

Abstract
Mitochondrial dysfunction is closely associated with the pathogenesis of nonalcoholic steatohepatitis (NASH). The aim of the present study was to comprehensively determine mitochondrial abnormalities in NASH by detecting the proteomics in liver mitochondria in a NASH rat model, which was induced for 16 weeks by the provision of a high fat and high cholesterol diet (HFD). Serum parameters, including triglycerides, total cholesterol, low-density lipoprotein cholesterol and high-density lipoprotein cholesterol were determined, and hematoxylin and eosin staining of liver tissues was examined to evaluate the NASH rat model. Various parameters associated with mitochondrial function were examined, including mitochondrial DNA (mtDNA) copy number, mitochondrial membrane potential (MMP) and mitochondrial respiratory chain complex (MRC) activity. The mitochondrial proteomics were analyzed and identified using isobaric tags for relative and absolute quantitation labeling coupled with two-dimensional liquid chromatography-tandem mass spectrometry. The identified proteins were classified and grouped using the Blast2GO program against the non-redundant protein database, the Kyoto Encyclopedia of Genes and Genomes database and the Cluster of Orthologous Groups of proteins database. Compared with the control, mtDNA copy number, MMP, and activities of MRC I and III were decreased markedly in the HFD group. A total of 18 upregulated and 13 downregulated proteins were identified, with a significant 1.2-fold difference between the control and NASH groups. The dysregulated proteins were closely involved in mitochondrial oxidative phosphorylation, the lipid metabolic process and fatty acid β-oxidation. The results of the present study provide important proteomic information regarding liver mitochondria in NASH and serve as a basis for further detailed investigations of the pathogenesis of NASH.
Keywords: mitochondrial function, nonalcoholic steatohepatitis, isobaric tags for relative and absolute quantitation, rat model
Introduction
Nonalcoholic fatty liver disease (NAFLD) is a common cause of chronic liver disease, and comprises a wide range of pathological changes in the liver, from non-progressive steatosis to nonalcoholic steatohepatitis (NASH), advanced fibrosis and cirrhosis, ultimately leading to hepatocellar carcinoma (HCC) (1). NASH is characterized by lobular inflammation, hepatocellular ballooning and fibrosis, and is key in the progression to cirrhosis and HCC (2). Day and James described a two-hit theory (3), regarding the hepatic triglyceride accumulation as the first hit and mitochondrial dysfunction, oxidative stress and inflammatory factors as the second hit (4). Substantial evidence has indicated that mitochondrial dysfunction is directly associated with the pathogenesis of NASH and has suggested that NASH is a mitochondrial disease (5,6). Mitochondrial dysfunction impairs lipid metabolism and induces the overproduction of reactive oxygen species (ROS), which triggers the peroxidation of lipids and apoptosis of hepatocytes (7). In addition, this can decrease the activity of the mitochondrial respiratory chain (MRC), which further impairs adenosine triphosphate (ATP) synthesis and enhances oxidative stress (7). However, the alterations in mitochondrial proteomics in response to NASH remain to be fully elucidated.
In our previous study, a high fat and high cholesterol diet (HFD) was used to establish a NASH rat model, which is more similar to dietary conditions, compared with the methionine-choline-deficient diet in humans (8). To date, using the NASH rat model, there has been no attempt to establish a comparative proteome profile of liver mitochondria using isobaric tags for relative and absolute quantitation (iTRAQ) technology coupled with two-dimensional liquid chromatography-tandem mass spectrometry (2-D LC-MS/MS) analysis.
Compared with traditional 2-D gel electrophoresis, it is easier to analyze eight samples simultaneously using iTRAQ technology, thus enhancing throughput, and increasing sensibility and accuracy (9). To the best of our knowledge, the present study using the NASH rat model is the first to perform proteomic analysis of liver mitochondria using iTRAQ technology, to provide novel insights into the pathogenesis and progression of NASH via mitochondrial protein profiling.
Materials and methods
Animal models of NASH
Eight-week-old male Sprague-Dawley rats weighing ~320 g were purchased from the Laboratory Animal Centre of Wenzhou Medical University (Wenzhou, China). Under a 12-h light-dark cycle and controlled temperature (23±2°C), all rats were raised under specific pathogen-free conditions with free access to water and food. The animals were acclimatized to the laboratory conditions for 1 week prior to the experiments and were randomly divided into two groups: Control group (n=6) and HFD group (n=10). The rats in the control group and the HFD group were respectively fed with a standard chow diet (SCD) and a high fat diet (1% cholesterol, 19% lard and 80% SCD) for 16 weeks. The body weights of the rats were measured weekly. The rats were sacrificed following overnight fasting. All protocols and procedures conformed to the guidelines of the Laboratory Animal Ethics Committee of Wenzhou Medical University, and all efforts were made in order to minimize the suffering and the number of animals used.
Blood and liver sample preparation
Following sacrifice, blood samples from the abdominal vein of the rats were collected in coagulation-promoting tubes and centrifuged at 1,500 g for 15 min at 4°C (Eppendorf 5810R; Eppendorf, Hamburg, Germany) to obtain the serum for biochemical analysis, which was stored at −80°C prior to analysis. The levels of glucose, total cholesterol (TC), triglyceride (TG), low-density lipoprotein (LDL-C) and high-density lipoprotein (HDL-C) were measured using an automated biochemistry analyzer (Hitachi, Tokyo, Japan). The livers were excised, cleaned with ice-cold phosphate-buffered saline and weighed immediately. The left lobes of each liver were then fixed in 4% paraformaldehyde solution for further morphological analysis using hematoxylin and eosin (H&E) staining, with images captured using a Nikon microscope (Nikon E-100 A12.0705; Nikon Corporation, Tokyo, Japan). Additionally, sections of the liver tissue were added to a volume of storage medium containing 20% dimethyl sulfoxide, 0.21 M mannitol and 0.07 M sucrose (pH 7.5) for mitochondrial separation, and other sections were directly snap frozen in liquid nitrogen and then preserved at −80°C until use (10).
Determination of relative mtDNA copy number
Total DNA from the liver tissues was extracted using a Blood and Cell Culture DNA Mini kit (Qiagen GmbH, Hilden, Germany). Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis was used to determine the relative mtDNA copy number. The RT-qPCR amplification reaction was performed via SYBR-Green chemistry using Bio-Rad CFX Manage 2.1 (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The mtDNA was synthesized and amplified using the following primers: ND1 forward, 5′-ATTCTAGCCACATCAAGTCTTT-3′ and reverse, 5′-GGAGGACGGATAAGAGGATAAT-3′; β-actin forward, 5′-GAAATCGTGCGTGACATTAAAG-3′ and reverse, 5′-ATCGGAACCGCTCATTG-3′. Each sample was analyzed in triplicate with a 20 µl final volume containing SYBR-Green Supermix PCR 1X Master mix (Bio-Rad Laboratories, Inc.), 0.5 µM forward and reverse primes and 100 ng DNA template. Following 3 min denaturation at 95°C, amplification was performed for 39 cycles, including 95°C for 10 sec for denaturation, and 55°C for 30 sec for annealing and extension. Melting curve analysis was performed at the end of each run to validate the specificity of the PCR products. The quantification of the relative mtDNA copy number was performed using the 2ΔΔCq method (11), normalized to β-actin.
Isolation of liver mitochondria
The frozen liver tissues of the rats were rapidly thawed using a preheated medium of 0.4% BSA (Beyotime Institute of Biotechnology, Haimen, China), 0.25 M sucrose and 0.01 M Tris-HCl (pH 7.5) at 45°C in a 4:1 ratio of medium to tissue. The mitochondria were isolated as described previously (12). In brief, the liver tissues were weighed, washed and homogenized at 4°C in isolation buffer containing 0.5% BSA, 225 mM mannitol, 75 mM sucrose, 30 mM Tris/HCl and 0.5 mM EGTA (pH 7.4). The homogenate was centrifuged twice at 4°C and 740 g for 10 min to remove unbroken cells and nuclei, and at 4°C and 9,000 g for 10 min three times to precipitate the mitochondrial pellet and obtain the crude mitochondria. The pellet was resuspended in MRB buffer containing 250 mM mannitol, 5 mM HEPES and 0.5 mM EGTA (pH 7.4), and gently layered on top of 30% (v/v) percoll. Following 50 min of centrifugation at 4°C and 95,000 × g, collection and washing of the mitochondrial fraction twice, centrifugation was performed at 4°C and 6,300 × g for 10 min with MRB to remove the residual percoll. Using the BCA protein assay kit (Thermo; Fisher Scientific, Inc., Waltham, MA, USA) the concentration of mitochondrial protein was measured. Mitochondria were stored in −80°C for proteomic analysis.
MRC enzymatic assays
As described above, the pellet (crude mitochondria) was resuspended in MRB and freeze-thawed three times using liquid nitrogen. All samples were measured in triplicate using a Varioskan Flash reader (Thermo Fisher Scientific, Inc.).
The specific activity of complex I, ubiquinone oxidoreductase (NADH) was analyzed according to the decrease in absorbance at 340 nm due to the oxidation of NADH. The isolated mitochondria (25 µg) were added into 200 µl of the reaction buffer (195 µM NADH, 50 mM potassium phosphate buffer, 10 µg/ml antimycin, 10 µM decylubiquinone and 5 mM NaN3) in a 96-well plate. Under the conditions of 340 nm and 30°C, the rate of oxidation of NADH was measured for 90 sec.
The specific activity of complex II (succinate dehydrogenase) was assessed by the decrease of absorbance at 600 nm due to reduction of dichlorophenol indophenols. The 200 µl reaction buffer (270 mM potassium phosphate buffer, 200 mM succinate, 12 µM rotenone, 7.5 µM NaN3, 5 µg/ml antimycin, 100 mM 2,6-dichlorophenolindophenolsodium salt and 20 µg mitochondria protein) was equilibrated for 10 min at 30°C. Following the addition of 4 µl of 1 mM decylubiquinone, the reaction was initiated and absorbance was measured at 600 nm for 90 sec at 30°C.
Complex III (decylubiquinol cytochrome coxidoreductase) was measured by the increase of absorbance at 550 nm due to the reduction of cytochrome c. The 200 µl reaction buffer (250 mM sucrose, 100 mM Tris/Hcl, 1 mM EDTA, 50 µM cytochrome c, 50 µM decylubiquinol, 45 µM n-dodecyl-β-d-maltoside and 7.5 µM NaN3) was incubated for 60 sec and the reaction was induced by adding 20 µg of mitochondrial protein. The absorbance was measured at 550 nm for 90 sec at 30°C.
Complex IV (cytochrome c oxidase) was measured by the decrease of absorbance at 550 nm due to oxidization of the reduced cytochrome c. The mitochondria (20 µg) were added into 200 µl reaction buffer, containing 9.4 mM potassium phosphate buffer, 50 µM reduced cytochrome c and 450 µm n-dodecyl-β-d-maltoside. The reaction was detected at 550 nm for 135 sec at 30°C.
The activity of citrate synthase was assessed by alterations of thionitrobenzoate anion formation. The mitochondrial protein (20 µg) was added to 200 µl reaction buffer (0.1 M Tris/Hcl, 0.1 M 5,5′-dithiobis-2-nitrobenzoate, 0.3 mM acetyl-CoA, 450 µM n-dodecyl-β-d-maltoside and 500 µM oxaloacetate). The absorbance was then measured at 412 nm for 270 sec. The activity of CS was expressed in nmol/min/mg, and normalized to total tissue protein content.
ATP synthase activity
According to the manufacturer's protocol of the ATP Synthase Enzyme Activity Microplate Assay kit (Abcam, Cambridge, UK), ADP and phosphate are produced by ATP synthase hydrolyzing ATP. The oxidation of NADH is coupled with the production of ADP and ultimately becomes NAD+. The reaction was detected at 340 nm for 90 sec at 30°C.
Mitochondrial membrane potential (MMP) analysis using JC-1
MMP was determined in the crude mitochondria freshly isolated from liver tissues using a JC-1 Mitochondrial Membrane Potential Detection kit (Beyotime Institute of Biotechnology). According to the manufacturer's protocol, 50 µg of mitochondria were stained by JC-1 and scanned at 490 nm excitation/530 nm emission and at 525 nm excitation/590 nm emission to detect green and red JC-1 fluorescence, respectively, using the Varioskan Flash reader.
Quantitative proteomics using the iTRAQ technique
Mitochondria were solubilized in lysis buffer (7 M urea, 2 M thiourea, 40 mM Tris, 2 mM EDTA, 1 mM PMSF, 0.2% SDS and 4% CHAPS), and sonicated at 200 W for 15 min on ice, followed by centrifugation at 4°C and 25,000 g for 20 min. The supernatant was added to 10 mM DTT (final concentration) and maintained at 56°C for 1 h, this step was for reducing the disulfide bonds in the proteins. The mixture was kept in the dark, and 55 mM IAM (final concentration) was added and incubated for 1 h in order to block the cysteines. To remove detergents, which may interfere with iTRAQ™ labeling, the protein was precipitated by the addition of five volumes of chilled acetone for 2 h at −20°C. Following centrifugation at 4°C at 25,000 g for 20 min, the pellet was dissolved in 500 µl of 0.5 M triethylammonium bicarbonate (Applied Biosystems; Thermo Fisher Scientific, Inc.) and sonicated again. The samples were then centrifuged at 25,000 g for 20 min at 4°C. The Bradford method (Thermo Fisher Scientific, Inc.) was used to quantify the supernatant. Protein of each sample (100 µg) was digested with trypsin (Promega Corporation, Madison, WI, USA), at 20:1 protein to trypsin ratio, overnight at 37°C. Vacuum centrifugation was performed to dry the peptides following the digestion with trypsin. According to the iTRAQ™ reagents protocol, using 8-plex iTRAQ reagent (Applied Biosystems; Thermo Fisher Scientific, Inc.), the peptides were dissolved and samples labeled as follows: Control (CON)-2 (117 tag), CON-4 (114 tag), CON-5 (119 tag), CON-8 (116 tag), HFD-1 (113 tag), HFD-2 (118 tag), HFD-4 (115 tag) and HFD-8 (121 tag) randomly selected from the control group and HF group, respectively. Vacuum centrifugation at 4°C and 12,000 g for 10 min was performed to pool and dry the labeled peptide mixtures.
The peptide mixtures were added to 4 ml solvent A, which contained 25 mM NaH2PO4 dissolved in 25% can (pH 2.7) and then injected into a 4.6×250 mm Ultremex SCX column, which worked with the LC-20AB HPLC Pump system (Shimadzu Corporation, Kyoto, Japan) and contained 5 µm particles (Phenomenex, Inc., Torrence, CA, USA). A gradient of solvents was used to elute the peptides at a 1 ml/min flow rate: 10 min of 100% solvent A, 7 min of 5% solvent B, which contained 1 M KCl and 25 mM NaH2PO4 dissolved in 25% ACN (pH 2.7), 20 min of 5–60% solvent B and 1 min of 60–100% buffer B. Finally, washing was performed for 1 min of 100% buffer B and equilibrated for 10 min in buffer A prior to the next loading. The absorbance of elution was monitored at 214 nm and the fractions were collected every 1 min. In total, 20 fractions were collected, which were desalted in a Strata X C18 column (Phenomenex, Inc.) and then dried them completely in a vacuum centrifuge.
Fractions were reconstituted with solvent A (2% CAN, 0.1% FA) and centrifuged for 10 min at 4°C and 20,000 g to remove the insoluble substance, and the final concentration of peptide was adjusted to 0.5 µg/µl. In each fraction, using the autosampler, 5 µg of peptide mixture was injected into a Shimadzu LC-20AD nano HPLC (Shimadzu Corporation), which had a 2 cm C18 trap column. Subsequently, an in-house packed analytical column (75 µm × 10 cm, C18) was used to elute the peptides. The peptides were loaded at 8 µl/min for 4 min and separated at a flow of 300 nl/min over 44 min with a gradient of solvent B (98% ACN and 0.1 FA). Subsequently, the linear gradient was increased to 80% within 2 min and maintained at 80% for 4 min, followed by a return to 5% for 1 min. The effluent was analyzed using a Q-Exactive mass spectrometer (Thermo Fisher Scientific, Inc.) with nanoelectrospray and voltage set at 1.6 KV. At a resolution of 7,000 in an Orbitrap mass analyzer, full MS scans were acquired from 350–2,000 m/z for the detection. A fragment ion spectrum was produced via high-energy collision dissociation and the mass range of 100–1,800 m/z was detected in by Orbitrap mass analyzer at a resolution of 17,500. In the MS survey scan, following a dynamic exclusion duration of 15 sec, MS/MS data were obtained through data-dependent acquisition, which used the 15 most abundant precursor ions above the threshold ion count of 20,000. The automatic gain control target for full MS was 3e6 and for MS/MS was 1e5, and were used to optimize the spectra generated by the Orbitrap analyzer.
Protein identifications were performed using Discoverer 1.2 (Thermo Electron, San Jose, CA, USA), compared with a database containing the UniprotRat sequences, using the Mascot search engine (version 2.3.02; Matric Science, London, UK). In the identification of proteins, which allowed for one missed cleavage in the trypsin digestion, the tolerance of the peptide mass in MS was 20 ppm and for fragmented ions was 0.05 Da. The conversion of N-terminal glutamine to pyroglutamic acid, oxidation of methionine and tyrosine labeled by iTRAQ-8-plex were set as the potential variable modifications. The carbamidomethylation of cysteine at the N-terminal of peptides and lysine labeled by iTRAQ-8-plex were considered to fix these modifications. Peptides with significance scores (≥20; P<0.01) were counted as identified in order to reduce false peptide identification. At least one unique peptide was involved in the identification of each confident protein and at least two unique peptides were required to quantify protein. Significant changes in the quantitative protein ratios were identified by setting cut off values of a fold change >1.2 and P<0.05. Using the Blast2GO program (www.blast2go.com), compared with the non-redundant protein database (NR; NCBI), functional annotations, which had differential proteins, were performed. The Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg/) and Cluster of Orthologous groups (http://www.ncbi.nlm.nih.gov/COG/) databases were used to classify and group these identified proteins.
Western blot analysis
The proteins levels of NADH dehydrogenase 1α subcomplex subunit 5 (Ndufa5), NADH dehydrogenase iron-sulfur protein 6 (Ndufs6), ATP synthase α subunit (ATP5A), transcription factor A, mitochondrial (TFAM) and cytochrome b (CytB) from the liver mitochondria of the control and HFD groups were measured using western blot analysis. The proteins were extracted as described above in the ‘Isolation of liver mitochondria’ section and quantified by BCA Protein assay kit (Thermo Fisher Scientific, Inc.). The proteins (20 µg) were separated on an SDS-PAGE gel and transferred onto nitrocellulose membranes (Bio-Rad Laboratories, Inc.), and then blocked with 5% nonfat milk buffer for 1.5 h. The membranes were then incubated overnight with anti-Ndufa5 (1:1,000; cat. no. 16640-1-AP; ProteinTech Group, Inc., Chicago, USA), anti-Ndufs6 (1:1,000; cat. no 14417-1-AP; ProteinTech Group, Inc.), anti-ATP5A (1:1,000; cat. no. 14676-1-AP; ProteinTech Group, Inc.), anti-TFAM (1:1,000; cat. no. ab131607; Abcam) and CytB (1:1,000; cat. no. 55090-1-AP; ProteinTech Group, Inc.) at 4°C. The membranes were washed once with TBST buffer and incubated with horseradish peroxidase-conjugated anti-rabbit secondary antibodies (1:3,000; cat. no. A0208; Beyotime Institute of Biotechnology) at room temperature for 1 h. The intensity of the bands was detected using ECL Western Blotting Substrate (Thermo Fisher Scientific, Inc.).
Statistical analysis
All results are presented as the mean ± standard deviation and data were analyzed using Student's t-test. Statistical analyses were performed with SPSS 20.0 (IMB SPSS, Armonk, NY, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Characterization of the NASH rat model
The 16 weeks of HFD feeding successfully induced hepatic steatohepatitis. The body weights of the rats, compared with those in the control group, were significantly increased in the HFD group (P<0.05; Table I). Of note, HFD chow resulted in a marked increase in the liver weight and hepatic index (liver weight/body weight %). Higher levels (P<0.01) of serum TC and LDL-C were also found in the HFD-fed rats, compared with those in the controls, although no significant differences were found in the TG, HDL-C or glucose levels (Table II). Following 16 weeks of HFD feeding, the livers sections showed that there were abundant macrovesicular fat droplets in the hepatocytes and increased quantities of ballooned hepatocytes in the centrilobular parenchyma, leading to destruction of the normal structure of numerous hepatic lobules (Fig. 1). Furthermore, foci of necrosis and infiltration of inflammatory cells were identified in the centrilobular region. The liver tissues of the control group showed no histological abnormalities (Fig. 1).
Table I.
Body weights and liver weights or rats.
| Parameter | Con (n=6) | HFD (n=10) |
|---|---|---|
| Initial body weight (g) | 318.83±19.31 | 321.30±15.25 |
| Final body weight (g) | 516.50±37.07 | 548.20±22.23a |
| Liver weight (g) | 11.71±1.17 | 16.56±3.26b |
| Liver weight/body weight (%) | 2.26±0.22 | 3.02±0.57b |
Values are expressed as the mean ± standard deviation.
P<0.05
P<0.01, vs. Con group. Con, control; HFD, high fat and high cholesterol diet.
Table II.
Serum biochemical parameters.
| Parameter (mM) | Con (n=6) | HFD (n=10) |
|---|---|---|
| Glucose | 10.92±0.90 | 10.49±2.10 |
| TG | 0.56±0.13 | 0.64±0.10 |
| TC | 1.66±0.41 | 2.44±0.48a |
| LDL-C | 1.07±0.35 | 1.86±0.43a |
| HDL-C | 0.34±0.11 | 0.29±0.13 |
Values are expressed as the mean ± standard deviation.
P<0.01, vs. Con. TC, total cholesterol; TG, triglyceride; LDL-C, low-density lipoprotein; HDL-C, high-density lipoprotein; Con, control; HFD, high fat and high cholesterol diet.
Figure 1.

Hepatic pathology. Liver sections from the Con and HFD groups were stained with hematoxylin and eosin. Representative images from (A) Con and (B) HFD groups at ×100 magnification. Representative images from (C) Con and (D) HFD groups at ×400 magnification. Fat droplets, ballooned hepatocytes and inflammatory cells are present in the HFD group. HFD, high fat and high cholesterol diet; Con, control; HFD, high fat and high cholesterol diet.
Effect of mtDNA copy number
The present study also evaluated alterations of mitochondrial biogenesis in rats with HFD-induced NASH using information on mtDNA copy number. HFD feeding induced a significant decrease (P<0.05) in mtDNA copy number, which was reflected in the ratio of ND1 DNA to actin DNA in the liver (Fig. 2).
Figure 2.
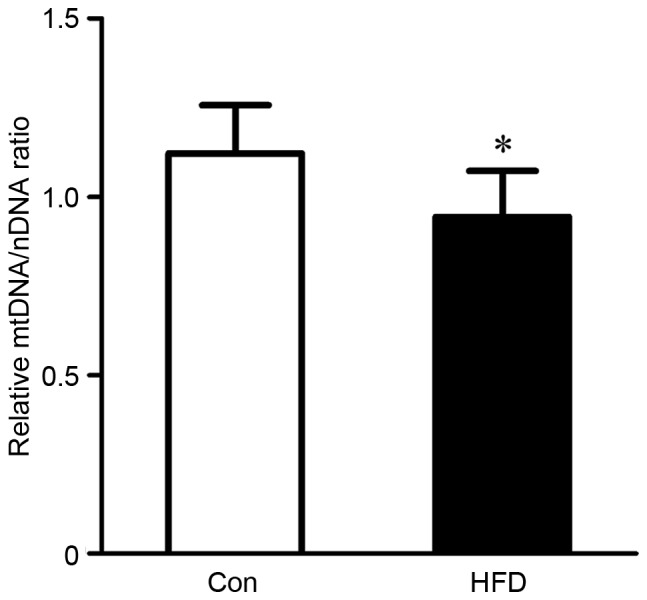
Evaluation of mtDNA copy numbers were evaluated in livers from rats in the Con and HFD groups via analysis of the ratio of ND1 DNA to actin DNA. The ND1 gene belongs to the mitochondrial genome, and actin gene belongs to the nuclear genome. Values are presented as the mean ± standard deviation. *P<0.05, vs. Con group. HFD, high fat and high cholesterol diet; Con, control; mtDNA, mitochondrial DNA.
Evaluation of MRC and MMP
As MRCs are pivotal in the generation of energy by oxidative phosphorylation, the present study measured whether the activity of the MRC was affected in the liver tissues of the HFD group. As shown in Fig. 3A, the activity of complex I was significantly reduced in the HFD group, compared with that in the control group (complex I/CS, 1.93±0.56 vs. 2.72±0.67; P<0.05). No difference was observed in complex II (Fig. 3B), however, the activity of complex III was also significantly lower in the HFD group, compared with that in the control group (complex III/CS, 1.39±0.29 vs. 1.81±0.24; P<0.05), as shown in Fig. 3C. No significant differences were observed in complex IV or ATP synthase (Fig. 3D and E). Citrate synthase is usually regarded as a quantitative marker enzyme for the content of intact mitochondria, and no significant change was detected between the HFD group and the control group (Fig. 3F). HFD chow also led to loss of MMP, which was associated with mitochondrial dysfunction (Fig. 4).
Figure 3.

Analysis of mitochondrial respiratory chain enzymes in liver mitochondria isolated from control and HFD groups. Complexes (A) I, (B) II, (C) III, and (D) IV. Complex activities were normalized to CS. (E) ATP synthase activity (F) CS activity. The values are expressed as the mean ± standard deviation. *P<0.05, vs. Con group. HFD, high fat and high cholesterol diet; Con, control; CS, citrate synthase.
Figure 4.
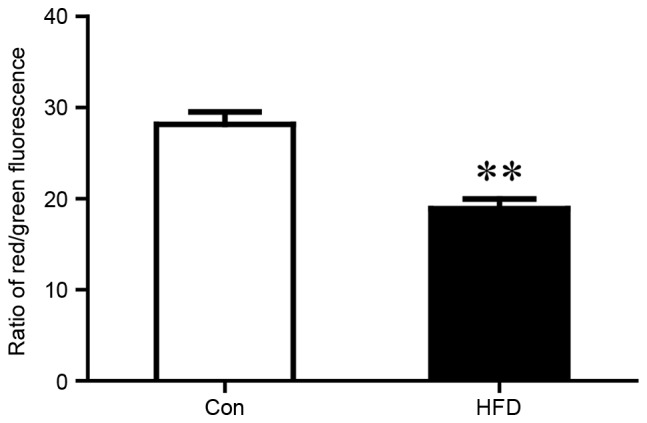
MMP was reflected by the ratio of red and green fluorescence. Liver mitochondria from the Con and HFD groups were isolated for MMP analysis. The values are presented as the mean ± standard deviation. **P<0.01 vs. Con group. MMP, mitochondrial membrane potential; HFD, high fat and high cholesterol diet; Con, control.
Mitochondrial proteomic profiling analysis
In order to fully understand the pivotal role of mitochondria dysfunction in NASH, the present study measured the mitochondrial protein profiles of the liver from the HFD group model using iTRAQ labeling technology. A total of 61 significantly differentially expressed proteins were identified and were synchronous with the comparisons of liver mitochondrial samples isolated from the control and HFD groups. Compared with the control group, 30 upregulated proteins with a fold change >1.0 and 31 downregulated proteins with a fold change <1.0 were observed in the HFD group, subsequently, 18 upregulated (Table III) with a fold change >1.2 and 13 downregulated proteins (Table IV) with a fold change ≤0.8 were selected. The biological processes were found to be associated with dysregulated proteins using the GO database. Of note, the majority of these differentially expressed proteins were closely involved, including mitochondrial oxidative phosphorylation, lipid metabolic process, acyl-CoA metabolic process and fatty acid β-oxidation (Table V). In the HFD group, proteins involved in mitochondrial oxidative phosphorylation were almost unanimously downregulated, compared with the control group. Among the dysregulated proteins, seven decreased proteins, including Ndufc2, Ndufs6, Ndufb3, Ndufa2, Ndufa5, Ndufb5 and NADH-ubiquinone oxidoreductase chain 5 (ND5), were component parts of mitochondrial complex II and the decreased proteins, ubiquinol-cytochrome c reductase, complex III subunit X (UQCR10) and cytochrome B (CytB) were the subunits of mitochondrial complex III. In the inner mitochondrial membrane, these proteins form the middle segment of the respiratory chain. The decrease in these proteins coincided with the significant decline in the activities of complexes I and III (Fig. 3A and B). These proteomics may provide novel insights into pathogenesis of NASH, although further functional investigations are required to specify proteins.
Table III.
Identification of 18 mitochondrial proteins upregulated in the high fat and high cholesterol diet group, compared with the control group, using isobaric tags for relative and absolute quantitation labeling technology.
| Accession no. | Protein name | Gene name | Fold change |
|---|---|---|---|
| tr|Q5U2U5|Q5U2U5_RAT | Perilipin 2 | Plin2 | 3.61 |
| sp|O70490|ACSM2_RAT | Acyl-coenzyme A synthetase | ACSM2 | 2.02 |
| sp|P61354|RL27_RAT | 60S ribosomal protein L27 | Rpl27 | 1.94 |
| sp|Q5EB77|RAB18_RAT | Ras-related protein Rab-18 | Rab18 | 1.90 |
| tr|D3ZSY4|D3ZSY4_RAT | Eosinophil peroxidase | Epx | 1.68 |
| tr|F1LRE2|F1LRE2_RAT | Insulin-like growth factor-binding protein complex acid labile subunit | Igfals | 1.68 |
| sp|P55159|PON1_RAT | Serum paraoxonase/arylesterase 1 | Pon1 | 1.64 |
| sp|P11915|NLTP_RAT | Non-specific lipid-transfer protein | Scp2 | 1.47 |
| sp|P07687|HYEP_RAT | Epoxide hydrolase 1 | Ephx1 | 1.45 |
| sp|P07871|THIKB_RAT | 3-ketoacyl-CoA thiolase B | Acaa1b | 1.44 |
| tr|G3V743|G3V743_RAT | Glucosidase 1 | Mogs | 1.35 |
| sp|Q5BJY9|K1C18_RAT | Keratin, type I cytoskeletal 18 | Krt18 | 1.32 |
| tr|D4ABM5|D4ABM5_RAT | Mitochondrial ribosomal protein S34 | Mrps34 | 1.31 |
| sp|Q4FZX5|MSRB2_RAT | Methionine-R-sulfoxide reductase B2 | Msrb2 | 1.30 |
| sp|P63095|GNAS2_RAT | Guanine nucleotide-binding protein G(s) subunit α isoforms | Gnas | 1.27 |
| sp|P41034|TTPA_RAT | α-tocopherol transfer protein | Ttpa | 1.25 |
| sp|Q5U3Z3|ISOC2_RAT | Isochorismatase domain-containing protein 2 | Isoc2 | 1.24 |
| tr|F6Q5K7|F6Q5K7_RAT | Mitochondrial ribosomal protein S18B | Mrps18b | 1.22 |
Fold-changes of >1.2 and P<0.05 for all statistical data.
Table IV.
Identification of 13 mitochondrial proteins downregulated in the high fat and high cholesterol diet group, compared with the control group, using isobaric tags for relative and absolute quantitation labeling technology.
| Accession no. | Protein name | Gene name | Fold change |
|---|---|---|---|
| tr|F1LMQ2|F1LMQ2_RAT | Farnesyl pyrophosphate synthase | FPPS | 0.46 |
| sp|O35760|IDI1_RAT | Isopentenyl-diphosphate δ-isomerase 1 | Idi1 | 0.47 |
| tr|Q5PQZ9|Q5PQZ9_RAT | NADH dehydrogenase [ubiquinone] 1 subunit C2 | Ndufc2 | 0.64 |
| sp|P11951|CX6C2_RAT | Cytochrome c oxidase subunit 6C-2 | Cox6c2 | 0.64 |
| tr|B2RYX1|B2RYX1_RAT | Cytochrome b-c1 complex subunit 9 | Uqcr10 | 0.65 |
| tr|D3ZCZ9|D3ZCZ9_RAT | NADH dehydrogenase [ubiquinone] iron-sulfur protein 6 | Ndufs6 | 0.65 |
| sp|Q63362|NDUA5_RAT | NADH dehydrogenase [ubiquinone] 1α subcomplex subunit 5 | Ndufa5 | 0.66 |
| tr|D4A4P3|D4A4P3_RAT | NADH dehydrogenase [ubiquinone] 1β subcomplex subunit 3 | Ndufb3 | 0.69 |
| tr|B0M1Q8|B0M1Q8_RAT | Cytochrome b | CytB | 0.72 |
| tr|D3ZS58|D3ZS58_RAT | NADH dehydrogenase [ubiquinone] 1α subcomplex subunit 2 | Ndufa2 | 0.72 |
| tr|D4A565|D4A565_RAT | NADH dehydrogenase (ubiquinone) 1β subcomplex, 5 | Ndufb5 | 0.72 |
| tr|Q06QA1|Q06QA1_RAT | NADH-ubiquinone oxidoreductase chain 5 | ND5 | 0.76 |
| sp|Q6PCT8|DHSD_RAT | Succinate dehydrogenase [ubiquinone] cytochrome b small subunit | Sdhd | 0.80 |
Fold changes of ≤0.8 and P<0.05 for all statistical data.
Table V.
Dysregulated proteins involved in biological processes, according to the Gene Ontology database.
| Biological process | Change in expression |
|---|---|
| Mitochondrial oxidative phosphorylation | |
| NADH dehydrogenase [ubiquinone] 1 subunit C2 | Downregulated |
| NADH dehydrogenase [ubiquinone] iron-sulfur protein 6 | Downregulated |
| NADH dehydrogenase [ubiquinone] 1β subcomplex subunit 3 | Downregulated |
| NADH dehydrogenase [ubiquinone] 1α subcomplex subunit 2 | Downregulated |
| NADH-ubiquinone oxidoreductase chain 5 | Downregulated |
| NADH dehydrogenase [ubiquinone] 1α subcomplex subunit 5 | Downregulated |
| Succinate dehydrogenase [ubiquinone] cytochrome b small subunit | Downregulated |
| Cytochrome b | Downregulated |
| Cytochrome b-c1 complex subunit 9 | Downregulated |
| Lipid metabolic process | |
| Perilipin2 | Upregulated |
| Acyl-coenzyme A synthetase | Upregulated |
| Arylsulfatase B | Upregulated |
| Estradiol 17-β-dehydrogenase 11 | Upregulated |
| Non-specific lipid-transfer protein | Upregulated |
| 3-ketoacyl-CoA thiolase B | Upregulated |
| Farnesyl pyrophosphate synthase | Downregulated |
| Isopentenyl-diphosphate δ-isomerase 1 | Downregulated |
| Cytochrome P450 2D18 | Downregulated |
| Acyl-CoA-binding protein | Downregulated |
| Acyl-CoA metabolic process | |
| Acyl-coenzyme A synthetase | Upregulated |
| Non-specific lipid-transfer protein | Upregulated |
| Acyl-CoA-binding protein | Downregulated |
| α-aminoadipic semialdehyde synthase | Downregulated |
| Fatty acid β-oxidation | |
| Non-specific lipid-transfer protein | Upregulated |
| 3-ketoacyl-CoA thiolase B | Upregulated |
Western blot analysis
The western blot analysis confirmed the differentially expressed proteins identified using iTRAQ technology. For the analysis, four proteins of interested were selected (Ndufa5, CytB, ATP5A and TFAM). Voltage-dependent anion-selective channel 1 (VDAC1) was used as the loading control. The expression levels of Ndufa5 and CytB were significantly decreased in the HFD group, compared with the control group, whereas no significant differences were observed in the levels of TFAM and ATP5A between the HFD group and control group. These findings were in accordance with the results obtained using iTRAQ technology (Fig. 5).
Figure 5.
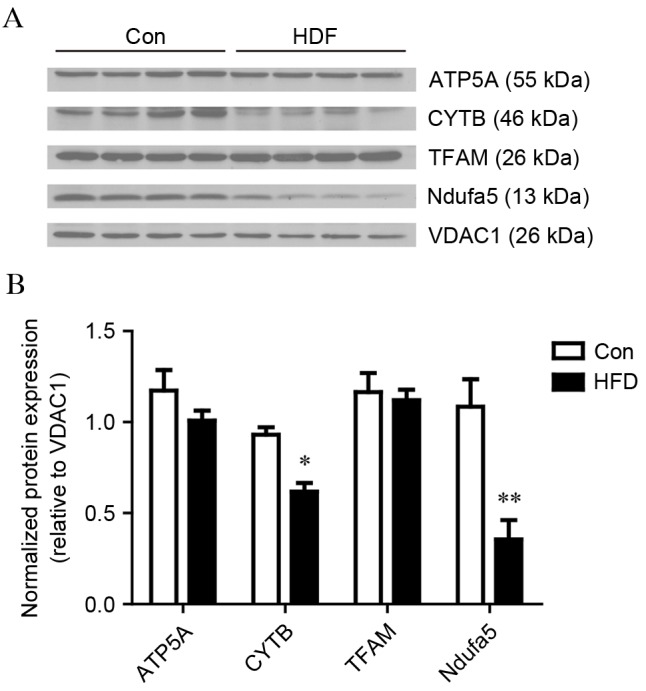
Western blot analysis validation of mitochondrial proteins. Liver mitochondrial expression levels of ATP5A, CytB, TFAM and Ndufa5 between the Con and HFD groups. VDAC1 was used as a loading control. (A) Images of the western blots from four biological replicates. (B) Bar graphs of the results of the western blot analysis. The values are presented as the mean ± standard deviation. *P<0.05 and **P<0.01, vs. Con group. HFD, high fat and high cholesterol diet; Con, control; ATP5A, ATP synthase α subunit; CYTB, cytochrome b; TFAM, transcription factor A, mitochondrial; Ndufa5, NADH dehydrogenase [ubiquinone] 1α subcomplex subunit 5; VDAC1, voltage-dependent anion-selective channel 1.
Discussion
NASH is considered to be important in the progression of NAFLD, which can progress into cirrhosis and subacute liver failure (1). However, the primary underlying mechanism contributing to the pathogenesis of NASH remains to be fully elucidated. The principal function of the mitochondria is to provide energy to maintain several cell functions. Previous studies have linked mitochondrial dysfunction and oxidative stress to the pathogenesis of NASH (7). Furthermore, mitochondrial structural abnormalities and decreased activity of MRCs have been reported in the livers of patients with NASH (13). In the present study, comprehensive analysis of liver mitochondrial proteomics was performed to provide a novel perspective for the pathophysiology of the condition.
In the proteomic analysis, a number of proteins involved in mitochondrial oxidative phosphorylation and the metabolism of lipids were significantly dysregulated, which may have contributed to the progression of NASH. For example, proteins in MRC complexes decreased significantly, including Ndufc2, Ndufs6, Ndufb3, Ndufa2, Ndufa5, Ndufb5, ND5 (complex I), Sdhd (complex II), Cox6c2, Uqcr10 and CytB (complex III). Ndufc2 is one of the accessory subunits of complex I. The downregulation of Ndufc2 can decrease MMP and interfere with complex I integrity (13). The expression of Ndufb3 and Ndufb5, which are members of mitochondrial complex I, can be decreased by HFD, resulting in the dysfunction of mitochondrial oxidative phosphorylation in skeletal muscle (14). Ndufa5 localizes to the inner mitochondrial membrane, and its expression is reduced in the brains of patients with autism and in ischemic heart failure (15,16), involving organs with high energy demand. The expression levels of ND5- and cytochrome b-encoded mtDNA were decreased, which can be correlated with mtDNA depletion. The changes of 10 proteins in the mitochondrial complex was consistent with decreased activities of complex II and III. However, the downregulation of Sdhd did not decrease the activity of complex II. Mitochondria complex I is critical in transferring electrons to ubiquinone. Complex I deficiency has been reported in several diseases, including heart failure, myopathies, encephalomyopathies and neurodegenerative disorders (17,18). Furthermore, complex I and III are two sites of ROS generation in cells (19). It is well established that the administration of a HFD in rats leads to the profound modification of mitochondrial lipid composition, causes the inhibition of fatty acid oxidation and the generation of mitochondrial ROS (20). In terms of MRC activity, ROS are overproduced following any significant reduction, which triggers oxidative stress (7). ROS has been implicated in the hepatic tissue injury associated with NASH (5). The depletion of mtDNA in the HFD group may be associated with ROS, which leads to the degradation of mtDNA nucleases. Complex I abnormality may result in increasing ROS production, and ROS conversely affects the activity of mitochondrial complex I through the oxidative damage of cardiolipin (21), which can exacerbate oxidative stress and potentially contribute to the pathogenesis of NASH. In the present study, the decrease in components of complex I and III were expected to decrease complex activity with increasing ROS, which may be relevant to the decrease of MMP and be involved in the progression of NASH.
The present study also found several dysregulated proteins associated with preventing the progression of NASH. Non-specific lipid transfer protein (Scp2) is located in the cytoplasm and mitochondria (22). It has high affinity to several lipid species, including fatty acid, fatty acyl CoAs, lysophosphatidic acid, and phosphatidylinositol, and is also involved in the mitochondrial oxidation of cholesterol in cells (23). Tumor necrosis factor and Fas, released by mitochondrial-free cholesterol, also induces NASH (24). Therefore, the upregulation of Scp2 may be a protective response to excess lipids in NASH. Medium-chain length fatty acids and xenobiotic carboxylic acids were catalyzed by the upregulation of acyl CoA synthetase (ACSM2) in the HFD group. In the liver, the decreased expression of ACSM2 can lead to an increase of free medium-chain fatty acids. Following treatment with medium-chain fatty acids, the protein level of the transcription factor, SREBP-1, is reduced and has a contrasting reaction to that of insulin in primary chicken hepatocytes (25). NASH is associated with insulin resistance, including lipolysis increase, free fatty acid delivery and hepatic fatty acid β oxidation, which lead to an increase in oxidative stress (6). Therefore, ACSM2 may indirectly enhance insulin function in hepatocytes, which may be a protective response to insulin resistance in the liver. However, the specific generating process requires further investigation.
Among the downregulated proteins, farnesyl pyrophosphate synthase and Isopentenyl-diphosphate δ-isomerase 1 are two enzymes of the mevalonate pathway, which catalyzes the synthesis of farnesyl pyrophosphate (FPP) in mitochondria (26). As with ubiquinones in the electron transport chain, mitochondrial isoprenoids are synthesized by FPP. However, the detailed association between FPP and the development of NASH remains to be fully elucidated.
In conclusion, the present study reports the protein profiling in NASH liver mitochondria using iTRAQ technology to perform detailed analysis of the pathogenesis of NASH for the first time. The results revealed 31 differentially expressed proteins in the NASH rat model, compared with the control rats. The dysregulation of proteins in NASH were predominantly associated with the MRC and lipid metabolic process. However, these proteins require further investigation for elucidating the molecular mechanism underlying the pathogenesis of NASH.
Acknowledgements
The present study was supported by a grant from the National Basic Research Program of China (grant no. 2013CB531702).
Glossary
Abbreviations
- NASH
nonalcoholic steatohepatitis
- HFD
high fat and high cholesterol diet
- iTRAQ
isobaric tags for relative and absolute quantitation
- 2D LC-MS/MS
two-dimensional liquid chromatography-tandem mass spectrometry
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- NAFLD
nonalcoholic fatty liver disease
- HCC
hepatocellar carcinoma
- ROS
reactive oxygen species
- ATP
adenosine triphosphate
- SCD
standard chow diet
- TC
total cholesterol
- TG
triglyceride
- LDL-C
low-density lipoprotein
- HDL-C
high-density lipoprotein
- H&E
hematoxylin and eosin
- RT-qPCR
reverse transcription-quantitative polymerase chain reaction
- NADH
ubiquinone oxidoreductase
- CS
citrate synthase
- MMP
mitochondrial membrane potential
- mtDNA
mitochondrial DNA
- MRC
mitochondrial respiratory complex
- ACSM2
acyl CoA synthetase
- FPP
farnesyl pyrophosphate
References
- 1.Melin Patrick AJ, Kalinski MI, Kelly KR, Haus JM, Solomon TP, Kirwan JP. Nonalcoholic fatty liver disease: Biochemical and therapeutic considerations. Ukr Biokhim Zh (1999) 2009;81:16–25. [PubMed] [Google Scholar]
- 2.Masuoka HC, Chalasani N. Nonalcoholic fatty liver disease: An emerging threat to obese and diabetic individuals. Ann N Y Acad Sci. 2013;1281:106–122. doi: 10.1111/nyas.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Day CP, James OF. Steatohepatitis: A tale of two ‘hits’? Gastroenterology. 1998;114:842–845. doi: 10.1016/S0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 4.Day CP. From fat to inflammation. Gastroenterology. 2006;130:207–210. doi: 10.1053/j.gastro.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 5.Pessayre D, Fromenty B. NASH: A mitochondrial disease. J Hepatol. 2005;42:928–940. doi: 10.1016/j.jhep.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 6.Sanyal AJ, Sargent Campbell C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, Clore JN. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183–1192. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 7.Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion. 2006;6:1–28. doi: 10.1016/j.mito.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 8.Anstee QM, Goldin RD. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int J Exp Pathol. 2006;87:1–16. doi: 10.1111/j.0959-9673.2006.00465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ye H, Sun L, Huang X, Zhang P, Zhao X. A proteomic approach for plasma biomarker discovery with 8-plex iTRAQ labeling and SCX-LC-MS/MS. Mol Cell Biochem. 2010;343:91–99. doi: 10.1007/s11010-010-0502-x. [DOI] [PubMed] [Google Scholar]
- 10.Pallotti F, Lenaz G. Isolation and subfractionation of mitochondria from animal cells and tissue culture lines. Methods Cell Biol. 2007;80:3–44. doi: 10.1016/S0091-679X(06)80001-4. [DOI] [PubMed] [Google Scholar]
- 11.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 12.Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, Pinton P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat Protoc. 2009;4:1582–1590. doi: 10.1038/nprot.2009.151. [DOI] [PubMed] [Google Scholar]
- 13.Carreras M Pérez, Del Hoyo P, Martin MA, Rubio JC, Martin A, Castellano G, Colina F, Arenas J, Herruzo Solis JA. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology. 2003;38:999–1007. doi: 10.1002/hep.1840380426. [DOI] [PubMed] [Google Scholar]
- 14.Sparks LM, Xie H, Koza RA, Mynatt R, Hulver MW, Bray GA, Smith SR. A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes. 2005;54:1926–1933. doi: 10.2337/diabetes.54.7.1926. [DOI] [PubMed] [Google Scholar]
- 15.Anitha A, Nakamura K, Thanseem I, Matsuzaki H, Miyachi T, Tsujii M, Iwata Y, Suzuki K, Sugiyama T, Mori N. Downregulation of the expression of mitochondrial electron transport complex genes in autism brains. Brain Pathol. 2013;23:294–302. doi: 10.1111/bpa.12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu T, Chen L, Kim E, Tran D, Phinney BS, Knowlton AA. Mitochondrial proteome remodeling in ischemic heart failure. Life Sci. 2014;101:27–36. doi: 10.1016/j.lfs.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang PT, Chen CL, Ren P, Guarini G, Chen YR. BCNU-induced gR2 defect mediates S-glutathionylation of Complex I and respiratory uncoupling in myocardium. Biochem Pharmacol. 2014;89:490–502. doi: 10.1016/j.bcp.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finsterer J, Rauschka H, Segal L, Kovacs GG, Rolinski B. Affection of the respiratory muscles in combined complex I and IV deficiency. Open Neurol J. 2017;11:1–6. doi: 10.2174/1874205X01711010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vial G, Dubouchaud H, Couturier K, Rousselle Cottet C, Taleux N, Athias A, Galinier A, Casteilla L, Leverve XM. Effects of a high-fat diet on energy metabolism and ROS production in rat liver. J Hepatol. 2011;54:348–356. doi: 10.1016/j.jhep.2010.06.044. [DOI] [PubMed] [Google Scholar]
- 21.Paradies G, Petrosillo G, Pistolese M, Ruggiero FM. Reactive oxygen species affect mitochondrial electron transport complex I activity through oxidative cardiolipin damage. Gene. 2002;286:135–141. doi: 10.1016/S0378-1119(01)00814-9. [DOI] [PubMed] [Google Scholar]
- 22.Gallegos AM, Atshaves BP, Storey SM, Starodub O, Petrescu AD, Huang H, McIntosh AL, Martin GG, Chao H, Kier AB, Schroeder F. Gene structure, intracellular localization, and functional roles of sterol carrier protein-2. Prog Lipid Res. 2001;40:498–563. doi: 10.1016/S0163-7827(01)00015-7. [DOI] [PubMed] [Google Scholar]
- 23.Schroeder F, Atshaves BP, McIntosh AL, Gallegos AM, Storey SM, Parr RD, Jefferson JR, Ball JM, Kier AB. Sterol carrier protein-2: New roles in regulating lipid rafts and signaling. Biochim Biophys Acta. 2007;1771:700–718. doi: 10.1016/j.bbalip.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mari M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, Enrich C, Checa Fernandez JC, Ruiz Garcia C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006;4:185–198. doi: 10.1016/j.cmet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Yin L, Hillgartner FB. SREBP-1 integrates the actions of thyroid hormone, insulin, cAMP, and medium-chain fatty acids on ACCalpha transcription in hepatocytes. J Lipid Res. 2003;44:356–368. doi: 10.1194/jlr.M200283-JLR200. [DOI] [PubMed] [Google Scholar]
- 26.Martin D, Piulachs MD, Cunillera N, Ferrer A, Bellés X. Mitochondrial targeting of farnesyl diphosphate synthase is a widespread phenomenon in eukaryotes. Biochim Biophys Acta. 2007;1773:419–426. doi: 10.1016/j.bbamcr.2006.11.015. [DOI] [PubMed] [Google Scholar]