

Abstract
A common hallmark of amyloids is their resistance to an array of proteases, highlighting the difficulty in degrading these disease-related aggregated proteinaceous materials. Here, we report on the potent activity of cathepsin L (CtsL), a lysosomal protease that proteolyzes the Parkinson’s disease-related amyloid formed by α-synuclein (α-syn). Using liquid chromatography mass spectrometry and transmission electron microscopy, an elegant mechanism is revealed on the residue and ultrastructural level, respectively. Specifically, CtsL always truncates α-syn fibrils first at the C-terminus before attacking the internal β-sheet-rich region between residues 30–100. This suggests that only upon removal of the α-syn C-terminus can CtsL gain access to residues within the amyloid core. Interestingly, three of the four mapped sites contain a glycine residue (G36, G41, and G51) which is likely to be involved in a β-turn in the fibril, where upon cutting would lead to solvent exposure of internal residues and allow further proteolysis. By closely inspecting the fibril morphology, products resulting from CtsL degradation show imperfections along the fibril axis, with missing protein density as though they have been cannibalized. The ability of CtsL to degrade α-syn amyloid fibrils offers a promising strategy in improving cellular clearance of aggregated α-syn through the modulation of protease levels and activity.
TOC Image
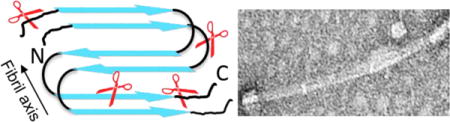
Parkinson’s disease (PD) afflicts more than 10 million people worldwide and usually affects people over the age of 50.1 A cellular hallmark of PD is the presence of cytoplasmic protein inclusions called Lewy bodies (LBs).2 A distinguishing feature of LBs is the abundance of amyloid fibrils of α-synuclein (α-syn).3 Significant research has been invested in understanding the biochemistry, structural biology, and cell biology of α-syn as it is a crucial player in PD etiology due to its strong genetic association to early-onset autosomal dominant PD.4, 5 Despite these efforts, the specific roles of α-syn amyloid fibrils in disease remain inconclusive. Both prefibrillar oligomers6 and fibrils7 have been shown to elicit cell death. A number of laboratories have shown that in vitro generated α-syn fibrils seed and amplify endogenous α-syn aggregation in cellular and animal models, indicating that fibril propagation may be an underlying mechanism for the spread of neurodegeneration in PD.8–10
Structurally, amyloid fibrils are defined by a cross-β structure where individual β-strands are aligned perpendicular to the fibril axis.11 Most recently, a high resolution structural model of full length α-syn fibrils has been reported,12 but because α-syn amyloids have been demonstrated to be polymorphic,13 other fibril structures may be possible and likely to be dependent on the conditions that they are formed.14 Biochemically, amyloid fibrils share similar properties such as they bind Congo red and Thioflavin T (ThT) and are highly protease- and detergent-resistant.15
A critical factor controlling the aggregation process of α-syn is its overall cellular level, regulated by the efficiency of degradation by the proteasome and lysosome.16 Generally, the lysosome is considered to be responsible to remove aggregation-prone species or excess levels of α-syn. Impaired lysosomal turnover of α-syn is associated with PD.16 In prior work, we discovered that cathepsin L (CtsL), an ubiquitous lysosomal protease, is essential in the lysosomal degradation of α-syn.17 Indeed, increased CtsL protein levels have been found within nigral neurons of PD patients, supporting a role for CtsL in disease.18 Notably, we also found that CtsL efficiently degrades α-syn amyloid fibrils (α-synf) at acidic pH.17 We showed that the reaction between CtsL and preformed α-synf resulted in optical clearance, loss of ThT fluorescence intensity, disappearance of fibrils in transmission electron microscopy (TEM) images, and loss of detectable full length protein and peptide fragments in SDS-PAGE analysis. As the lysosome is a cellular site for α-syn proteolysis, modulation of CtsL levels and activity offers a promising strategy in improving cellular clearance of α-syn. Consequently, in this study we employed liquid chromatography mass spectrometry (LC-MS) and sought to understand how CtsL degrades α-synf, particularly to investigate whether there are key cleavage sites that destabilize fibril structure and lead to fibril disassembly.
The substrate specificity of purified human CtsL (Sigma) was first assessed using recombinant monomeric N-terminally acetylated α-syn (α-synm),19 where the degradation process was peptide mapped as a function of time. α-Synm (15 μM) was incubated with CtsL (15 nM) at pH 5 buffer in sealed, glass vials and monitored every 15 min. The reaction was stopped by the addition of guanidinium hydrochloride (2 M) and subsequently analyzed by LC-MS (30 μL). Time points were taken between 0–105 min and after 23 h (Figure S1). Measured masses and peptide assignments are reported in Tables S1–4. Cleavage sites (denoted by X/X, where / indicates the cut, Figure 1A (red)) taken after 15 min incubation are at M5/K6, S9/K10, A17/A18, A27/E28, V40/G41, G41/S42, H50/G51, A53/T54, G67/G68, T75/A76, and N103/E104 (Table S1). These initial cut sites generate peptide fragments 1–17, 18–140, 1–103, 104–140, 1–41, 42–140, 1–50, 51–140, 1–53, 54–140, 1–75, 76–140, 6–140, 10–140, 28–140, 1–40, and 68–140 (Figure 1B).
Figure 1.
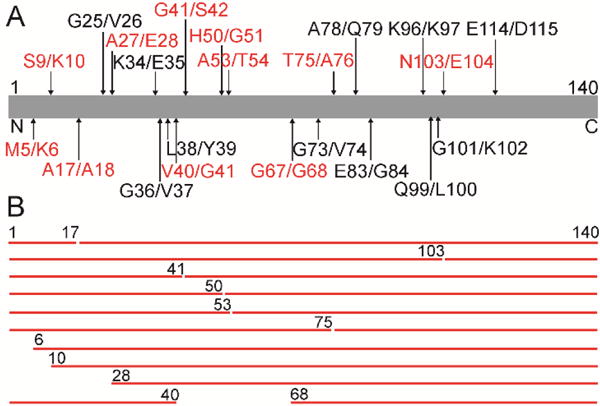
CtsL degradation of soluble α-syn at pH 5. (A) Schematic representation of the primary amino acid sequence of α-syn (residues 1 to 140) with LC-MS mapped cleavage sites from CtsL activity after 15 min (red) and 23 h (black) incubation. (B) Observed peptide fragments derived from CtsL degradation of α-synm (15 μM) after 15 min incubation.
Continued incubation between 30–105 min and after 23 h resulted in additional cleavage sites G25/V26, K34/E35, G36/V37, L38/Y39, G73/V74, A78/Q79, E83/G84, K96/K97, Q99/L100, G101/K102 and E114/D115 (Figure 1A (black), Table S1–4). After 23 h incubation most peptide fragments were < 4 kDa and assigned mostly to the N-terminal region of α-synm (Table S4). The exception was largely intact C-terminal regions corresponding to residues 84–140, 97–140, 102–140 and 104–140. TEM images show no fibrils formed (data not shown), supporting previous data that the peptides are not amyloidogenic under these conditions.17
For the fibril experiment, the amount of CtsL was increased by 10-fold to 150 nM. This allowed us to compare the reactions on comparable timescales. Fibrils were generated by incubating protein samples (15, 29, or 40 μM in 50 mM sodium acetate, 20 mM NaCl, pH 5) in glass vials (with a magnetic stir bar) at 37 °C with orbital shaking (600 rpm) for several days using a benchtop shaker and were visualized by TEM (Figure S2). Degradation reactions was monitored by LC-MS after 0.5, 1, 2, 3, 4, and 23 h (Figure S3). After 30 min incubation, C-terminal cleavage sites at G101/K102, N103/E104, E114/D115, N122/E123, E126/M127 and Q134/D135 were generated (Figure 2A (red), Table S5), corresponding to the release of C-terminal truncated fragments: 1–134, 1–126, 1–122, 1–114, 1–103, and 1–101 (Figure 2B). A minor population of an N-terminal cleavage at M5/K6 was also observed, generating the fragment, 6–114.
Figure 2.
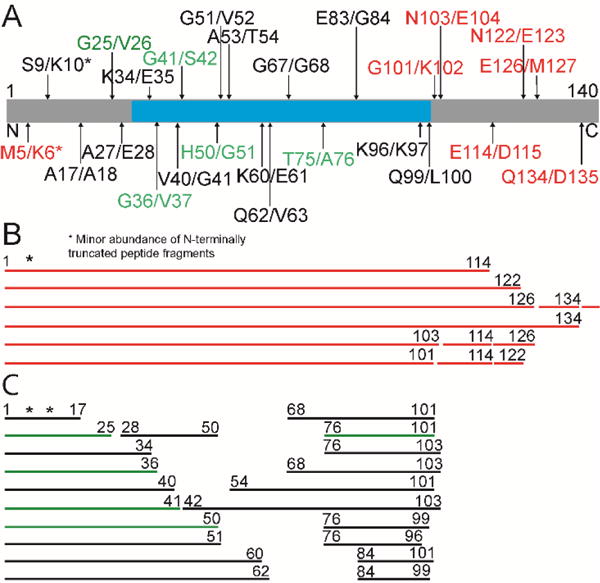
CtsL degradation of α-syn fibrils at pH 5. (A) Schematic representation of the primary amino acid sequence of α-syn (residues 1 to 140) with LC-MS mapped cleavage sites from CtsL activity after 30 min (red), 1 h (red and green) and 23 h (green and black) incubation. Consensus β-sheet-rich region (residues 30–100) identified from various solid-state NMR experiments of α-syn fibrils is shown in cyan.20, 21 (B) Observed peptide fragments derived from CtsL degradation of α-syn fibrils after 30 min incubation. (C) Additional peptide fragments observed after CtsL activity for 1 h (green) and 4 h (green and black).
Albeit a minor population, prolonging the reaction to 1 h revealed new masses in addition to C-terminal truncated species (Figure 2C (green)). These new peptides 1–25, 1–36, 1–41, 1–50 and 76–101 are derived from cut positions: G25/V26, G36/V37, G41/S42, H50/G51, and T75/A76, respectively (Figure 2A (green), Table S5). After 4 h incubation, an increase in the signal for these peptides was observed (Figure S3), in addition to new peptide masses (Figure 2C (black)) that correspond to cuts at S9/K10, A17/A18, A27/E28, K34/E35, V40/G41, G51/V52, A53/T54, K60/E61, Q62/V63, G67/G68, E83/G84, K96/K97 and Q99/L100 (Figure 2A (black), Table S6). Extensive incubation (23 h) resulted in complete loss of full-length and C-terminal truncated species, while small peptide masses, some of which resemble those observed for monomer degradation incubated under similar conditions were populated (Figure S3, Table S7). These peptides were previously shown to be non-amyloidogenic.17
Comparing the initial degradation behavior of α-syn monomer versus fibril by CtsL, it is evident that the amyloid-forming region (residues 30–100) (Figure 2A (cyan)) is inaccessible in the fibrillar state during short exposure to CtsL. This is in stark contrast to the monomer data, where cleavage occurs throughout this region (Figure 1B). Furthermore, in the fibrillar state, CtsL initially attacks the acidic C-terminus (defined here as residues 110–140) at cleavage sites that are generally not observed for the monomer (Figure 2B). However, there were two initial overlapping sites between monomer and fibril data, N103/E104 and a small population of N-terminal cutting at M5/K6 that was found on the C-terminal truncated fragments derived from the fibrils (Figure 2B, Table S5). It is only after longer exposure of the fibrils to CtsL, cleavage sites identical to those observed during early monomer degradation are seen, although the generated fragments differ substantially; degradation of α-syn monomer makes fragments with an intact C-terminus (Figure 1B), while fibril degradation results in fragments that loses the last ~37–44 C-terminal residues (Figure 2C).
To observe changes in the ultrastructure of fibrils during degradation, TEM images were taken under identical conditions to those during LC-MS analysis. Due to the propensity of these fibrils to clump at acidic pH, brief bath sonication was used to obtain a more homogeneous sample for imaging purposes. α-Synf in the absence of CtsL were shown to be composed of single and multi-bundled fibrils with variable widths (> 13 nm, Figures 3A and B). Interestingly, the differences in width of single fibrils suggest they are likely composed of multiple smaller protofibrils. This can be observed infrequently and varies from sample to sample (Figure S2).
Figure 3.

TEM snapshots of CtsL degradation of α-syn fibrils. α-Syn fibrils (15 μM) formed at pH 5 before (A and B) and after (C-G) Cts L proteolysis ([CtsL] = 150 nM at 37 °C for 4 h). Jagged fibril ends (arrows), fibril hole (circle) and missing fibril density (line bar) are shown.
During early time points of CtsL treatment of α-synf where C-terminal truncated peptides are first seen, TEM images show a greater number of shorter and thinner (< 13 nm) fibrils (Figure S4). While protofibrils appear more evident under these conditions, the heterogenous nature of these samples which still included full-length α-synf makes it difficult to definitively assign the nature of the fibrils. However, after 4 h of degradation when C-terminal truncations are in high abundance, obvious changes in fibril structures are revealed in the TEM images (Figure 3C–G). Firstly, protofibrils are clearly observed with an average diameter of ~2.5 nm. Figure 3C exemplifies this by showing three protofibrils adjacent to one another within a fibril. Secondly, many of these CtsL-treated fibrils have defects along the fibril axis with some appearing thinner in diameter. For example, Figure 3E shows fibrils composed of several protofibrils that have jagged fibril ends (arrows) of uneven lengths. Here, a hole within one of these fibrils is also seen (circle), suggesting CtsL is directly eating away at the fibril structure. Lastly, prolonged exposure to CtsL is clearly visible in Figure 3F where large fractions of the fibril is missing density (line bar). While the ends of this fibril look largely intact, the middle portion has a translucent appearance containing remnants of what once was a fibril. Collectively, the eclectic appearance of these fibrils (Figure 3C–G) highlights the heterogeneous nature of CtsL activity on fibrils.
In summary, tracking the degradation revealed intact fibrils are first truncated at the C-terminus. This observation is in strong agreement with prior literature reporting this region as unstructured and not directly part the amyloid core.22–24 However, it has been shown that both the N- and C-terminus play a role in amyloid formation.25 Interestingly, many of these C-terminal derived cleavages (e.g. N122/E123, E126/M127, and Q134/D135) (Figure 2A) are absent from monomer degradation (Figure 1A). This may imply CtsL is either being forced to proteolyze at a region that is not a preferred substrate site, or the C-terminus is structurally different and inaccessible24 during monomer degradation. The small abundance of N-terminal cleavage at M5/K6, originating from C-terminal truncations (Figure 2B) suggests access to this region occurs only after removal of the C-terminus.
Once ΔC truncations have occurred, CtsL access to substrate sites within the putative amyloid core of α-syn (Figure 2A, (cyan)), such as G41/S42, H50/G51, and T75/A76, is revealed. The resulting peptides shown in Figure 2C do not include any large C-terminal regions, indicating a direct origin from one of the C-terminally truncated peptide fragments, e.g. 1–134, 1–126, 1–122, 1–114, 1–103 or 1–101 (Figure 2B). Many of these N-terminal cleavage sites contain a glycine, which are known to participate in β-turns in an amyloid structure.20 Cutting at these turns is likely more feasible than a direct attack on β-sheets, which are more solvent-excluded and thus less accessible to CtsL. Interestingly, many of the solid-state NMR experiments on α-syn fibrils find residue G51 to reside in a β-turn.20 Also, the crystal structure of peptide composed of 47GVVHGVTTVA56, shows two tightly interdigitated β- sheets,26 termed steric zippers,27 with the β-strand kinked at residue G51. The consequence of severing β-turns would likely remodel the fibril structure, potentially making it more susceptible to further proteolytic attack. Precedence for β-turn disruption and fibril restructuring comes from the enzymatic digestion of Aβ(1–42) fibrils with protease XIV,28 where hydrolysis at the K28/G29 turn region linking the two β-sheets results in random assembly of β-sheets.
The observed CtsL cleavage site at T75/A76 is central to the critical region implicated in α-syn aggregation29,30 and toxicity.31 In fact, the peptide 68GAVVTGVTAVA78 is shown to aggregate and form fully extended β-strands that stack in-register into β-sheets.26 Accessing and breaking up this amide bond would disrupt the alignment of β-strands in a β-sheet, resulting in fibril disassembly. However, since the LC-MS analysis showed the concomitant build-up of peptides that are derived from this cleavage site and the putative β-turns, it is not possible to discern a single critical cut site.
Another example for enzymatic degradation of amyloid comes from the serine protease HRTA1, which was reported to degrade Tau and Aβ fibrils by a mechanism that couples disintegrase activity to destabilize fibrils followed by proteolytic attack.32 Expanding on our observation of CtsL degrading α-syn fibrils, it will be instrumental in seeing if other amyloidogenic proteins such as Aβ, Tau and/or huntingtin can be degraded. From a lysosomal perspective, the potency of CtsL activity exemplifies the capability for the organelle to deal with such disease-related structures. In PD patients, lysosomal dysfunction has been implicated33 and therapeutic pathways are generally centered outside of the lysosome.34 From our work, further enhancing lysosomal activity by CtsL offers a new therapeutic agent to combat PD.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program at the National Institutes of Health, National Heart, Lung, Blood Institute. We acknowledge the NHLBI Biochemistry and Electron Microscopy Core Facilities for use and maintenance of the LC-MS equipment and electron microscope, respectively.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. Materials and methods, Table S1–S7, Figures S1–S4.
Notes
The authors declare no competing financial interests.
References
- 1.http://www.pdf.org/en/parkinson_statistics
- 2.Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VMY, Trojanowski JQ, Iwatsubo T. Am J Pathol. 1998;152:879–884. [PMC free article] [PubMed] [Google Scholar]
- 3.Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, Goedert M. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 4.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et al. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 5.Lashuel HA, Overk CR, Oueslati A, Masliah E. Nat Rev Neurosci. 2013;14:38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bemporad F, Chiti F. Chem Biol. 2012;19:315–327. doi: 10.1016/j.chembiol.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 7.Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VMY. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo JL, Lee VMY. Nat Med. 2014;20:130–138. doi: 10.1038/nm.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jucker M, Walker LC. Nature. 2013;501:45–51. doi: 10.1038/nature12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walsh DM, Selkoe DJ. Nat Rev Neurosci. 2016;17:251–260. doi: 10.1038/nrn.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shewmaker F, McGlinchey RP, Wickner RB. J Biol Chem. 2011;286:16533–16540. doi: 10.1074/jbc.R111.227108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tuttle MD, Comellas G, Nieuwkoop AJ, Covell DJ, Berthold DA, Kloepper KD, Courtney JM, Kim JK, Barclay AM, Kendall A, et al. Nat Struct Mol Biol. 2016;23:409–415. doi: 10.1038/nsmb.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dearborn AD, Wall JS, Cheng NQ, Heymann JB, Kajava AV, Varkey J, Langen R, Steven AC. J Biol Chem. 2016;291:2310–2318. doi: 10.1074/jbc.M115.698787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tycko R. Neuron. 2015;86:632–645. doi: 10.1016/j.neuron.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGlinchey RP, Yap TL, Lee JC. Phys Chem Chem Phys. 2011;13:20066–20075. doi: 10.1039/c1cp21376h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xilouri M, Brekk OR, Stefanis L. Mol Neurobiol. 2013;47:537–551. doi: 10.1007/s12035-012-8341-2. [DOI] [PubMed] [Google Scholar]
- 17.McGlinchey RP, Lee JC. Proc Natl Acad Sci U S A. 2015;112:9322–9327. doi: 10.1073/pnas.1500937112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li LY, Wang XX, Fei XF, Xia LP, Qin ZH, Liang ZQ. Neurosci Lett. 2011;489:62–67. doi: 10.1016/j.neulet.2010.11.068. [DOI] [PubMed] [Google Scholar]
- 19.Human α-syn is acetylated at its α-amino terminus, achievable in E coli through co-expression of the acetylation enzyme, NatB.
- 20.Meier BH, Bockmann A. Curr Opin Struct Biol. 2015;30:43–49. doi: 10.1016/j.sbi.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 21.Vilar M, Chou HT, Luhrs T, Maji SK, Riek-Loher D, Verel R, Manning G, Stahlberg H, Riek R. Proc Natl Acad Sci U S A. 2008;105:8637–8642. doi: 10.1073/pnas.0712179105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McClendon S, Rospigliosi CC, Eliezer D. Protein Sci. 2009;18:1531–1540. doi: 10.1002/pro.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eliezer D. J Mol Biol. 2013;425:2393–2396. doi: 10.1016/j.jmb.2013.03.031. [DOI] [PubMed] [Google Scholar]
- 24.Wu KP, Weinstock DS, Narayanan C, Levy RM, Baum J. J Mol Biol. 2009;391:784–796. doi: 10.1016/j.jmb.2009.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yap TL, Pfefferkorn CM, Lee JC. Biochemistry. 2011;50:1963–1965. doi: 10.1021/bi2000824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodriguez JA, Ivanova MI, Sawaya MR, Cascio D, Reyes FE, Shi D, Sangwan S, Guenther EL, Johnson LM, Zhang M, et al. Nature. 2015;525:486–490. doi: 10.1038/nature15368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJW, McFarlane HT, et al. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 28.Numata K, Kaplan DL. Biochemistry. 2010;49:3254–3260. doi: 10.1021/bi902134p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Waxman EA, Mazzulli JR, Giasson BI. Biochemistry. 2009;48:9427–9436. doi: 10.1021/bi900539p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giasson BI, Murray IVJ, Trojanowski JQ, Lee VMY. J Biol Chem. 2001;276:2380–2386. doi: 10.1074/jbc.M008919200. [DOI] [PubMed] [Google Scholar]
- 31.Bodles AM, Guthrie DJS, Greer B, Irvine GB. J Neurochem. 2001;78:384–395. doi: 10.1046/j.1471-4159.2001.00408.x. [DOI] [PubMed] [Google Scholar]
- 32.Poepsel S, Sprengel A, Sacca B, Kaschani F, Kaiser M, Gatsogiannis C, Raunser S, Clausen T, Ehrmann M. Nat Chem Biol. 2015;11:862–869. doi: 10.1038/nchembio.1931. [DOI] [PubMed] [Google Scholar]
- 33.Nixon RA. Nat Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- 34.Mazzulli JR, Zunke F, Isacson O, Studer L, Krainc D. Proc Natl Acad Sci U S A. 2016;113:1931–1936. doi: 10.1073/pnas.1520335113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.