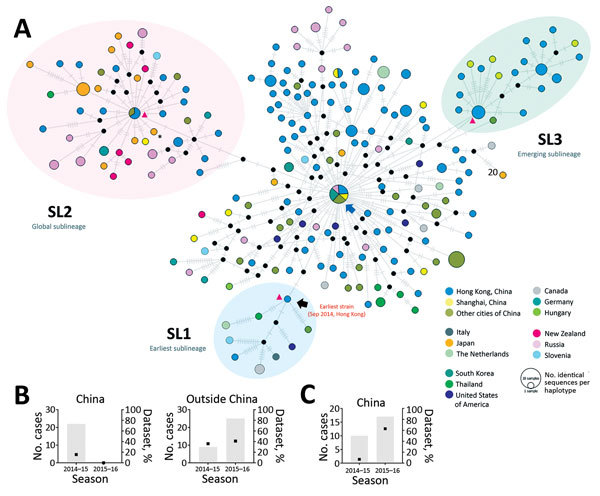
Martin CW Chan
Martin CW Chan
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Yunwen Hu
Yunwen Hu
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Haili Chen
Haili Chen
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Alexander T Podkolzin
Alexander T Podkolzin
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Ekaterina V Zaytseva
Ekaterina V Zaytseva
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Jun Komano
Jun Komano
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Naomi Sakon
Naomi Sakon
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Yong Poovorawan
Yong Poovorawan
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Sompong Vongpunsawad
Sompong Vongpunsawad
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Thanundorn Thanusuwannasak
Thanundorn Thanusuwannasak
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Joanne Hewitt
Joanne Hewitt
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Dawn Croucher
Dawn Croucher
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Nikail Collins
Nikail Collins
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Jan Vinjé
Jan Vinjé
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Xiaoli L Pang
Xiaoli L Pang
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Bonita E Lee
Bonita E Lee
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Miranda de Graaf
Miranda de Graaf
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Janko van Beek
Janko van Beek
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Harry Vennema
Harry Vennema
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Marion PG Koopmans
Marion PG Koopmans
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Sandra Niendorf
Sandra Niendorf
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Mateja Poljsak-Prijatelj
Mateja Poljsak-Prijatelj
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Andrej Steyer
Andrej Steyer
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Peter A White
Peter A White
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Jennifer H Lun
Jennifer H Lun
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Janet Mans
Janet Mans
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Tin-Nok Hung
Tin-Nok Hung
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Kirsty Kwok
Kirsty Kwok
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Kelton Cheung
Kelton Cheung
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Nelson Lee
Nelson Lee
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,
Paul KS Chan
Paul KS Chan
1The Chinese University of Hong Kong, Hong Kong, China (M.C.W. Chan, T.-N. Hung, K. Kwok, K. Cheung, N. Lee, P.K.S. Chan);
2Fudan University, Shanghai, China (Y. Hu, H. Chen);
3Central Research Institute of Epidemiology, Moscow, Russia (A.T. Podkolzin, E.V. Zaytseva);
4Nagoya Medical Center, Nagoya, Japan (J. Komano);
5Osaka Prefectural Institute of Public Health, Osaka, Japan (N. Sakon);
6Chulalongkorn University, Bangkok, Thailand (Y. Poovorawan, S. Vongpunsawad, T. Thanusuwannasak);
7The Institute of Environmental Science and Research, Porirua, New Zealand (J. Hewitt, D. Croucher);
8Centers for Disease Control and Prevention, Atlanta, Georgia, USA (N. Collins, J. Vinjé);
9University of Alberta, Edmonton, Alberta, Canada (X.L. Pang, B.E. Lee);
10Alberta Provincial Laboratory for Public Health, Edmonton, Canada (X.L. Pang); Erasmus MC, Rotterdam, the Netherlands (M. de Graaf, J. van Beek, M.P.G. Koopmans);
11National Institute for Public Health and the Environment, Bilthoven, the Netherlands (J. van Beek, H. Vennema, M.P.G. Koopmans);
12Robert Koch-Institute, Berlin, Germany (S. Niendorf);
13University of Ljubljana, Ljubljana, Slovenia (M. Poljsak-Prijatelj, A. Steyer);
14University of New South Wales, Sydney, New South Wales, Australia (P.A. White, J.H. Lun); University of Pretoria, Pretoria, South Africa (J. Mans)
1,2,3,4,5,6,7,8,9,10,11,12,13,14,✉