

Abstract
Background:
Progressive bulbar palsy (PBP) is a classic phenotype of bulbar onset amyotrophic lateral sclerosis (ALS) with more rapid progression and worse prognosis. However, as an often under-understood variant of ALS, isolated bulbar palsy (IBP) appears to progress more slowly and has a relatively benign prognosis. This study aimed to investigate the natural course and clinical features of IBP in Chinese population and to compare them with those of PBP.
Methods:
The clinical data of patients with bulbar onset ALS were collected from January 2009 to December 2013. Revised ALS Functional Rating Scale (ALSFRS-R), forced vital capacity (FVC), and follow-up evaluation were performed, and the differences in basic clinical features, ALSFRS-R, FVC, and primary outcome measures between IBP and PBP were analyzed. The independent t-test, Chi-square test, Mann-Whitney U-test, and Kaplan-Meier analysis were used.
Results:
Totally 154 patients with bulbar onset ALS were categorized into two groups, 33 with IBP and 121 with PBP. In the IBP group, the male to female ratio was 0.7 to 1.0, and the mean onset age was 58.5 years. The mean duration from the onset was 16.0 months, and the mean ALSFRS-R score was 43.4 at patients’ first visit to our hospital. In 14 IBP patients performing FVC examination, the mean FVC value was 90.5% and there were only two cases with abnormal FVC. In 26 IBP patients completing follow-up, 15 (58%) suffered death or tracheotomy and the mean survival time was 40.5 months. Significant differences were noted in sex ratio, onset age, ALSFRS-R score, upper motor neuron limb signs, pure lower motor neuron (LMN) bulbar signs, FVC, and survival time between IBP and PBP.
Conclusions:
IBP was evidently different from PBP, which was characterized with the predominance of female, pure LMN bulbar signs, an older onset age, a relative preservation of respiratory function, and a better prognosis.
Keywords: Amyotrophic Lateral Sclerosis, Bulbar Palsy, Prognosis, Vital Capacity
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive disease characterized with degeneration of motor neurons in the brain and spinal cord.[1,2] ALS is invariably fatal with a median survival period of 3–5 years.[2,3] Progressive bulbar palsy (PBP), an ALS phenotype which manifests bulbar onset and develops progressive limb symptoms and signs in short-term, generally portends a worse prognosis with shorter survival time than other ALS phenotypes.[4]
However, isolated bulbar palsy (IBP), an often under-understood variant of ALS, has symptoms confined to bulbar region for extended periods.[5,6] IBP is characterized with insidious onset of dysarthria or dysphagia, which aggravates slowly, and relative preservation of limb and respiration function initially. IBP patients appear to have a relatively benign prognosis and longer survival compared to PBP.[5,6] It is important to distinguish IBP from PBP for prognosis prediction, patient care, and even treatment options.
Only limited case series to date tried to delineate IBP while other studies did not identify any case of IBP.[5,6,7,8] This study was to investigate the natural course and clinical features of IBP in Chinese population and to compare them with those of PBP.
METHODS
Ethical approval
The study was conducted in accordance with the Declaration of Helsinki and was approved by the Ethical Committee of Peking University Third Hospital (PUTH) (No. 2008009). All patients gave informed consent for the present study.
Patients
We reviewed the clinical data of all sporadic ALS patients who were enrolled into the Department of Neurology of PUTH from January 2009 to December 2013. Patients were diagnosed according to the Airlie House diagnostic criteria.[9] All patients were interviewed and examined by board-certified neurologists from the study group who had experience with motor neuron diseases. Each patient was independently examined by two neurologists, and if their diagnoses or disease categories differed, a third and more experienced neurologist examined the patient to make a final determination. For all cases, baseline demographic information and clinical data were collected during the patient's first visit to PUTH. All patients diagnosed with bulbar onset ALS were selected, who had complaints of insidious onset of dysarthria, dysphagia, or both. Alternative diagnoses were excluded by means of detailed investigations including magnetic resonance imaging of the brain and spinal cord, electromyography (EMG), neurophysiological, and serum autoantibody tests. Spinal and bulbar muscular atrophy (SBMA) was excluded with genetic test for exon 1 of androgen receptor gene if SBMA was difficult to clinically distinguish from bulbar onset ALS.
From all bulbar onset ALS cases, IBP was diagnosed if patients had no significant limb involvement or evidence of progression over the initial disease course of 6 months. Significant limb involvement was defined as symptomatic limb weakness that contributed to the clinical presentation.[5,6] Moreover, EMG showed denervation changes (fibrillation potentials, positive sharp waves, or large polyphasic motor units) isolated to the bulbar region. Patients with bulbar onset who manifested or developed progressive limb symptoms and signs were diagnosed with PBP.[5,6] Those cases were excluded if their clinical durations were <6 months and meanwhile, there were no symptoms, signs, or neurogenic EMG changes in any other regions except the bulb.
Investigations
Revised ALS Functional Rating Scale (ALSFRS-R) and forced vital capacity (FVC) were simultaneously assessed during patients’first visit to PUTH. ALSFRS-R was graded by an experienced neurologist to each patient and the total score was 48. FVC values were expressed as percentages of the predicted values.[10,11] FVC values <80% were considered as abnormal, representing ventilation dysfunction.[10,11] Diseases related to the cardiovascular system and lungs were excluded from the study.
Each patient was given a follow-up evaluation by telephone every 3 or 6 months from the first visit till December 2014. Death or tracheotomy was predefined as primary outcome measures.
The differences in basic clinical features, ALSFRS-R, FVC, and primary outcome measures between IBP and PBP were analyzed.
Statistical analysis
Data were analyzed using SPSS version 18.0 software for Windows (SPSS Inc., Chicago, IL, USA). Kolmogorov-Smirnov test was used to determine whether data were normally distributed. Quantitative data that were normally distributed were expressed as mean ± standard deviation and nonnormally distributed data were expressed as median (minimum, maximum). Differences in categorical variables were assessed using Chi-square test or Fisher's exact test and continuous variables were evaluated using independent t-test or Mann-Whitney U-test on parametric or nonparametric nature of the data. Survival curves were estimated using Kaplan-Meier analysis and log-rank test were used for comparison of survival between groups. The value of P < 0.05 (two-sided) was considered statistically significant.
RESULTS
Clinical characteristics
From a total cohort of 1177 consecutive sporadic ALS patients, 154 patients (89 males, 65 females, mean age 56.5 ± 10.5 years, range 33–80 years) with bulbar onset were retrospectively recruited.
The 154 bulbar onset ALS patients exhibited a mean onset age of 55.4 ± 10.6 years (range 33–79 years), a median disease duration of 12 (2, 56) months from the initial symptom and a mean ALSFRS-R score of 39.8 ± 6.1 (range 18–48) at the first visit to PUTH.
From the total cohort of bulbar onset ALS, 33 patients were identified as IBP (14 males, 19 females, mean onset age 58.5 ± 8.2 years, median duration 12 [6, 35] months, mean ALSFRS-R score 43.4 ± 2.4) and 121 patients were categorized as typical PBP (75 males, 46 females, mean onset age 54.6 ± 11.0 years, median duration 12 [2, 56] months, mean ALSFRS-R score 38.8 ± 6.4) [Table 1]. Female was more frequent in IBP and PBP was the opposite (IBP, 58%; PBP, 38%; P < 0.05). Onset age was older (P < 0.05) and ALSFRS-R score was higher (P < 0.001) in IBP group than in PBP group and there was no significant difference in duration in both groups.
Table 1.
Clinical characteristics of 154 patients with bulbar onset ALS
| Groups | Gender, n (male:female) | Onset age (years) | Duration (months) | ALSFRS-R score | Limb weakness, n (%) | Brisk tendon reflex, n (%) | Babinski’s sign, n (%) | Pure UMN bulbar signs, n (%) | Pure LMN bulbar signs, n (%) | UMN and LMN bulbar signs, n (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| IBP (n = 33) | 14:19 | 58.5 ± 8.2 | 12 (6, 35) | 43.4 ± 2.4 | 0 | 17 (52) | 3 (9) | 0 | 14 (42) | 19 (58) |
| PBP (n = 121) | 75:46* | 54.6 ± 11.0* | 12 (2, 56) | 38.8 ± 6.4* | 99 (82)* | 96 (79)* | 44 (36)* | 6 (5) | 24 (20)* | 91 (75)* |
| Statistics | 4.066† | 2.203‡ | 1.103§ | 6.457‡ | 75.600† | 10.275† | 9.095† | – | 7.119† | 3.949† |
| P | 0.044 | 0.031 | 0.270 | <0.001 | <0.001 | 0.001 | 0.003 | 0.342 | 0.008 | 0.047 |
Values are presented as n (%), mean ± SD, or median (minimum, maximum). *P value for contrast of variates between patients with IBP and PBP; P<0.05, as compared with patients with IBP, †χ2 values; ‡t values; §Z values. ALS: Amyotrophic lateral sclerosis; IBP: Isolated bulbar palsy; PBP: Progressive bulbar palsy; ALSFRS-R: Revised ALS Functional Rating Scale; UMN: Upper motor neuron; LMN: Lower motor neuron; –: Not applicable; SD: Standard deviation.
Clinically, there were 99 (82%) PBP patients with limb weakness, and by definition, no limb weakness was detected in IBP [Table 1]. Brisk tendon reflex was less frequent in IBP patients (IBP, 52%; PBP, 79%; P = 0.001), as was Babinski's sign (IBP, 9%; PBP, 36%; P = 0.003). Pure lower motor neuron (LMN) bulbar signs, including tongue amyotrophy or fasciculation, or neurogenic damages of EMG in tongue, sternocleidomastoid or upper trapezius muscles,[12,13] were more common in IBP compared to PBP (IBP, 42%; PBP, 20%; P = 0.008).
Forced vital capacity
FVC was checked in 14 IBP patients (6 males, 8 females, mean onset age 58.6 ± 8.0 years, median duration 8 [6, 33] months, ALSFRS-R score 43.7 ± 2.2) and 41 PBP (31 males, 10 females, mean onset age 52.4 ± 9.7 years, median duration 9 [2, 54] months, mean ALSFRS-R score 39.6 ± 6.5) [Table 2]. Significant differences were noted in gender, onset age, and ALSFRS-R score except duration between the two groups. The mean FVC value was 90.5% ± 8.2% (range 76–102%) of the predicted in IBP and 81.9% ± 13.7% (range 49–103%) in PBP. Two IBP patients displayed FVC <80% and 19 in the other group. Both the mean FVC value and the frequency of FVC <80% were significantly different between the two groups. Nineteen IBP and 80 PBP patients were not capable of finishing FVC assessment because of weakness or amyotrophy of bulbar muscles, or refusal of patients for some other reasons such as inspection fee or time of appointment.
Table 2.
FVC of 55 patients with bulbar onset ALS
| Groups | Gender, n (male:female) | Onset age (years) | Duration (months) | FVC (%) | FVC <80%, n | ALSFRS-R score | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| IBP (n = 14) | 6:8 | 58.6 ± 8.0 | 8 (6, 33) | 90.5 ± 8.2 | 2 | 43.7 ± 2.2 | ||||
| PBP (n = 41) | 31:10* | 52.4 ± 9.7* | 9 (2, 54) | 81.9 ± 13.7* | 19* | 39.6 ± 6.5* | ||||
| Statistics | – | 2.162† | 0.068‡ | 2.188† | 4.543§ | 3.495† | ||||
| P | 0.045 | 0.035 | 0.946 | 0.033 | 0.033 | 0.001 | ||||
Values are presented as n (%), mean ± SD, or median (minimum, maximum). *P value for contrast of variates between patients with IBP and PBP; P<0.05, as compared with patients with IBP. †t values; ‡Z values; §χ2 values. ALS: Amyotrophic lateral sclerosis; IBP: Isolated bulbar palsy; PBP: Progressive bulbar palsy; FVC: Forced vital capacity; ALSFRS-R: Revised ALS Functional Rating Scale; –: Not applicable; SD: Standard deviation.
Primary outcome measures
Twenty-six IBP and 106 PBP patients finished the telephone follow-up till December 2014. The lost follow-up rate was 21% in IBP and 12% in PBP. A smaller proportion of IBP suffered primary outcome events compared to PBP during the follow-up (IBP, 15 [58%]; PBP, 83 [78%]). The median survival time was 38.5 (12.0, 80.0) months in IBP and 29.0 (11.0, 89.0) months in PBP. The Kaplan-Meier curves of the two groups were different [Figure 1]. Log-rank test showed that the median survival time of patients with IBP was significantly longer than that with PBP (Log rank = 9.863, P = 0.002).
Figure 1.
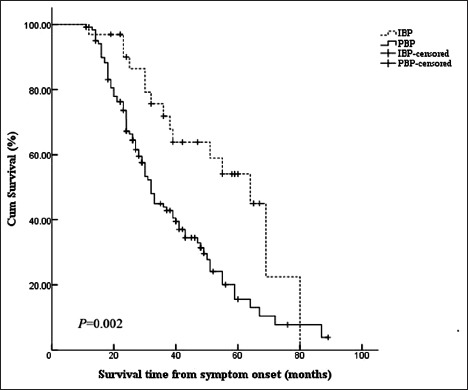
Kaplan-Meier survival curves of 154 patients with bulbar onset ALS according to phenotype. ALS: Amyotrophic lateral sclerosis; IBP: Isolated bulbar palsy; PBP: Progressive bulbar palsy.
DISCUSSION
ALS patients with bulbar onset often have a worse prognosis with a shorter survival period of about 24 months than those with limb onset.[4] However, it is clear that a small group of patients with bulbar onset do not progress as rapidly as typical ALS, although limited reports to date have tried to delineate this variant of ALS.[5,6] There is still controversy about the clinical manifestation and classification of bulbar onset ALS. Some consider PBP as the sole phenotype of bulbar onset ALS and eventually progressing into ALS after some months[14] while others consider that some subtype exists besides PBP.[5,6]
IBP is an uncommon regional variant of ALS that seems to progress more slowly than typical bulbar-onset ALS.[5,6] Recent reports on IBP showed its lower morbidity, which was estimated about 1–4% of ALS.[5,6,15,16] Onset age was slightly younger than typical bulbar-onset ALS, with a mean onset age of 61 years reported.[5] Female gender was predominant in IBP with a female to male of 3 to 1.[5] Patients with IBP might have upper motor neuron (UMN) and/or LMN signs in the bulbar region.[5,6,14] Respiratory function was generally preserved in a relatively long period.[5] Percutaneous endoscopic gastrostomy (PEG) tube placement might be required at early phase because of dysphagia.[5,6]
The present study described the natural course and several important clinical features of IBP in the mainland of China. Patients with IBP represented about 3% of sporadic ALS and 21% of bulbar-onset ALS. IBP was evidently different from PBP, a classic phenotype of bulbar onset ALS with more rapid progression and worse prognosis. Patients with IBP were predominantly female and limb function preserved, and PBP was the opposite. IBP was characterized with older onset age and less UMN signs in both limb and bulb with comparison to PBP, such as brisk tendon reflex, Babinski's sign, palmomental reflex, sucking reflex. LMN bulbar signs, such as tongue amyotrophy and fasciculation and neurogenic damages of EMG in bulbar region, were more common in IBP compared to PBP.
The diagnosis of ALS depends on signs of UMN and LMN lesion and the number of regions involved according to the Airlie House diagnostic criteria.[9] Given patients’ limbs uninvolved, IBP does not meet criteria for probable or definite ALS and is diagnosed merely as possible ALS when UMN and LMN bulbar dysfunction are found. IBP with only LMN bulbar involvement is unclassifiable or considered as “suspected ALS”, which was included in the original EI Escorial criteria and deleted from the Airlie House diagnostic criteria.[9,17] IBP might be reclassified as probable or definite ALS if limb symptoms and signs developed. However, these diagnostic criteria do not take prognosis into account, potentially leading to difficulties when classifying patients for treatment trials.[5] In this study, LMN bulbar signs were predominant in IBP with comparison to PBP. Burrell et al.[5] found that UMN bulbar signs were more frequent in IBP and Karam et al.[14] reported that UMN and LMN bulbar signs did not differ significantly in both groups. ALS, including IBP, is a disorder with great clinical heterogeneity. Moreover, differences in races, criteria of patients’ selection and items examined might give increase to the divergence among the studies.
This study showed that at each patient's first visit to our department, ALSFRS-R score, mean FVC value, and the frequency of patients with FVC ≥80% were significantly higher in IBP with comparison to PBP and no significant difference found in duration in both groups. These results coincide with the viewpoint that IBP progresses and damages respiration more slowly than PBP in view of the fact that ALS patients’ motor functions including respiration usually aggravate gradually with the duration prolonging.[5,6] The results of follow-up on primary outcome measures revealed that the incidence of death or tracheotomy was lower and survival time longer in IBP. ALS is characterized with focal and regional susceptibility in the pathophysiologic mechanism, which implies that the region adjacent to onset site is apt to be prior involved.[18] The medulla oblongata and the diaphragm, which is innervated by LMNs of C3–C5 spinal cord anterior horn, are tightly adjoined and their motor neurons may be concomitantly involved in ALS.[19] Consequently, dyspnea and respiratory failure, which is the main cause of death in ALS, may occur earlier in patients with typical bulbar-onset ALS. However, motor neurons damage in IBP is confined to the bulbar region for extended periods for unknown reasons, which may partially explain the phenomenon of respiration preserved and a relatively benign prognosis in IBP. The feature of predominance in female gender in IBP indicates that genetic factor may play a role. However, IBP has been almost reported as sporadic cases and genetic susceptibility in IBP needs to be further studied.
The study described the predominance of female gender and pure LMN bulbar signs, an older onset age, a relative preservation of respiratory function and a better prognosis in IBP, which somewhat distinguished IBP from more typical PBP clinically. Understanding the characteristics of IBP may help to yield important information about the pathogenesis of ALS and provide guidance to treatment options. Studies showed that noninvasive positive pressure ventilation (NIPPV) could extend the survival time of ALS patients for 1 year when FVC <75%.[20] Accordingly, patients with IBP may need NIPPV less and later than PBP. The viewpoint that Riluzole might take better effects in ALS patients with slower progression may apply to IBP.[21]
It is acknowledged that there were several limitations in this study. The group size was comparatively small, especially the group performing FVC examination, and the clinical significance of the study might be limited by selection bias. The information about patients using PEG or not were not collected in telephone follow-up.
Financial support and sponsorship
This study was supported by a grant from the National Natural Science Foundation of China (No. 81030019).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Yuan-Yuan Ji
REFERENCES
- 1.Chiò A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, et al. Prognostic factors in ALS: A critical review. Amyotroph Lateral Scler. 2009;10:310–23. doi: 10.3109/17482960802566824. doi: 10.3109/17482960802566824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14:248–64. doi: 10.1038/nrn3430. doi: 10.1038/nrn3430. [DOI] [PubMed] [Google Scholar]
- 3.Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, et al. Amyotrophic lateral sclerosis. Lancet. 2011;377:942–55. doi: 10.1016/S0140-6736(10)61156-7. doi: 10.1016/S0140-6736(10)61156-7. [DOI] [PubMed] [Google Scholar]
- 4.Chiò A, Calvo A, Moglia C, Mazzini L, Mora G PARALS Study Group. Phenotypic heterogeneity of amyotrophic lateral sclerosis: A population based study. J Neurol Neurosurg Psychiatry. 2011;82:740–6. doi: 10.1136/jnnp.2010.235952. doi: 10.1136/jnnp.2010.235952. [DOI] [PubMed] [Google Scholar]
- 5.Burrell JR, Vucic S, Kiernan MC. Isolated bulbar phenotype of amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2011;12:283–9. doi: 10.3109/17482968.2011.551940. doi: 10.3109/17482968.2011.551940. [DOI] [PubMed] [Google Scholar]
- 6.Jawdat O, Statland JM, Barohn RJ, Katz JS, Dimachkie MM. Amyotrophic lateral sclerosis regional variants (Brachial Amyotrophic Diplegia, Leg Amyotrophic Diplegia, and Isolated Bulbar Amyotrophic Lateral Sclerosis) Neurol Clin. 2015;33:775–85. doi: 10.1016/j.ncl.2015.07.003. doi: 10.1016/j.ncl.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cerero Lapiedra R, Moreno López LA, Esparza Gómez GC. Progressive bulbar palsy: A case report diagnosed by lingual symptoms. J Oral Pathol Med. 2002;31:277–9. doi: 10.1034/j.1600-0714.2002.310505.x. doi: 10.1034/j.1600-0714.2002.310505.x. [DOI] [PubMed] [Google Scholar]
- 8.Becker A, Hardmeier M, Steck AJ, Czaplinski A. Primary lateral sclerosis presenting with isolated progressive pseudobulbar syndrome. Eur J Neurol. 2007;14:e3. doi: 10.1111/j.1468-1331.2007.01699.x. doi: 10.1111/j.1468-1331.2007.01699.x. [DOI] [PubMed] [Google Scholar]
- 9.Brooks BR, Miller RG, Swash M, Munsat TL. World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–9. doi: 10.1080/146608200300079536. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 10.Criée CP, Baur X, Berdel D, Bösch D, Gappa M, Haidl P, et al. Standardization of spirometry: 2015 update. Published by German Atemwegsliga, German Respiratory Society and German Society of Occupational and Environmental Medicine. Pneumologie. 2015;69:147–64. doi: 10.1055/s-0034-1391345. doi: 10.1055/s-0034-1391345. [DOI] [PubMed] [Google Scholar]
- 11.Rochester CL, Vogiatzis I, Holland AE, Lareau SC, Marciniuk DD, Puhan MA, et al. An official American Thoracic Society/European Respiratory Society Policy Statement: Enhancing Implementation, Use, and Delivery of Pulmonary Rehabilitation. Am J Respir Crit Care Med. 2015;192:1373–86. doi: 10.1164/rccm.201510-1966ST. doi: 10.1164/rccm.201510-1966ST. [DOI] [PubMed] [Google Scholar]
- 12.Kang DX, Fan DS. The electrophysiological study of differential diagnosis between amyotrophic lateral sclerosis and cervical spondylotic myelopathy. Electromyogr Clin Neurophysiol. 1995;35:231–8. [PubMed] [Google Scholar]
- 13.Xu YS, Zheng JY, Zhang S, Fan DS. Upper trapezius electromyography aids in the early diagnosis of bulbar involvement in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2011;12:345–8. doi: 10.3109/17482968.2011.582647. doi: 10.3109/17482968.2011.582647. [DOI] [PubMed] [Google Scholar]
- 14.Karam C, Scelsa SN, Macgowan DJ. The clinical course of progressive bulbar palsy. Amyotroph Lateral Scler. 2010;11:364–8. doi: 10.3109/17482960903513159. doi: 10.3109/17482960903513159. [DOI] [PubMed] [Google Scholar]
- 15.Logroscino G, Beghi E, Zoccolella S, Palagano R, Fraddosio A, Simone IL, et al. Incidence of amyotrophic lateral sclerosis in Southern Italy: A population based study. J Neurol Neurosurg Psychiatry. 2005;76:1094–8. doi: 10.1136/jnnp.2004.039180. [Google Scholar]
- 16.Argyriou AA, Polychronopoulos P, Papapetropoulos S, Ellul J, Andriopoulos I, Katsoulas G, et al. Clinical and epidemiological features of motor neuron disease in South-Western Greece. Acta Neurol Scand. 2005;111:108–13. doi: 10.1111/j.1600-0404.2004.00362.x. doi: 10.1111/j.1600-0404.2004.00362.x. [DOI] [PubMed] [Google Scholar]
- 17.Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors. J Neurol Sci. 1994;124(Suppl):96–107. doi: 10.1016/0022-510x(94)90191-0. doi: 10.1016/0022-510X(94)90191-0. [DOI] [PubMed] [Google Scholar]
- 18.Ravits J, Appel S, Baloh RH, Barohn R, Brooks BR, Elman L, et al. Deciphering amyotrophic lateral sclerosis: What phenotype, neuropathology and genetics are telling us about pathogenesis. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(Suppl 1):5–18. doi: 10.3109/21678421.2013.778548. doi: 10.3109/21678421.2013.778548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang HG, Zhang S, Xu YS, Zhang N, Fan DS. Association between rectus abdominis denervation and ventilation dysfunction in patients with amyotrophic lateral sclerosis. Chin Med J. 2016;129:2063–6. doi: 10.4103/0366-6999.189070. doi: 10.4103/0366-6999.189070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carratù P, Spicuzza L, Cassano A, Maniscalco M, Gadaleta F, Lacedonia D, et al. Early treatment with noninvasive positive pressure ventilation prolongs survival in amyotrophic lateral sclerosis patients with nocturnal respiratory insufficiency. Orphanet J Rare Dis. 2009;4:10. doi: 10.1186/1750-1172-4-10. doi: 1186/1750-1172-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valentino P, Conforti FL, Pirritano D, Nisticò R, Mazzei R, Patitucci A, et al. Brachial amyotrophic diplegia associated with a novel SOD1 mutation (L106P) Neurology. 2005;64:1477–8. doi: 10.1212/01.WNL.0000158679.47281.03. doi: 10.1212/01.WNL.0000158679.47281.03. [DOI] [PubMed] [Google Scholar]