

Abstract
Alzheimer's disease (AD) is the most common type of senile dementia, which often develops in elderly or presenile individuals. As one of the pathological features of AD, amyloid β-protein (Aβ) causes energy dysmetabolism, thereby inducing cellular damage and apoptosis. Salidroside is the main active component of the traditional Chinese medicine Rhodiola. Previous studies have demonstrated that salidroside exerts a regulatory role in energy metabolism. However, the role and the mechanism of action of salidroside in AD remain unclear. Therefore, the present study used Aβ1–40 to induce damage in PC12 cells, thereby establishing a cell model of AD. In addition, salidroside treatment was performed to investigate the protective effect of salidroside and the underlying mechanisms. Aβ1-40-induced neuronal toxicity reduced cell viability and caused cellular damage. As a result, the expression level of nicotinamide phosphoribosyltransferase (NAMPT) decreased, the synthesis of nicotinamide adenine dinucleotide (NAD+; an energy metabolism-associated coenzyme) became insufficient, and the NAD+/nicotinamide adenine dinucleotide hydride ratio was reduced. Administration of salidroside alleviated Aβ-induced cell damage and increased the expression level of the key protein NAMPT and the synthesis of NAD+. The results of the present study demonstrate that salidroside exerts a protective effect on Aβ1-40-damaged PC12 cells. The underlying mechanism may be associated with the regulation of energy metabolism that relies predominantly on the NAMPT signaling pathway.
Keywords: Alzheimer's disease, salidroside, amyloid β-protein1–40, nicotinamide phosphoribosyltransferase, nicotinamide adenine dinucleotide
Introduction
Alzheimer's disease (AD) is the most common form of dementia that affected an estimated 44 million individuals in 2015. This figure is expected to increase to 135 million by the year 2050 throughout the world (1). The main clinical symptoms of AD include memory impairment, decreased self-care ability, aphasia and visuospatial dysgnosia (2). Neuropathological studies have confirmed that extracellular senile plaque deposition, intracellular accumulation of neurofibrillary tangles and neuronal degeneration loss are the main pathological hallmarks of AD (3,4). Deposition of amyloid β-protein (Aβ) in the brain leads to the formation of senile plaques, which represent a primary pathological characteristic of AD (5). Aβ refers to a class of amyloid precursor protein (APP)-derived polypeptides that are formed under the action of β- and γ-secretase. The common subtypes of Aβ are Aβ1–40 and Aβ1–42, and each exert neurotoxic effects and induce apoptosis. Therefore, reducing Aβ-induced neuron injury and intervening in AD progression are critical for the treatment of AD.
Studies have shown that energy dysmetabolism contributes significantly to the pathogenesis of a variety of ageing-associated diseases and degenerative diseases of the nervous system, including AD (6). Homeostatic imbalance of energy metabolism develops gradually in the brain with age, which promotes Aβ production. Aβ further induces energy dysmetabolism in the brain, resulting in impaired neuronal synaptic function (7,8). Nicotinamide adenine dinucleotide (NAD+) is an important coenzyme in the in vivo redox reaction. It participates in energy synthesis through glycolysis, the tricarboxylic acid cycle and mitochondrial oxidative phosphorylation (9). NAD+ may be synthesized de novo, although the majority of NAD is synthesized via the salvage pathway from the conversion of nicotinamide into nicotinamide mononucleotide, which is later converted to NAD+ (10). The NAD+/nicotinamide adenine dinucleotide hydride (NADH) ratio is a fundamental indicator of the cellular redox status (11). Nicotinamide phosphoribosyltransferase (NAMPT) is the rate-limiting enzyme in the NAD+ salvage pathway, and it has been demonstrated that NAMPT participates in energy metabolism and maintains the homeostasis of energy metabolism in the brain by regulating NAD+ synthesis; thus, increased expression levels of NAMPT have been shown to positively regulate NAD+ levels (12). Knockout of the NAMPT gene enhances the formation of Aβ plaques in the brain (13). Therefore, recent studies have focused on regulation of NAMPT expression for the purpose of protecting against AD and other neurodegenerative diseases (13–15).
Rhodiola rosea is a perennial herb that belongs to the family Crassulaceae and genus Rhodiola. Rhodiola is a traditional herbal medicine commonly used across Asia and grows at altitudes of 1,800–2,500 m in cold, pollution-free zones. Rhodiola stimulates the nervous system, improves work efficiency, alleviates fatigue and prevents mountain sickness. Salidroside is the primary active ingredient of Rhodiola. Various studies have shown that salidroside inhibits the inflammatory response (16), reduces oxidative stress-induced damage (17) and exerts an anticancer effect (18). Certain studies demonstrated that salidroside has a neuroprotective effect, for example, salidroside inhibited Aβ-induced neurotoxicity by regulating the phosphatidylinositol 3-kinase/Akt signaling pathway in primary cultured neurons (19). In a rat model of AD, salidroside improved memory impairment of rats (20), and salidroside reduced mitochondrial swelling and attenuated the injury in the central nervous system (21). However, whether the neuroprotective effects of salidroside involve regulation of the NAMPT/NAD+ signaling pathway in PC12 cells remains unknown.
To gain further insights into the neuroprotective functions of salidroside, a PC12 model was used in the present study and the underlying mechanisms were investigated. Whether the neuroprotective effects of salidroside increasing NAD levels against Aβ1–40-induced neurotoxicity in PC12 cells were elucidated in the current study. Furthermore, the possible underlying mechanisms were examined by investigating NAMPT expression levels.
Materials and methods
Preparation of Aβ1–40
The lyophilized powder of Aβ1–40 (cat no. A1075; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was diluted with sterile distilled water to a concentration of 200 µmol/l. Subsequently, Aβ1–40 was incubated at 37°C for 7 days and stored at 4°C for further assays.
Cell culture and treatment
Highly differentiated PC12 rat adrenal pheochromocytoma cells (Type Culture Collection of the Chinese Academy of Sciences, Shanghai, China) were maintained in Dulbecco's modified Eagle's medium (DMEM; cat no. 12100046) containing Gibco 10% fetal bovine serum (cat no. 10099-141) (both from Thermo Fisher Scientific, Inc., Waltham, MA, USA) and 1% penicillin-streptomycin. The cells were maintained in a 37°C incubator in an atmosphere of 5% CO2 and grew adherently. The culture medium was replaced daily. Upon reaching 80–90% confluence, the cells were trypsinized for 1–2 min. Once the cells became round, 2 ml of culture medium was added to the cells. The cells were then centrifuged, 1,500 × g for 5 min at room temperature, resuspended, and split into culture dishes. The cells were divided into the following main groups: Normal control group, the Aβ1–40 group, the Aβ1–40 + salidroside (cat no. B20504; Shanghai Yuanye Biological Technology Co., Ltd., Shanghai, China) group, and the salidroside group. The normal control group was cultured conventionally in DMEM and did not receive any treatment. The Aβ1–40 group was subjected to 24 h of treatment with 5 µmol/l Aβ1-40. The Aβ1–40 + salidroside group was subjected to simultaneous Aβ1–40 (5 µmol/l) and salidroside (100 µmol/l) treatments for 24 h. The salidroside group was treated with 100 µmol/l salidroside for 24 h.
Cell viability assay
PC12 cells were seeded into 96-well plates and cultured at 37°C in DMEM for 24 h. Once the cells attached stably to the culture surface, various concentrations of Aβ1–40 (1, 5 or 10 µmol/l) or salidroside (12.5, 25, 50, 100 or 200 µmol/l) were added to the cells. Following a 24-h treatment, each well of cells was overlaid with 10 µl MTT solution (5 mg/ml; cat no. C0009) obtained from Beyotime Institute of Biotechnology (Haimen, China) and incubated at 37°C for 4 h. The supernatant was discarded, and 150 µl dimethyl sulphoxide was added to each well of cells to fully dissolve the crystals. Subsequently, absorbance was measured at a wavelength of 490 nm using a PowerWave XS microplate reader (BioTek Instruments, Inc., Winooski, VT, USA).
Lactate dehydrogenase (LDH) activity assay
The cells were subjected to different treatments (medium only, 5 µmol/l Aβ1-40, 5 µmol/l Aβ1–40 + 100 µmol/l salidroside or 100 µmol/l salidroside). Subsequently, the activity of released LDH was examined according to the instructions provided with the LDH Activity Assay kit (cat. no. G1780; Promega Corporation, Madison, WI, USA). Absorbance was measured at a wavelength of 490 nm. The LDH release was expressed as a percentage relative to the positive control group.
NAD+/NADH analysis
An NAD+/NADH quantification kit was used to determine NAD+ and NADH levels (cat no. k337-100; BioVision, Inc., Milpitas, CA, USA) according to the manufacturer's instructions. The cells were washed with pre-cooled phosphate-buffered saline (PBS), homogenized in 400 µl NAD+/NADH extraction buffer, and then centrifuged for 5 min at 18,000 × g and 4°C. The resulting supernatants were transferred to fresh Eppendorf (EP) tubes and labelled as NADt samples. Subsequently, 200 µl NADt sample was transferred to fresh EP tubes and heated at 60°C for 30 min (all NAD+ decomposed, leaving only NADH to be analyzed). After cooling on ice, the samples were quickly centrifuged 12,000 × g for 30 sec at 4°C, and the resulting pellets were discarded. The supernatants were collected and labelled as NADH samples for further assays. Standards (0, 2, 4, 6, 8 and 10 µl) and 40 µl samples were loaded into the wells of a microtiter plate, and appropriate volumes of NAD+/NADH Extraction Buffer were added to bring the volume to 50 µl. Subsequently, 100 µl enzyme reaction mix and 10 µl NADH developer were added to each well. The mixtures were allowed to react at room temperature (RT) for 1–4 h, and the absorbance was measured using a microplate reader (wavelength, 450 nm).
Immunofluorescence assay
Following various treatments and interventions, all groups of cells were washed with PBS for 5 min. The cells were then fixed with 4% paraformaldehyde for 15 min, blocked with a blocking solution containing 10% calf serum for 45 min at RT, incubated with anti-NAMPT antibody (dilution, 1:200; cat no. ab45890; Abcam, Cambridge, MA, USA) at 37°C for 1 h, and placed in a 4°C refrigerator overnight. After washing with PBS, the cells were incubated with fluorescein isothiocyanate-labelled goat anti-rabbit IgG (dilution, 1:500; cat no. A0562; Beyotime Institute of Biotechnology) for 1 h in a 37°C incubator. After 3 rinses with PBS, the cells were covered with DAPI staining solution (cat no. C1005; Beyotime Institute of Biotechnology) and incubated at RT for 5 min. The cells were rinsed 3 more times with PBS (5 min each time). Subsequently, the cells were mounted onto glass slides in PBS and glycerol (1:1). The fluorescence signal was examined using an Olympus BX51 biological microscope at a magnification of ×200.
Western blot analysis
At the end of the treatment period, all groups of cells were collected. Following centrifugation 1,500 × g for 5 min at RT, the cells were lysed in lysis buffer. The resulting supernatants were extracted and the protein concentrations in the supernatants were determined using the bicinchoninic acid (BCA) method. Subsequently, the supernatants were mixed thoroughly with 5X sodium dodecyl sulphate (SDS) loading buffer, boiled at 100°C for 5 min, cooled on ice and then stored at −20°C. Immediately before the experiments, the protein samples were thawed at RT. The protein samples (50 µg each) were heated at 95°C for 5 min, then separated by SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. The membranes were blocked with 2% bovine serum albumin for 1 h and then incubated with the primary antibody (anti-NAMPT antibody, dilution, 1:250; cat no. ab45890; Abcam) at 4°C overnight. Subsequently, the membranes were washed with PBS for 45 min, incubated with horseradish peroxidase-labelled secondary antibody (dilution, 1:2,000; cat no. SA00001-2; ProteinTech Group, Inc., Chicago IL, USA) at 37°C for 1 h, and washed again in PBS for 45 min. Protein bands were examined using an ImageQuant LAS 4000 mini chemiluminescence detector (GE Healthcare Life Sciences, Little Chalfont, UK). GAPDH (dilution, 1:1,000, cat no. 5174S; Cell Signaling Technology, Inc., Danvers, MA, USA) served as a loading control.
Statistical analysis
The results were statistically analyzed using GraphPad Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA, USA). All data are expressed as the mean ± standard error. One-way analysis of variance was used to assess multiple groups of data and Student's t-test was used to compare between two groups and P<0.05 was considered to indicate a statistically significant difference.
Results
Impact of Aβ1–40 treatment on the viability of PC12 cells and the protective effect of salidroside
PC12 cells were treated with various concentrations of Aβ1–40 (1, 5 or 10 µmol/l) for 24 h, which enabled the analysis of changes in PC12 cell viability under the action of different concentrations of Aβ1-40. The results demonstrated that cell viability declined as the Aβ1–40 concentration increased. The viability of PC12 cells decreased by ~15% after treatment with 5 µmol/l Aβ1-40. Therefore, a concentration of 5 µmol/l Aβ1–40 was selected to establish the cell model of AD (Fig. 1A).
Figure 1.
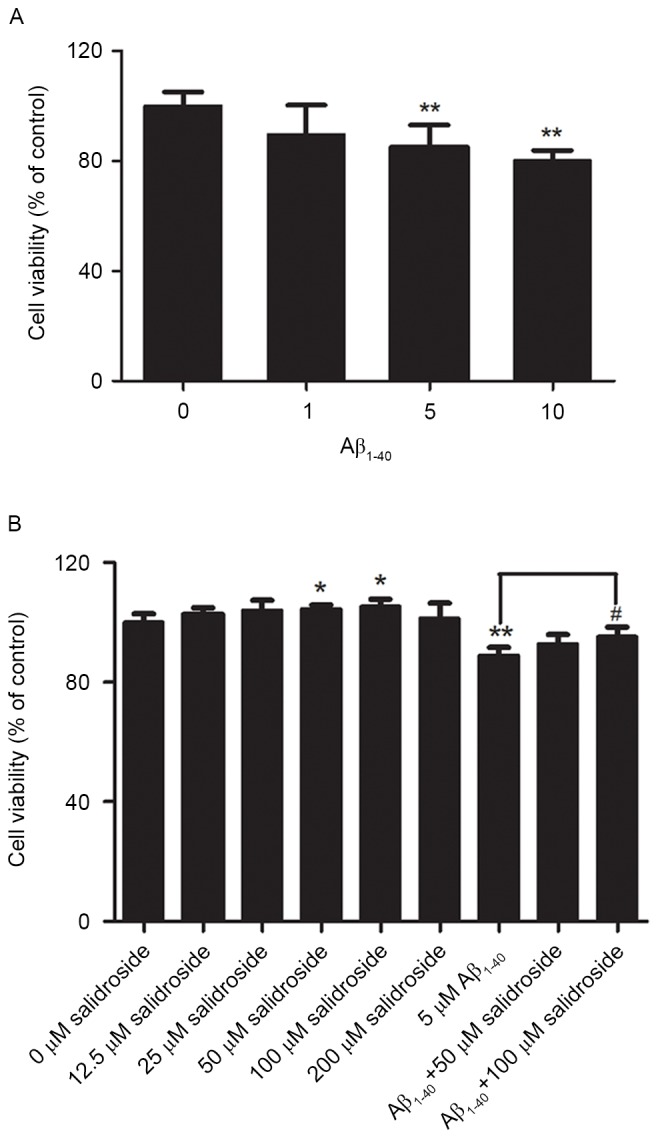
Impact of Aβ1–40 treatment on the viability of PC12 cells and the effect of salidroside. (A) Effect of the cytotoxicity of Aβ1–40 on PC12 cell viability. PC12 cells were treated with various concentrations of Aβ1–40 (1, 5 and 10 µmol/l) for 24 h. (B) Effects of salidroside treatment on the viability of PC12 cells. PC12 cells were treated with various concentrations of salidroside (12.5, 25, 50, 100 and 200 µmol/l) for 24 h, and with different concentrations of salidroside (0, 50, and 100 µmol/l) and exposed to Aβ simultaneously. All data are expressed as the mean ± standard error from four independent experiments. *P<0.05 and **P<0.01 vs. control group; #P<0.05 vs. Aβ1–40 group. Aβ, amyloid β-protein.
Before investigation of the protective effect of salidroside, the present study first examined whether salidroside would exert a neurotoxic effect on normal PC12 cells at certain concentrations. PC12 cells were treated with various concentrations of salidroside (12.5, 25, 50, 100 or 200 µmol/l). Compared with the normal control group, 24-h incubation with 12.5, 25 or 200 µmol/l salidroside exerted no significant effect on normal PC12 cells, whereas incubation with 50 and 100 µmol/l salidroside improved cell viability (P<0.05). Therefore, the two concentrations of salidroside (50 and 100 µmol/l) were selected for further investigation of the effect of salidroside on the viability of PC12 cells that were damaged by Aβ1–40 exposure. The results indicated that cell viability was significantly decreased in the Aβ1–40 group compared with the normal control group (P<0.01). Compared with the Aβ1–40 group, treatment with 50 µmol/l salidroside failed to significantly improve the viability of Aβ1–40-damaged cells, whereas treatment with 100 µmol/l salidroside improved the viability of cells damaged by Aβ1–40 (P<0.05). The results demonstrated that 100 µmol/l salidroside exerted an effect on cell viability and alleviated the Aβ-induced PC12 cell injury. Therefore, this salidroside concentration (100 µmol/l) was selected for further experiments (Fig. 1B).
Salidroside reduces the LDH level of Aβ1–40-treated cells
The LDH level of a culture supernatant indirectly reflects the effects of Aβ1–40 and salidroside on cells. As shown in Fig. 2, the Aβ1–40 group displayed a significantly increased LDH level compared with that of the normal control group (P<0.01). By contrast, the LDH level was markedly reduced in the Aβ1–40 + salidroside group compared with the Aβ1–40 group (P<0.05). The results demonstrated that salidroside reduced the Aβ1-40-induced cellular damage.
Figure 2.
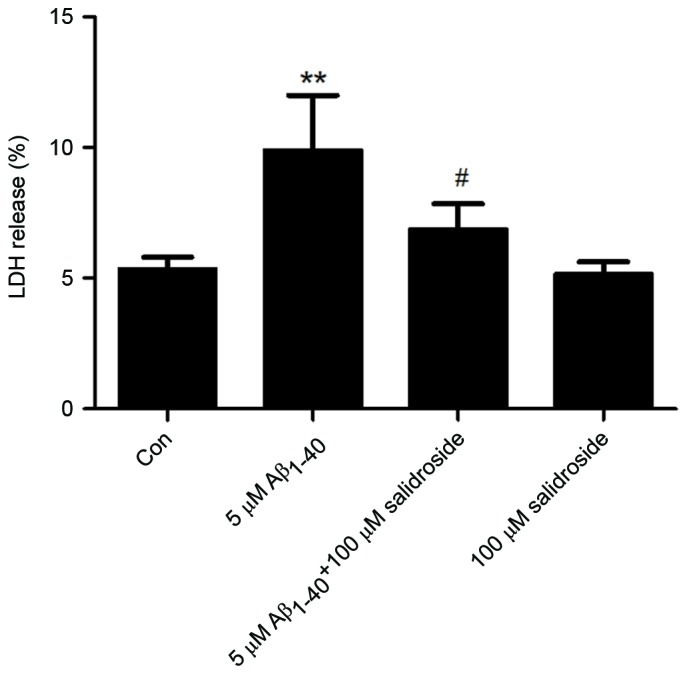
Effect of salidroside treatment on the LDH level of Aβ1–40-treated cells. PC12 cells were subjected to different treatments (medium only, 5 µmol/l Aβ1–40, 5 µmol/l Aβ1–40 + 100 µmol/l salidroside, and 100 µmol/l salidroside) for 24 h. Subsequently, the cells were examined using an LDH Activity Assay kit. All data are presented as the mean ± standard error from four independent experiments (n=4). **P<0.01 vs. control group. #P<0.05 vs. Aβ1–40 group. LDH, lactate dehydrogenase; Aβ, amyloid β-protein; Con, control.
Salidroside increases the NAD+ level and NAD+/NADH ratio of Aβ1–40-treated cells
NAD+ and NADH are coenzymes that are necessary for cellular energy metabolism. Under normal physiological conditions, the NAD+/NADH ratio reflects the cellular redox status and is in a constant state of dynamic equilibrium. Disorders of cellular energy metabolism are characterized by changes in NAD+ and NADH levels, and an imbalance in the NAD+/NADH ratio (22). In the present study, the Aβ1–40 group exhibited a decreased level of NAD+ compared with that of the normal control group (P<0.01; Fig. 3A). However, the NADH level did not change significantly in the Aβ1–40 group compared with the normal control group (Fig. 3B). As a result, the NAD+/NADH ratio was reduced in the Aβ1–40 group (P<0.01; Fig. 3C). No significant difference was identified in the NADH level between the Aβ1–40 + salidroside group and the Aβ1–40 group (Fig. 3B). By contrast, the Aβ1–40 + salidroside group demonstrated a significant increase in the NAD+ level (P<0.05; Fig. 3A) and the NAD+/NADH ratio (P<0.05; Fig. 3C).
Figure 3.

Effect of salidroside treatment on the NAD+ level and NAD+/NADH ratio of Aβ1–40-treated PC12 cells. (A) Effect of salidroside treatment on the NAD+ level in the PC12 cells damaged by Aβ1–40 exposure. (B) Effect of salidroside treatment on the NADH level in the PC12 cells. (C) Effect of salidroside treatment on the NAD+/NADH ratio in Aβ1–40-induced damaged PC12 cells. All data are expressed as the mean ± standard error from four independent experiments (n=4). **P<0.01 vs. control group. #P<0.05 and ##P<0.01 vs. Aβ1–40 group. NAD+, nicotinamide adenine dinucleotide; NADH, nicotinamide adenine dinucleotide hydride; Aβ, amyloid β-protein; Con, control.
Salidroside upregulates the expression level of NAMPT
NAMPT is the rate-limiting enzyme in the salvage pathway of NAD+ biosynthesis and is important in regulating the homeostasis of energy metabolism. The results of an immunofluorescence assay demonstrated that the NAMPT-derived fluorescence signal was reduced following 24 h of Aβ1–40 treatment, whereas salidroside treatment increased the NAMPT fluorescence signal (Fig. 4A and B). In addition, western blot analysis demonstrated that NAMPT expression was decreased in the Aβ1–40 group compared with the normal control group. Salidroside treatment significantly increased the expression level of NAMPT (Fig. 4C and D). Thus, the results indicate that salidroside regulates NAMPT protein expression.
Figure 4.

Salidroside upregulates the expression level of NAMPT. (A) NAMPT were detected by immunofluorescence staining and a representative photomicrograph is presented (scale bar, 10 µm). (B) The fluorescence intensity of the NAMPT protein. All data are expressed as the mean ± standard error from three independent experiments. **P<0.01 vs. control group. #P<0.05 vs. Aβ1–40 group. (C) Western blot analyses of PC12 lysates from the control (without any treatment), Aβ1–40 (with 5 µmol/l Aβ1–40 treatment), Aβ1–40 + salidroside (5 µmol/l Aβ1–40 and 100 µmol/l salidroside simultaneously) and salidroside (100 µmol/l salidroside only) groups. (D) Quantitation of NAMPT protein expression levels. Data are presented as the mean ± standard error of three independent experiments. **P<0.01 vs. control group. ##P<0.01 vs. Aβ1–40 group. NAMPT, nicotinamide phosphoribosyltransferase; Aβ, amyloid β-protein; Con, control.
Discussion
AD is the most common cause of dementia in the elderly, but the exact pathogenetic mechanisms underlying AD remain uncertain. In previous studies, one extensively investigated mechanism is Aβ-mediated neurotoxicity; the aggregates of Aβ peptides exhibit cytotoxicity in vivo and in vitro (23). Aβ1–40 and Aβ1–42 are two major forms of Aβ, although Aβ1–42 is markedly more prone to aggregation and more toxic to neurons than Aβ1–40; however, Aβ1–40 was the predominant isoform of the most abundant cleaved form of APP (~90%) and exerts a toxic effect on neurons in the AD brain (24). In addition, the deposition of Aβ1–40 is required for the development of mature amyloid plaques from the initial deposition of Aβ1–42, which was regarded as an early pathological process of AD, and Aβ1–40 is often used to establish the model of AD in vivo and in vitro (25). Injecting 5 mg/ml Aβ1–40 caused hippocampal neuronal loss or functional impairment and is thus considered as an indication of energy metabolism (26). As Aβ1–40 possesses neuronal toxicity and is capable of inducing neuronal neurotoxicity, PC12 cells treated with Aβ1–40 were used as the cell model of AD in the current study. The neuronal toxicity of Aβ1–40 is reflected in its ability to cause energy dysmetabolism, and induce cell damage and death (27,28). A number of previous studies used PC12 cells to investigate the molecular mechanisms underlying the development of degenerative diseases of the nervous system (29–32). Therefore, the present study employed the PC12 cells that were damaged by Aβ1–40 exposure as the cell model of AD and the significance of NAMPT in AD was evaluated.
The present study demonstrated that Aβ1–40 treatment reduced the NAD+ expression level. The underlying reason may be associated with decreased NAMPT expression. As the rate-limiting enzyme in the NAD+ salvage pathway, NAMPT promotes the production of NAD+ (33). Previous studies demonstrated that an elevated NAMPT level reduces cell death (34,35), which is consistent with the results of the present study, which indicate that salidroside increased NAMPT expression and improved cell survival. In addition, the present study identified that salidroside significantly reduced the level of LDH released when compared with that in the Aβ1–40 group. The results of the LDH assay were consistent with those of the MTT assay, demonstrating that salidroside treatment attenuated the Aβ1–40-induced PC12 cell injury and exerted a protective effect on PC12 cells.
The expression level of Aβ increases as AD progresses, which causes mitochondrial dysfunction and energy metabolism disorders. The elevated Aβ expression level represents one of the reasons behind neuronal damage and apoptosis (36). NAD+ participates in energy metabolism. Furthermore, loss of NAD+ homeostasis leads to decreased sirtuin-1 (SIRT1) activity. Consequently, SIRT1-mediated deacetylation of signaling molecules (including transcription factors and enzymes) is reduced (37,38). Peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α) is a coactivator of the nuclear hormone receptor peroxisome proliferator-activated receptor γ, which also undergoes SIRT1-mediated deacetylation and participates in mitochondrial biosynthesis and energy homeostasis (39). The decreased SIRT1 activity in the brain affects the function of the downstream protein PGC-1α in energy synthesis, resulting in a brain energy crisis (40). As NAMPT is important in NAD+ synthesis, the present study investigated whether salidroside exerts its regulatory effect on energy metabolism pathways via NAMPT. Namely, salidroside attenuates the neuronal toxicity of Aβ by further regulating NAD+/NADH levels. Studies have demonstrated that the expression level of NAMPT decreases gradually with age, with the decrease in NAMPT level particularly evident in the brain of AD mice compared with aged healthy mice. Such a decrease exacerbates neuronal apoptosis (34,41,42). By contrast, increased NAMPT expression levels in the hippocampus and cortex improve cognitive function (35). Therefore, novel treatment strategies for AD may involve the administration of salidroside to regulate NAMPT expression and reduce the severity of AD.
The present study demonstrated that salidroside exerted a protective effect on PC12 cells damaged by Aβ1–40 exposure. An AD model was established by Aβ1–40 treatment. The viability of PC12 cells decreased and the level of LDH release significantly increased. In addition, the protein expression of NAMPT, the level of NAD+ and the ratio of NAD+/NADH were decreased. The above-mentioned effects of Aβ1–40 were attenuated following salidroside treatment. In addition, the present study demonstrated that the decrease in NAMPT/NAD+ levels may represent one of the molecular pathological mechanisms of AD.
In conclusion, the present study demonstrated that salidroside exerted a protective effect on PC12 damaged by Aβ1–40 treatment. The protective effect of salidroside may reside in its regulatory effect in energy metabolism, such as the increases in NAMPT expression levels and NAD+ production. Therefore, NAMPT and NAD+ are considered to be key factors in AD, as well as other neurodegenerative diseases. Thus, regulation of NAMPT and NAD+ may serve as novel therapeutic strategies for the treatment of AD.
Acknowledgements
The present study was supported by grants from the National Natural Science Foundation of China (grant no. 81503626) and the Shanghai Health Bureau Youth Fund (grant nos. 201540254 and 20134331).
References
- 1.Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet Med. 2016;18:421–430. doi: 10.1038/gim.2015.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S, Van der Flier WM. Alzheimer's disease. Lancet. 2016;388:505–517. doi: 10.1016/S0140-6736(15)01124-1. [DOI] [PubMed] [Google Scholar]
- 3.Perl DP. Neuropathology of Alzheimer's disease. Mt Sinai J Med. 2010;77:32–42. doi: 10.1002/msj.20157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1:a006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sadigh-Eteghad S, Sabermarouf B, Majdi A, Talebi M, Farhoudi M, Mahmoudi J. Amyloid-beta: A crucial factor in Alzheimer's disease. Med Princ Pract. 2015;24:1–10. doi: 10.1159/000369101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yin F, Boveris A, Cadenas E. Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid Redox Signal. 2014;20:353–371. doi: 10.1089/ars.2012.4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang R, Li JJ, Diao S, Kwak YD, Liu L, Zhi L, Büeler H, Bhat NR, Williams RW, Park EA, Liao FF. Metabolic stress modulates Alzheimer's β-secretase gene transcription via SIRT1-PPARγ-PGC-1 in neurons. Cell Metab. 2013;17:685–694. doi: 10.1016/j.cmet.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu MF, Yin JH, Hwang CS, Tang CM, Yang DI. NAD attenuates oxidative DNA damages induced by amyloid beta-peptide in primary rat cortical neurons. Free Radic Res. 2014;48:794–805. doi: 10.3109/10715762.2014.907889. [DOI] [PubMed] [Google Scholar]
- 9.Ussher JR, Jaswal JS, Lopaschuk GD. Pyridine nucleotide regulation of cardiac intermediary metabolism. Circ Res. 2012;111:628–641. doi: 10.1161/CIRCRESAHA.111.246371. [DOI] [PubMed] [Google Scholar]
- 10.Rongvaux A, Shea RJ, Mulks MH, Gigot D, Urbain J, Leo O, Andris F. Pre-B-cell colony-enhancing factor, whose expression is up-regulated in activated lymphocytes, is a nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved in NAD biosynthesis. Eur J Immunol. 2002;32:3225–3234. doi: 10.1002/1521-4141(200211)32:11<3225::AID-IMMU3225>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 11.Ying W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid Redox Signal. 2008;10:179–206. doi: 10.1089/ars.2007.1672. [DOI] [PubMed] [Google Scholar]
- 12.Revollo JR, Grimm AA, Imai S. The regulation of nicotinamide adenine dinucleotide biosynthesis by Nampt/PBEF/visfatin in mammals. Curr Opin Gastroenterol. 2007;23:164–170. doi: 10.1097/MOG.0b013e32801b3c8f. [DOI] [PubMed] [Google Scholar]
- 13.Villela D, Schlesinger D, Suemoto CK, Grinberg LT, Rosenberg C. A microdeletion in Alzheimer's disease disrupts NAMPT gene. J Genet. 2014;93:535–537. doi: 10.1007/s12041-014-0399-3. [DOI] [PubMed] [Google Scholar]
- 14.Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24:464–471. doi: 10.1016/j.tcb.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herrera-Arozamena C, Martí-Marí O, Estrada M, de la Fuente Revenga M, Rodríguez-Franco MI. Recent advances in neurogenic small molecules as innovative treatments for neurodegenerative diseases. Molecules. 2016;21 doi: 10.3390/molecules21091165. pii: E1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qi Z, Qi S, Ling L, Lv J, Feng Z. Salidroside attenuates inflammatory response via suppressing JAK2-STAT3 pathway activation and preventing STAT3 transfer into nucleus. Int Immunopharmacol. 2016;35:265–271. doi: 10.1016/j.intimp.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 17.Yang ZR, Wang HF, Zuo TC, Guan LL, Dai N. Salidroside alleviates oxidative stress in the liver with non- alcoholic steatohepatitis in rats. BMC Pharmacol Toxicol. 2016;17:16. doi: 10.1186/s40360-016-0059-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun KX, Xia HW, Xia RL. Anticancer effect of salidroside on colon cancer through inhibiting JAK2/STAT3 signaling pathway. Int J Clin Exp Pathol. 2015;8:615–621. [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang B, Wang Y, Li H, Xiong R, Zhao Z, Chu X, Li Q, Sun S, Chen S. Neuroprotective effects of salidroside through PI3K/Akt pathway activation in Alzheimer's disease models. Drug Des Devel Ther. 2016;10:1335–1343. doi: 10.2147/DDDT.S99958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao J, Zhou R, You X, Luo F, He H, Chang X, Zhu L, Ding X, Yan T. Salidroside suppresses inflammation in a D-galactose-induced rat model of Alzheimer's disease via SIRT1/NF-κB pathway. Metab Brain Dis. 2016;31:771–778. doi: 10.1007/s11011-016-9813-2. [DOI] [PubMed] [Google Scholar]
- 21.Zhang W, Peng M, Yang Y, Xiao Z, Song B, Lin Z. Protective effects of salidroside on mitochondrial functions against exertional heat stroke-induced organ damage in the rat. Evid Based Complement Alternat Med. 2015;2015:504567. doi: 10.1155/2015/504567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Y, Sauve AA. NAD+ metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim Biophys Acta. 2016;1864:1787–1800. doi: 10.1016/j.bbapap.2016.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Selkoe DJ, Schenk D. Alzheimer's disease: Molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol. 2003;43:545–584. doi: 10.1146/annurev.pharmtox.43.100901.140248. [DOI] [PubMed] [Google Scholar]
- 24.Cheng YF, Wang C, Lin HB, Li YF, Huang Y, Xu JP, Zhang HT. Inhibition of phosphodiesterase-4 reverses memory deficits produced by Aβ25–35 or Aβ1–40 peptide in rats. Psychopharmacology (Berl) 2010;212:181–191. doi: 10.1007/s00213-010-1943-3. [DOI] [PubMed] [Google Scholar]
- 25.Miguel-Hidalgo JJ, Cacabelos R. Beta-amyloid(1–40)-induced neurodegeneration in the rat hippocampal neurons of the CA1 subfield. Acta Neuropathol. 1998;95:455–465. doi: 10.1007/s004010050825. [DOI] [PubMed] [Google Scholar]
- 26.Yue T, Shanbin G, Ling M, Yuan W, Ying X, Ping Z. Sevoflurane aggregates cognitive dysfunction and hippocampal oxidative stress induced by β-amyloid in rats. Life Sci. 2015;143:194–201. doi: 10.1016/j.lfs.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 27.He F, Cao YP, Che FY, Yang LH, Xiao SH, Liu J. Inhibitory effects of edaravone in β-amyloid-induced neurotoxicity in rats. Biomed Res Int. 2014;2014:370368. doi: 10.1155/2014/370368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carrillo-Mora P, Luna R, Colín-Barenque L. Amyloid beta: Multiple mechanisms of toxicity and only some protective effects? Oxid Med Cell Longev. 2014;2014:795375. doi: 10.1155/2014/795375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han XH, Cheng MN, Chen L, Fang H, Wang LJ, Li XT, Qu ZQ. 7,8-Dihydroxyflavone protects PC12 cells against 6-hydroxydopamine-induced cell death through modulating PI3K/Akt and JNK pathways. Neurosci Lett. 2014;581:85–88. doi: 10.1016/j.neulet.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 30.Guo M, Li D, Shen H, Jin B, Ren Y, Li M, Xing Y. Leptin-Sensitive JAK2 activation in the regulation of Tau phosphorylation in PC12 cells. Neurosignals. 2016;24:88–94. doi: 10.1159/000442615. [DOI] [PubMed] [Google Scholar]
- 31.Chen CL, Tsai WH, Chen CJ, Pan TM. Centella asiatica extract protects against amyloid β1-40-induced neurotoxicity in neuronal cells by activating the antioxidative defence system. J Tradit Complement Med. 2015;6:362–369. doi: 10.1016/j.jtcme.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qian MC, Liu J, Yao JS, Wang WM, Yang JH, Wei LL, Shen YD, Chen W. Caspase-8 mediates amyloid-β-induced apoptosis in differentiated PC12 cells. J Mol Neurosci. 2015;56:491–499. doi: 10.1007/s12031-015-0498-5. [DOI] [PubMed] [Google Scholar]
- 33.Jieyu H, Chao T, Mengjun L, Shalong W, Xiaomei G, Jianfeng L, Zhihong L. Nampt/Visfatin/PBEF: A functionally multi-faceted protein with a pivotal role in malignant tumors. Curr Pharm Des. 2012;18:6123–6132. doi: 10.2174/138161212803582531. [DOI] [PubMed] [Google Scholar]
- 34.Ghosh D, Levault KR, Brewer GJ. Relative importance of redox buffers GSH and NAD(P)H in age-related neurodegeneration and Alzheimer disease-like mouse neurons. Aging Cell. 2014;13:631–640. doi: 10.1111/acel.12216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stein LR, Wozniak DF, Dearborn JT, Kubota S, Apte RS, Izumi Y, Zorumski CF, Imai S. Expression of Nampt in hippocampal and cortical excitatory neurons is critical for cognitive function. J Neurosci. 2014;34:5800–5815. doi: 10.1523/JNEUROSCI.4730-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cabezas-Opazo FA, Vergara-Pulgar K, Pérez MJ, Jara C, Osorio-Fuentealba C, Quintanilla RA. Mitochondrial dysfunction contributes to the pathogenesis of Alzheimer's disease. Oxid Med Cell Longev. 2015;2015:509654. doi: 10.1155/2015/509654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu XH, Lu M, Lee BY, Ugurbil K, Chen W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc Natl Acad Sci USA. 2015;112:2876–2881. doi: 10.1073/pnas.1417921112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y. Molecular links between caloric restriction and Sir2/SIRT1 activation. Diabetes Metab J. 2014;38:321–329. doi: 10.4093/dmj.2014.38.5.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodgers JT, Lerin C, Gerhart-Hines Z, Puigserver P. Metabolic adaptations through the PGC-1 alpha and SIRT1 pathways. FEBS Lett. 2008;582:46–53. doi: 10.1016/j.febslet.2007.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katsouri L, Parr C, Bogdanovic N, Willem M, Sastre M. PPARγ co-activator-1α (PGC-1α) reduces amyloid-β generation through a PPARγ-dependent mechanism. J Alzheimers Dis. 2011;25:151–162. doi: 10.3233/JAD-2011-101356. [DOI] [PubMed] [Google Scholar]
- 41.Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghosh D, LeVault KR, Barnett AJ, Brewer GJ. A reversible early oxidized redox state that precedes macromolecular ROS damage in aging nontransgenic and 3xTg-AD mouse neurons. J Neurosci. 2012;32:5821–5832. doi: 10.1523/JNEUROSCI.6192-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]