

Abstract
High-throughput screening (HTS) is one of the major techniques for discovering promising molecules for drug development. Rhodopsin mutations cause the most common autosomal dominant form of retinitis pigmentosa, an inherited retinal degenerative disease that currently has no effective treatment. To find an optimal pharmacological treatment for rhodopsin-associated retinitis pigmentosa, we performed two cell-based HTSs with mammalian cells expressing the P23H rod opsin mutant and identified two sets of novel compounds for further validation and characterization. The first HTS screen identified pharmacological chaperones of P23H opsin that increased its translocation from the endoplasmic reticulum to the plasma membrane. The second HTS screen selected small molecules that enhanced the clearance of the mutant opsin while vision could be sustained by the healthy gene allele expressing wild-type rhodopsin. Here we describe the methodology of these two HTS assays in detail.
Keywords: P23H rhodopsin, Retinitis pigmentosa, High-throughput screen, Drug discovery
1 Introduction
High-throughput screening (HTS) has been employed for drug discovery since the 1990s and associated technologies have evolved to the third generation [1–3]. Until 2008, among the 58 FDA approved drugs with their starting compounds documented, 19 drugs were developed from hit compounds identified in HTS [3]. Compared to other drug discovery methods such as structure-based or ligand-based virtual screening [4] and fragment-based drug design [5], HTS requires little knowledge of the target structure or the availability of an active model compound [6]. Resulting from a state-of-the-art automated facility, optimized drug-like compound libraries, and multiple quality control algorithms, hit compounds from about 50 % of HTS projects have led to the successful development of drugs in pharmaceutical pipelines [3].
Retinitis pigmentosa (PR) is a retinal degenerative disease with a heterogeneous genetic background. Mutations of the gene encoding rhodopsin, the visual pigment of rod photoreceptor cells, are found in about 25 % of individuals with autosomal dominant retinitis pigmentosa (adRP) [7, 8]. The P23H rhodopsin mutation causes the most common form of adRP, accounting for 12 % of cases in the United States [7]. To date, there is no effective treatment for this disease, although multiple experimental efforts have been reported [9–17]. Currently, active compounds which showed protective effects in P23H rhodopsin-associated adRP models are limited to two categories: (1) the native chromophore and its analogs with high light sensitivity, low stability, and relatively high toxicity [18–21] and (2) natural antioxidant substances which require high dosages for efficacy and are not suitable for human treatment [12–14].
Two models have been proposed for the molecular basis of P23H opsin-triggered photoreceptor cell death: (1) the overwhelmed unfolded protein response (UPR) model. Here the Pro to His mutation at codon 23 disrupts the local hydrophobic cluster in the rod opsin protein, leading to its immature glycosylation, misfolding, and resulting activation of the UPR in the endoplasmic reticulum (ER). Consistent expression of the misfolded P23H opsin could overwhelm the UPR system and result in apoptosis of photoreceptor cells, the first step in the progression of RP [21–26]. (2) The disrupted rod outer segment (ROS) model. In the P23H knock-in mouse model wherein most of the P23H opsin is degraded, a residual amount of the mutant rhodopsin pigment could be transported to the ROS where it disrupts disc organization and causes photoreceptor death [27–29].
Based on these two models, we designed two different HTS discovery strategies as follows: (1) identify small-molecule chaperones that stabilize the proper folding of P23H opsin and increase its translocation from ER to the plasma membrane (equivalent to ROS in rod photoreceptor cells) thereby reducing the UPR and (2) find small-molecule compounds that clear the mutant opsin, leaving only normal opsin derived from the healthy gene copy to maintain retinal structure and vision.
For the HTS of active compounds that increase the translocation of P23H opsin from the ER to plasma membrane, we generated a stable cell line (PathHunter U2OS mRHO(P23H)-PK total GPCR translocation cells) which expressed two active recombinant fusion proteins (Fig. 1a): (1) mRHO(P23H)-PK, the mouse P23H opsin fused with a small subunit of β-galactosidase (β-Gal), and (2) PLC-EA, a membrane-associated peptide (the PH domain of phospholipase C-δ, PLC) fused with a large subunit of β-Gal. Without treatment, misfolded mRHO(P23H)-PK accumulates in the ER, whereas PLC-EA associates with the plasma membrane. Hence, the separation of the two subunits of β-Gal into different cell compartments results in little β-Gal activity when substrate is added. But upon the treatment with an active compound, mRHO(P23H)-PK is transported from ER to the plasma membrane, leading to reconstitution of intact β-Gal. The restored activity of β-Gal can be measured by luminescence after the addition of substrate.
Fig. 1.
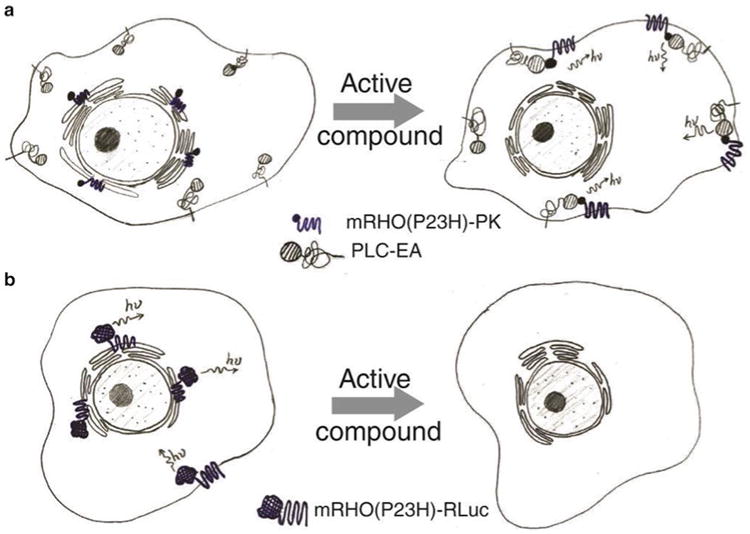
Diagrams of the β-Gal complement assay for the P23H opsin translocation HTS (a) and the RLuc assay for the P23H opsin clearance HTS (b)
For the HTS of active compounds that promote the P23H opsin clearance, we generated another stable cell line (Hek293 mRHO(P23H)-RLuc total GPCR quantification cells) using Renilla luciferase (RLuc) as a reporter for the mutant opsin (Fig. 1b). Here the P23H opsin is fused with RLuc expressed in human embryonic kidney 293 (Hek293) cells. The amount of the P23H-RLuc protein is correlated with the RLuc activity, which can be read by the luminescence recorded by a microplate reader.
Both HTS assays have been optimized with respect to cell seeding number, substrate conditions, and dimethyl sulfoxide (DMSO) tolerance to ensure that they are reliable and reproducible as indicated by the quality control parameters Z′ (i.e., Z′ > 0.5) and signal-to-background (S/B) ratio (i.e., S/B ratio > 3) [30] (see Subheading 3.1.4 for more detailed description).
Here the two HTSs are described in three tiers: primary HTS, hit confirmation screen, and dose-response screen. Initially, each compound from a Diversity Set of compound library is tested at a single concentration. Then identified “hit” compounds with the desired effect are retested at the same concentration in triplicate. Finally, each confirmed hit compound is tested at 10 concentrations in triplicate. EC50 values for the final hit compounds are obtained from their dose-response curves fitted by the Hill function.
2 Materials
2.1 Cells
PathHunter U2OS mRHO(P23H)-PK total GPCR translocation cells for the P23H opsin translocation screen. U2OS cells expressing mRHO(P23H)-PK (the 40 amino acid PK subunit of β-Gal fused on the C-terminus of the P23H mouse opsin mutant) and PLC-EA (the EA subunit of β-Gal fused on the C-terminus of PLC peptide) recombinant proteins were generated by a collaboration between Dr. Nevin A. Lambert (Georgia Regents University, GA) and DiscoveRx, CA. A total of 4 × 108 cells were collected at passage 7 and frozen in liquid nitrogen.
Hek293 mRHO(P23H)-RLuc total GPCR quantification cells for the P23H opsin clearance screen. The cDNA of RLuc (RLuc8, a spectrally shifted mutant of luciferase from Renilla reniformis, was fused on the C-terminus of the P23H mouse opsin (mRHO(P23H)-RLuc)) constructed in the pcDNA3.1/Zeo vector provided by Dr. Nevine A. Lambert (Georgia Regents University, GA). The DNA vector was transfected into Hek293 cells with polyethylenimine (see Note 1) to generate cells continuously expressing the P23H opsin-RLuc recombinant protein (see Note 2).
2.2 Tissue Culture
Heat-inactivated fetal bovine serum (FBS, HI): Thaw a 500 ml bottle of FBS (HyClone) at room temperature and heat- inactivate by incubation at 65 °C for 1 h. Then aliquot into 50 ml conical tubes and store at −20 °C. Thaw at 37 °C before use.
Cell growth medium: Dulbecco's modified Eagle medium (DMEM), 12 % FBS, 5 μg/ml Plasmocin. To a 500 ml bottle of DMEM high glucose medium (HyClone), add 60 ml of thawed FBS, HI, and 100 μl of 25 μg/ml Plasmocin (InvivoGen) in a tissue culture hood. Store medium at 4 °C and warm to 37 °C before use.
Cell plate medium: DMEM, 10 % FBS, 1 unit/ml penicillin, 1 μg/ml streptomycin and 2.92 μg/ml L-glutamine. To a 500 ml bottle of DMEM high glucose medium add, 50 ml of thawed FBS, HI, and 5 ml of 100× penicillin-streptomycin- glutamine (HyClone) in a tissue culture hood. Store medium at 4 °C and warm to 37 °C before use.
0.05 % trypsin solution (HyClone).
Sterile 1× phosphate buffered saline 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4, 137 mM NaCl and 2.7 mM KCl, pH 7.4.
Growth-enhanced treated 150 mm tissue culture plates (TPP).
70 μm cell strainer (BD Falcon).
15 ml and 50 ml conical tubes (BD Falcon).
200 ml conical bottom centrifuge bottles with adaptors (Thermo Scientific).
Hemocytometer (Fisher Scientific).
2.3 HTS
β-Gal Assay Substrate Buffer (for P23H opsin translocation screen): 4 % of Gal Screen Substrate and 96 % Gal Screen Buffer A. To prepare 12 ml of β-Gal Assay Substrate Buffer for one 384-well plate (25 μl/well), add 0.48 ml Gal Screen Substrate and 11.52 ml Gal Screen Buffer A from the Gal Screen System (Applied Biosystems) to a 15 ml tube and vortex to mix (see Note 3).
RLuc Assay Substrate Buffer (50 μM ViviRen) (for P23H opsin clearance screen): Dissolve 3.7 mg of ViviRen (Promega) in 100 μl of DMSO to prepare 60 mM ViviRen stock solution. Add 33.3 μl of ViviRen stock solution to 40 ml of PBS in a 50 ml conical tube to prepare 40 ml 50 μM RLuc Assay Substrate Buffer.
2 % n-dodecyl β-d-maltoside (DDM) in PBS.
Positive Control Solution for the P23H opsin translocation screen: 25 μM 9-cis-retinal in cell growth medium. In a dark room with dim red light, dissolve 1 mg 9-cis-retinal powder (Sigma) in 178 μl of DMSO to prepare a 20 mM stock solution. For a 25 μM 9-cis-retinal working solution, dilute 6.25 μl of 9-cis-retinal stock solution into 5 ml of growth medium, and vortex to mix. Wrap the tube with aluminum foil to protect from light.
Positive Control Solution for P23H opsin clearance screen: 1 mM Evans Blue solution. To prepare Evans Blue 25 mM stock solution, dissolve 24 mg Evans Blue powder (Sigma) in 1 ml of ddH2O. For 1 mM working solution, dilute 200 μl of 25 mM Evans Blue stock solution into 4.8 ml of cell growth medium.
Neutral control for both the β-Gal and RLuc assays: cell Plate medium (see Note 4).
Assay plate: A 384-well, white-walled, clear flat bottomed, sterile plate with lid (BD Falcon) or a 384-well, white-walled, clear flat bottomed, sterile plate with lid (Greiner Bio-One).
Compound plate: A 384-well, polypropylene, flat bottomed plate (BD Falcon).
2.4 Instruments
Perkin Elmer plate∷explorer™ uHTS system: This consists of an HW workstation (plate storage and incubation), an LW workstation (liquid handling), and an RW workstation (signal detection). It includes the following components: two Multidrop dispensers (dispenser A and B), a CyBi-Well dispenser, a microplate washer, a Rotanta 46 microplate centrifuge, a plate Lift storage device, a tissue culture incubator I189 (37 °C in 5 % CO2 with 95 % humidity), a storage incubator I30, turn tables, a bar-code reader, conveyor belt, and a Perkin Elmer Plate∷Vision™ Detector.
Stand-alone Perkin Elmer EnVision™: A plate reader for luminescence signal detection.
2.5 Compound Library
The 25,000 Diversity Set of University of Cincinnati Compound Collections (used for the P23H opsin translocation screen).
The 10,000 Diversity Set of University of Cincinnati Compound Collection (used for P23H opsin clearance screen) (see Note 5).
3 Methods
3.1 The P23H Opsin Translocation HTS
The 25,000 Diversity Set including 83 compound plates is separated into 2 cycles of HTS. Each cycle of HTS tests 42 or 41 compound plates that are further broken down into two sets of 21 or 20 plates for an automated experiment.
3.1.1 Preparation for HTS
Design and test-run the workflow of an automated experiment with used plates. Figure 2 shows the plate map of a P23H opsin translocation assay. Defined by the compound plate design, each assay plate has a maximum of 4 columns for controls (Columns 21–24).
Calibrate the pin tools (50 nl pin tool for primary HTS and hit confirmation screens and a 200 nl pin tool for the dose-response screen). Calculate the actual dispensing volume of each pin tool and the corresponding final concentration of the tested compounds for each screen (see Note 6).
Before starting each experiment, sterilize the dispenser tubes by priming with 70 % ethanol. Rinse the dispenser tubes again with sterile PBS buffer or cell plate medium to wash out residual ethanol.
Fig. 2.
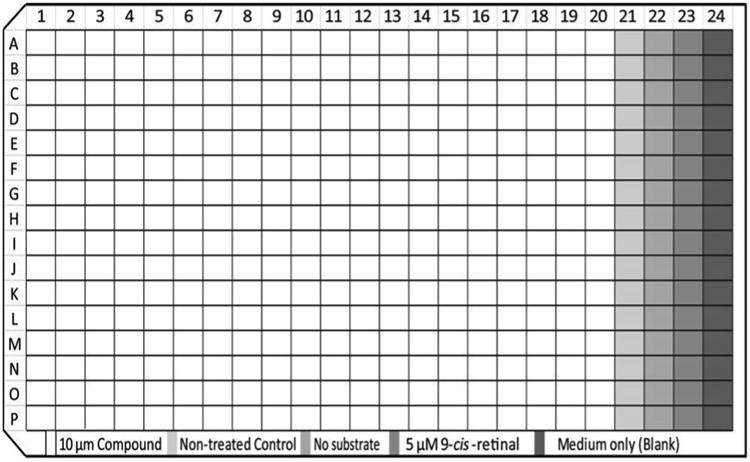
Plate map of the HTS for the correction of P23H opsin localization
3.1.2 Cell Preparation (See Note 7)
The following procedures are performed in a tissue culture hood under sterile conditions:
Day 1 Revival of frozen cells
Prewarm cell growth medium in a 37 °C water bath.
Place 25 cryo-vials with frozen PathHunter U2OS mRHO(P23H)-PK total GPCR translocation cells (see Note 8) in a 37 °C water bath until only small ice crystals remain and the cell pellet is almost completely thawed (30 s to 1 min).
Transfer every five vials of thawed cells to a 50 ml conical tube containing 25 ml of prewarmed cell growth medium. Centrifuge at 300 × ℊ for 4 min to pellet cells. Remove medium.
Resuspend cell pellet with 12.5 ml prewarmed cell growth medium (add 0.5 ml cell growth medium to cells from each cryo-vial). Combine cell suspensions into one conical tube and mix well.
Add 1 ml of cell suspension and 29 ml of prewarmed cell growth medium to each 150 mm cell culture plate. Gently rotate the plate to mix well. Incubate at 37 °C in 5 % CO2 with 95 % humidity for 24 h.
Day 2 Cell growth medium replacement
6. Gently remove medium from the 150 mm cell culture plate and replace with 30 ml of prewarmed cell growth medium.Incubate plates at 37 °C in 5 % CO2 with 95 % humidity until cells reach more than 90 % confluence on Day 5 (see Note 9).
Day 5 Preparation of cell suspensions for HTS plate seeding
7. Confirm the full confluence of cells under a light microscope. Prewarm the cell plate medium in a 37 °C water bath.
8. Remove growth medium and wash each plate with 20 ml of PBS. Remove PBS, and add 2 ml of trypsin (0.05 %) to each plate. Incubate at 37 °C for 5–8 min until cells are detached. Add 8 ml of cell plate medium to each plate, mix well, and filter through a 70 μm cell strainer to capture cell clusters. Collect the flow-through in a 200 ml conical bottomed centrifuge bottle.
9. Count cells with a hemocytometer and dilute them to 25 × 104 cells/ml in cell plate medium to prepare a cell suspension for HTS plate seeding. The total volume of cell suspension should be more than 190 ml, sufficient for distribution in 21 × 384-well plates at 20 μl/well (21 × 400 × 20 μl = 168,000 μl = 168 ml) leaving 22 ml as the dead volume of dispenser (see Notes 10 and 11).
10. Revive and prepare the cells for the 2nd cycle of HTS experiment (e.g., revive cells on Day 5 if the 2nd cycle of HTS starts on Day 9).
3.1.3 Primary HTS Screen
Day 5 Cell seeding
Place 21 (or 20 for the last experiment set of the 2nd HTS cycle) assay plates in the stackers of Plate∷Life storage.
Prime and fill the tubes of dispenser A with cell plate medium and confirm that liquid flows through each of the 8 tubes of the dispenser without air bubbles in the tubes.
Similarly, prime and fill the tubes of dispenser B with cell suspension prepared in Subheading 3.1.2 for plate seeding.
Perform steps 4–8 by automation (see Note 12):
4. Add 25 μl of cell plate medium to Column 24 of each assay plate with dispenser A.
5. Add 20 μl of the cell suspension (prepared in Subheading 3.1.2) to Columns 1–23 of each assay plate with dispenser B.
6. Centrifuge the assay plates at 300 × ℊ for 30 s to bring down cells to the bottom of each well.
7. Place assay plates in incubator I189.
8. Incubate assay plates at 37 °C in 5 % CO2 with 95 % humidity overnight.
9. Repeat steps 1–8 to seed cells in another set of 21 assay plates (or 20 plates for the last experiment of the 2nd HTS cycle).
Day 6 Compound treatment
10. Thaw 42 (or 41 for the 2nd HTS cycle) compound plates from the 25,000 Diversity Set (stored at −20 °C) at room temperature (see Note 13). Centrifuge each plate at 450 × ℊ for30 s to bring down liquid to the bottom of each well.
11. Remove the sealing foil of 21 compound plates. Record the plate IDs with a handheld bar-code scanner and input the compound plate ID into a spreadsheet to be paired with the assay plates (see Note 14).
12. Place 21 (or 20 for the last experiment of the 2nd HTS cycle) compound plates in the stackers of Plate∷Lift storage.
Perform steps 13–17 by automation:
13. Add 5 μl of cell plate medium to Columns 1–22 of assay plates with dispenser A, to achieve a final volume of 25 μl in each well.
14. In dim light, transfer compounds from compound plates to assay plates with the 50 nl pin tool (11.26 μM of each compound should be added according to the pin tool calibration).
15. Add 5 μl 9-cis-retinal working solution (positive control) to Column 23 of assay plates with dispenser B. Shake the assay plates for 3 s to mix the compounds with medium.
16. Place the assay plates in incubator I189.
17. Place the compound plates back in the stackers of Plate∷Lift storage.
18. Incubate assay plates at 37 °C in 5 % CO2 with 95 % humidity overnight.
19. Seal the tested compound plates with sealing foil and re-store them at −20 °C.
20. Repeat steps 11–19 to finish the treatment of another set of 21 assay plates (or 20 if for the last experiment of the 2nd HTS cycle) (see Note 15).
Day 7 luminescence reading
Perform steps 21 and 22 by automation:
21. Add 25 μl β-Gal Assay Substrate Buffer to each well of assay plates under dim light.
22. Incubate the assay plates at 25 °C in incubator I30 for 60 min.
23. Take out the assay plates sequentially and read their luminescence with the Perkin Elmer EnVision detector (100 ms integration time).
24. Repeat steps 21–23 to obtain luminescence readings for the 2nd set of 21 (or 20 for the 2nd HTS cycle) assay plates. In total, 42 out of the 83 compound plates from the 25,000 Diversity Set of compounds should be screened in the 1st HTS cycle.
Repeat Subheadings 3.1.2 and 3.1.3 to perform primary HTS for the other 41 compound plates of the 250,000 Diversity Set.
3.1.4 Data Analysis for Primary HTS Screen
Analyze the HTS screen data with Genedata Screener Assay Analyzer Software (10.0.2 Standard). Normalize the measured luminescence intensity to the controls for each assay plate. The normalized activity score (%) for compounds facilitating proper localization of P23H rhodopsin is calculated as shown in Table 1 (see Note 16).
-
Calculate the HTS quality control parameters for each assay plate:
(a) where STD represents the standard deviation of luminescence intensities and mean represents the average luminescence intensities:
(b) Export the results of normalized compound activity and quality control parameters from the Genedata Screener. Sort the data by activity score from high to low in Excel. Define “hits”as those with activity scores (%) equal or higher than 15.Import the data for hits (including their compound ID and activity score from the Genedata Screener) into Accelrys Pipeline Pilot software to incorporate the chemical structure, simplified molecular-input line-entry (SMILE) specification, and predicted physiochemical properties of hit compounds into the data set.
Table 1. Calculation of normalized activity scores for the P23H opsin translocation HTS.
| Control | Measurement | Activity score (%) | |
|---|---|---|---|
| Untreated control | Minimum amount of P23H opsin on the plasma membrane | 0 | |
| Positive control (5 μM 9-cis-retinal) | Maximum amount of P23H opsin on the plasma membrane | 100 | |
|
| |||
3.1.5 Dose-Response Screen (See Note 17)
Prepare compound plates for dose-response screen.
Based on the IDs of hit compounds, pull out the stock vials of these compounds in the UC Compound Collection. Transfer 40 μl of each hit (10 mM in DMSO) from the stock vial to a well in Column 1 or 11 of a 384-well Falcon polypropylene plate.
Add 20 μl of DMSO to Columns 2–10 and 12–20 and perform a twofold dilution of each compound. Use a multichannel pipette to transfer 20 μl of each compound from Column 1 to Column 2. Mix well by pipetting up and down 3 times. Then transfer 20 μl from Column 2 to Column 3, and so on, until each well of Column 10 is flled with 40 μl of diluted compound. Remove and discard 20 μl of liquid in each well of Column 10.
-
Repeat step 2 to make a twofold dilution series for compounds in Columns 11.
In summary, a dose-response compound plate is prepared by serial twofold dilution of each compound to achieve a total of ten concentrations.
Repeat steps in Subheadings 3.1.1–3.1.3 to perform the dose-response screen with freshly prepared compound plates. Instead of a 50 nl pin tool, use a 200 nl pin tool for compound transfer here to obtain higher concentrations for the dose-response curve. Each compound plate is tested in triplicate with three assay plates.
3.1.6 Data Analysis for Dose-Response Screen
Analyze the dose-response screen data with Genedata Screener Assay Analyzer Software (10.0.2 Standard). Normalize measured luminescence intensities to the controls for each assay plate as described in Table 1. Calculate Z′ and the S/B ratio of each assay plate for quality control.
Generate a dose-response curve of each tested compound using Genedata Screener Condoseo Software (10.0.2 Standard). Use Smart Fit Model to fit the dose-response curves. Set Sinf and S0 to +100 and 0, respectively (for definition of Sinf and S0 see Note 18). Define AC50 value as the concentration of compound (μM) that causes an activity score of 50 (see Note 19).
Define the final hits as compounds with AC50≤100 μM. Use Accelrys Pipeline Pilot software to incorporate their chemical structures and related properties (see Note 20).
3.2 The P23H Opsin Clearance HTS
The 10,000 Diversity Set includes 32 × 384-well compound plates which are separated into two cycles of HTS. Each cycle of HTS tests 16 compound plates.
3.2.1 Preparation for HTS
Follow procedures described in Subheading 3.1.1.
3.2.2 Cell Preparation
Day 1 Revival of frozen cells
Revive 3 vials of Hek293 mRHO(P23H)-RLuc total GPCR quantification cells (see Note 21) in 7 × 150 mm plates by procedures described in Subheading 3.1.2 Day 1.
Day 2 Replacement of cell growth medium
2. Follow procedures described in Subheading 3.1.2 Day 2.
Day 4 Preparation of cell suspensions for HTS plate seeding
3. Harvest confluent cells following procedures described in Subheading 3.1.2 Day 5. Count cells and dilute them to 25 × 104 cells/ml, so that the cell seeding number is 8,000 cells/well (32 μl/well). Prepare a total of 250 ml cell suspension in two sterile conical bottomed bottles (see Note 22).
4. Three days before the 2nd cycle of the primary HTS for P23H opsin clearance, revive 3 vials of P23H-RLuc Hek293 cells in 7 × 150 mm plates as described in steps 1–3 (e.g., revive cells on Day 4 if cells are to be seeded on Day 7).
3.2.3 Primary HTS Screen
Day 4 Cell seeding
Place 16 assay plates in the stackers of Plate∷Lift storage.
Prime and fill the tubes of dispenser A with cell plate medium.
-
Prime and fill the tubes of dispenser B with the cell suspension prepared in Subheading 3.2.2.
Perform steps 4–9 by automation.
Add 8 μl of cell plate medium to Columns 1–22 of each assay plate with dispenser A.
Add 40 μl of cell plate medium to Column 24 of each assay plate with dispenser A.
Add 32 μl of cell suspension to Columns 1–23 with dispenser B.
Centrifuge the plate at 300 × ℊ for 30 s to bring down cells to the bottom of the plate.
Place assay plates in incubator I189.
Incubate plates at 37 °C in 5 % CO2 with 95 % humidity for24 h.
Day 5 Compound treatment
10. Thaw 16 compound plates from the 10,000 Diversity Set (stored at −20 °C) at room temperature. Centrifuge each plate at 450 × ℊ for 30 s to bring down liquid to the bottom of each well.
11. Tear off the sealing foil of 16 compound plates. Record the plate ID using a handheld bar-code scanner and input the compound plate ID into a spreadsheet to be paired with the assay plates.
12. Place 16 compound plates in the stackers of Plate∷Lift storage.
-
13. Prime and fill the tubes of dispenser A with Evans Blue working solution and confirm that no air bubbles are visible in the dispensing tubes.
Perform steps 14–19 by automation:
14. Transfer compounds from compound plates to assay plates with the 50 nl pin tool (final concentration of each compound should be 9.93 μM according to pin tool calibration).
15. Add 8 μl Evans Blue working solution to Column 23 with dispenser A.
16. Shake for 3 s to mix the compound with the medium.
17. Replace the compound plates in the stackers of Plate∷Lift storage.
18. Replace the assay plates in incubator I189.
19. Incubate the assay plates at 37 °C in 5 % CO2 with 95 % humidity for 24 h.
20. Bring out the compounds plates from stackers of Plate∷Lift storage and cover them with sealing foil.
21. Re-store the 16 compound plates at −20 °C.
Day 6 Luminescence measurement
22. Prime and fill the tubes of dispenser A with 2 % DDM in PBS and confirm there are no air bubbles in any of the dispensing tubes (see Note 23).
-
23. Prime and fill the tubes of dispenser B with RLuc Assay Substrate Buffer (see Note 24).
Perform steps 24–29 by automation:
24. Add 5 μl of 2 % DDM in PBS to Columns 1–24 of assay plates with dispenser A.
25. Shake each assay plate for 5 s to mix well.
26. Incubate assay plates at 25 °C in the I30 incubator for 5 min.
27. Add 5 μl of RLuc Assay Substrate Buffer (final concentration, 5 μM) to Columns 1–24 with dispenser B.
28. Shake each assay plate for 5 s to mix well.
29. Incubate assay plates at room temperature in the stackers of Plate∷Lift storage for 60 min.
30. Take out the assay plates sequentially and read luminescence with the Perkin Elmer EnVision detector (100 ms integration time).
Repeat Subheadings 3.2.2 and 3.2.3 to perform primary HTS to test the remaining 16 compound plates in the 10,000 Diversity Set (see Note 25).
3.2.4 Data Analysis for Primary HTS
Analyze the HTS screen data with Genedata Screener Assay Analyzer Software (10.0.2 Standard). Normalize the luminescence intensity to the controls for each assay plate. Calculate the normalized activity score for each compound according to Table 2.
Calculate quality control parameters Z′ and S/B values as described in Subheading 3.1.4.
Incorporate compound IDs, activity scores, chemical structures, and SMILEs into a list. Sort the data by activity score from low to high. Export the data as an Excel file. Hit compounds are defined as those with activity scores ≤−50.
Table 2. Calculation of normalized activity scores for the P23H opsin clearance HTS.
| Control | Measurement | Activity score (%) | |
|---|---|---|---|
| Untreated control | Unaffected amount of P23H opsin reporter signal | 0 | |
| Evans Blue (200 μM) | Minimum amount of P23H opsin reporter signal | −100 | |
|
| |||
3.2.5 Hit Confirmation Screen
Generate compound plate maps on an Excel sheet. Based on the IDs of hit compounds, pull out the corresponding stock vials of hit compounds in the UC Compound Collection.
Transfer 20 μl of each hit compound (10 mM) into a well of a 384-well compound plate.
Repeat Subheadings 3.2.2 and 3.2.3 to perform the P23H clearance assay with freshly prepared compound plates. Each compound plate is tested in triplicate with 3 assay plates.
3.2.6 Data Analysis for Hit Confirmation Screen
Calculate the normalized activity score of each compound, Z′ and S/B values as described in Subheading 3.1.4.
Calculate the average and standard deviation of the three activity scores for each compound.
Sort the data set by average activity scores from low to high. Select compounds with average activity scores≤−50 as confirmed hits.
3.2.7 Dose-Response Screen
Generate compound plate maps for dose-response screen. “Cherry-pick” confirmed hit compounds from hit compound plates used in Subheading 3.2.5 and transfer 18 μl of each compound into a well in Column 1 or 11 of blank compound plates. Add 18 μl of DMSO to Columns 1–20 and perform serial twofold dilutions of each compound as described in Subheading 3.1.5, step 2.
Repeat Subheading 3.2.2 and 3.2.3 to perform the P23H clearance assay with freshly prepared compound plates. Test each compound plate in triplicate with 3 assay plates.
3.2.8 Data Analysis for Dose-Response Screen
Analyze data and generate a dose-response curve for each tested compound as described in Subheading 3.1.6. Set Sinf and S0 to −100 and 0, respectively.
Define the EC50 value as the concentration of the compound (μM) that causes an activity score of −50. Define the final hits as compounds with EC50 ≤ 20 μM.
Use Accelrys Pipeline Pilot software to incorporate their chemical structures, SMILEs, and predicted chemical properties in an Excel file (see Note 26).
4 Notes
- Protocol of DNA transfection with polyethylenimine (PEI).
- Prepare a 150 mM NaCl solution. Filter the solution through a 0.45 μm filter in a tissue culture hood to sterilize it. Store at 4 °C.
- Prepare a 1 mg/ml PEI solution. Add 33.75 mg PEI (Sigma-Aldrich) into 100 ml ddH2O in a 250 ml glass beaker, and stir vigorously after the addition of 0.5 ml of 12.1 M HCl until all powder is dissolved. Adjust pH to 7 by careful titration with 10 M NaOH. Filter the solution through a 0.45 μm filter in a tissue culture hood and collect 10 ml aliquots in 15 ml conical tubes. Store at −20 °C.
- Seed 0.5 × 106 Hek293 cells in a 6-well plate containing 2 ml of DMEM/10 % FBS medium 1 day before transfection and incubate plate at 37 °C in 5 % CO2 with 95 % humidity in an incubator, so that the cells reach >90 % confluence.
- Bring all reagents to room temperature prior to transfection.
- Add 1 μg DNA to 100 μl of 150 mM NaCl solution in a sterile 1.5 ml tube and then add 8 μl of PEI solution to 92 μl of 150 mM NaCl solution in another sterile 1.5 ml tube (8 μl of PEI is added per 1 μg DNA; if the amount of DNA is increased, increase also PEI). Vortex both tubes briefy.
- Add the 100 μl diluted PEI to the 100 μl DNA solution and gently mix by using a finger to tip the bottom of the tube. Let the mixture sit at room temperature for 15–30 min.
- Add the 200 μl mixture of PEI and DNA to cells in a well of a 6-well plate drop by drop. Shake the plate gently. Incubate the plate at 37 °C in 5 % CO2 with 95 % humidity.
- Protocol for the generation of a stable cell line expressing the P23H opsin-RLuc recombinant protein.
- After 2 days of transfection, subculture the cells from a 6-well plate in a 100 mm plate with DMEM + 10 % FBS + 500 μg/ml Zeocin (Life Technologies) for a week and incubate at 37 °C in 5 % CO2 with 95 % humidity.
- Count cells with a hemocytometer and seed 1,000 cells in a 100 mm plate in 15 ml of DMEM + 10 % FBS + 500 μg/ml Zeocin and incubate at 37 °C in 5 % CO2 with 95 % humidity.
- Positive cell clones should appear after 3–7 days. Pick up 10–20 clones with trypsin-immersed filter paper pieces (3 mm in diameter). Place each clone of cells in a well of a 24-well plate containing 1 ml DMEM + 10 % FBS + 500 μg/ml Zeocin solution and incubate 37 °C in 5 % CO2 with 95 % humidity.
- Select 10 clones after they reach 90 % confluence and subculture each clone in a well of a 6-well plate and a well of 96-well plate (20,000 cells/well). Cultures in the 6-well plate are for collection of a large amount of cells, whereas cultures in the 96-well plate are for positive clone confirmation. Incubate both plates at 37 °C in 5 % CO2 with 95 % humidity overnight.
- Test each clone for its RLuc activity by adding coelenterazine h (5 μM) (NanoLight Technology) and read luminescence intensity after 5 s with an integration time of 0.2 s. Retain only the RLuc positive clones (luminescence reading > 106 RLU) in the 6-well plate and subculture each clone in a 100 mm plate.
- When each RLuc positive clone reaches confluence in a 100 mm plate, detach and resuspend cells of each clone in 10 ml DMEM in a 15 ml conical tube. Transfer 1 ml of cells from each clone into a 1.5 ml tube and pellet both the 1 ml and 9 ml fractions of cells with a 300 × ℊ centrifugation. Remove medium from both tubes. Suspend the pellet from the 9 ml fraction in 0.9 ml DMEM + 10 % FBS + 10 % DMSO in a cryo-tube for storage. Freeze cells slowly in a Styrofoam box at −80 °C overnight. Then transfer the cryotubes to a liquid nitrogen tank for long-term storage.
- Wash the cell pellet of each clone from the 1 ml fraction with PBS and resuspend in 50 μl PBS. Confirm the expression of P23H-RLuc by immunoblots using the cell lysates in 50 μl PBS. Use mouse monoclonal B6-30 antibody recognizing the N-terminus of rod opsin for immunoblotting. Select the positive clone with highest expression level of P23H-RLuc for large scale culture and assay optimization for the P23H opsin clearance HTS.
Each well requires 25 μl of substrate buffer. For an HTS assay of 25,000 compounds (83 plates), we need 25 μl × 384 × 83 = 7 96,800 μl = 796.8 ml of substrate buffer plus ∼10 % dead volume (876.48 ml in total) in a 1 L glass bottle. Thus, we will add 876.48 ml × 4 % = 35.06 ml of Gal Screen Substrate and 841.42 ml of Gal Screen Buffer A to the solution in the 1 L glass bottle.
DMSO tolerance at concentrations ranging from 0.1 % to 1 % has been tested. The DMSO vehicle does not affect the assay performance within the test range.
The 25,000 and 10,000 Diversity Sets of the University of Cincinnati Compound Collection are representative subsets of its total number of ∼340,000 compounds. Different Diversity Sets contain different compound collections, while each Diversity Set was designed to uniformly fill up the “drug-like” space.
-
Protocol of pin tool calibration.
Liquid volumes and properties in an assay plate affect the volume of compounds transferred by a pin tool from a compound plate to an assay plate. Therefore, the pin tool needs to be calibrated under the same conditions used for the HTS experiments. For pin tool calibration prepare 10 mM Tamara stock solution dissolved in DMSO and use following plates: a 384-well, polypropylene, flat bottomed plate (BD Falcon) to represent a compound plate and a 384-well, polystyrene, black walled, ultra-clear bottomed plate (Greiner Bio-One) to represent an assay plate.- Prepare 10 ml of 50 μM TAMRA in DMSO by adding 50 μl of 10 mM TAMRA stock solution to 9.95 ml of DMSO in a 15 ml conical tube.
- Add 15 μl of 50 μM TAMRA to a well of a 96-well plate containing 285 μl cell plate medium and prepare a twofold dilution series: Add 150 μl of cell plate medium to Wells A2–A10 of the same plate. Mix the solution in Well A1 and transfer 150 μl to Well A2 and mix well. Transfer 150 μl of solution in Well A2 to Well A3 and repeat this transfer-and-mix step from well to well, until Well A10 has 300 μl of diluted TAMRA. Discard 150 μl of solution from Well A10.
- Add 25 μl of each dilution of TAMRA and cell plate medium in triplicate to a 384-well Greiner polystyrene plate (11 columns in total). Read fluorescence intensity at 530 nm/580 nm excitation/emission with a PerkinElmer Plate∷Vision detector (1 % light intensity and a 200 ms integration time). Generate a standard curve. A linear correlation of fluorescence intensity vs. TAMRA concentration should be obtained.
- Dispense 20 μl of 50 μM TAMRA solution into all wells of a 384-well Falcon polypropylene plate to represent a compound plate.
- Dispense 25 μl of cell plate medium into all wells of a 384-well Greiner polystyrene plate to represent an assay plate.
- Transfer compounds from the compound plate to assay plate using a 50 nl or 200 nl pin tool.
- Centrifuge assay plates at 450 × ℊ for 30 s to bring liquid down to the bottom of the assay plate. Read fluorescence of the assay plate at 530/580 nm with a PerkinElmer Plate∷Vision detector.
- Calculate the average concentration of TAMRA (xnM) in the assay plate by using the standard curve generated in step (d). Calculate the average pin tool transfer volume by the function: ynl =25 μl × (x) nM/50 μM. The average pin tool transfer volume is the calibrated pin tool volume and will be used to calculate the final concentration of a test compound. The final concentration of a test compound is zμM = (y × 10−3 μl × 10 × 103 μM)/(𝒶 μl) = 10y/𝒶 μM, where “ 𝒶 μl” is the total volume of liquid in each well of assay plate after compound treatment.
The β-Gal assay has been tested with cells seeded directly from thawed cryo-vials and also with cells revived and grown on 150 mm cell culture plates for one passage. The β-Gal assay data showed much less variation when the cells revived and grown for one passage were used.
For HTS screen of 25,000 Diversity Set (26,120 compounds in total), a total of 83 plates should be screened since each plate contains 320 compounds. The entire screen is divided into 2 cycles with each cycle further separated into two sets of automated experiments (21 or 20 plates per automated experiment). Each experiment needs 25 vials of cryo-frozen cells (4 × 106 cells/vial) that were revived and grown in 13 of 150 mm plates to reach confluence.
The timing for revived cells to reach confluence needs to be tested on-site before the HTS to ensure that enough cells are obtained at a scheduled time.
To prevent cells from dying or settling down, an autoclaved stir bar can be placed into the cell suspension bottle which is bathed in water at 37 °C in a glass beaker. Stabilize the water temperature with a heated plate and magnetic stirring.
The cell seeding number needs to be optimized during assay development. Cell numbers were tested from 1,000 to 10,000 cells/well under four conditions. Results showed that 5,000 cells/well provided the highest assay quality as suggested by Z′ and S/B ratio values.
Each assay plate contains a lid. Thus, in the HTS program, assay plate lids are removed and replaced before and after each dispensing step.
Each 384-well compound plate has 320 compounds (10 mM in DMSO) in Column 1–20 (Fig. 2). A compound plate map contains information about compound IDs, compound position in the plate, and the plate bar code.
It is important to ensure that the bar code of a compound plate and the bar code of its corresponding assay plate match and are recorded correctly.
Do not open the door of incubator I189 after the end of Day 6, because the positive control 9-cis-retinal regenerates P23H isorhodopsin which is light-sensitive. Any leakage of light after treatment with 9-cis-retinal and before substrate addition will compromise assay quality.
If an obvious plate pattern is observed, e.g., very high or very low activity scores show up in several adjacent rows and/or columns of an assay plate, record the plate bar codes of the assay plate and the corresponding compound plate. Retest the compound plate at the end of the primary HTS.
A hit confirmation screen (single dose of each compound tested in triplicate) normally is performed after a single-point HTS to reduce the number of hits to be tested in the dose-response screen. However, the primary HTS of P23H translocation yielded only 16 hit compounds. Due to this low hit number, a dose-response screen was performed directly after the primary HTS screen.
Sinf is defined as infinite activity, namely, the fitted activity score at infinite test compound concentration, whereas S0 is defined as zero activity, i.e., the fitted activity score at zero concentration. The Smart Fit Model automatically changes parameters of the Hill equation to optimize the fitting curve.
Due to cytotoxicity, atypical dose-response curves can be observed (Fig. 3) such as an increase followed by a decrease of activity score upon increasing dosage of a compound. Decreased activity scores due to cytotoxicity must be masked before curve fitting, as they will affect the dose-response curve.
The final hit compounds identified from HTS must be further validated by orthogonal screens and other assays to confirm their activity in correcting the P23H opsin localization. A neighboring search for selected hit compounds and activity tests of these similar compounds will enhance understanding of the structure-activity relationship (SAR) of the pharmacophore that is critical for lead optimization.
Each cryo-vial contains confluent cells harvested from a 150 mm plate.
For one cycle of primary HTS with 16 × 384-well assay plates, a total of 16 × 400 × 32 μl = 204,800 μl of cell suspension is expected to be dispensed. About 30 ml of dead volume will be needed for the dispenser. Therefore, a total of 250 ml of cell suspension should be prepared for seeding 16 assay plates.
Air bubbles are readily generated in DDM solutions. Thus, the 10 % DDM stock should be prepared at least a day before use, so that bubbles will have disappeared before the working solution is prepared. It also is important to make certain that there are no air bubbles in the dispenser tubes. Therefore, if air bubbles are observed, prime the dispenser with more DDM solution until they are washed out of the tubes.
Add 5 μl RLuc Assay Substrate Buffer working solution to each well. For 16 × 384-well plates (5 μl RLuc Assay Substrate Buffer per well) 16 × 400 × 5 μl = 32,000 μl = 32 ml plus about 6 ml dead volume, i.e., a total of 38 ml ViviRen working solution needs to be prepared.
Cells for the 2nd cycle of primary HTS should be revived on Day 4, so that cells are confluent on the 150 mm plates and ready for cell seeding immediately after the completion of the 1st primary HTS on Day 7.
RLuc is used as a reporter for P23H opsin clearance HTS. Compounds interfering with RLuc activity would be identified as hits as well [31]. Follow-up with a counter screen testing RLuc activity and a hit validation assay (such as immunoblotting) should be carried out to remove false positives.
Fig. 3.
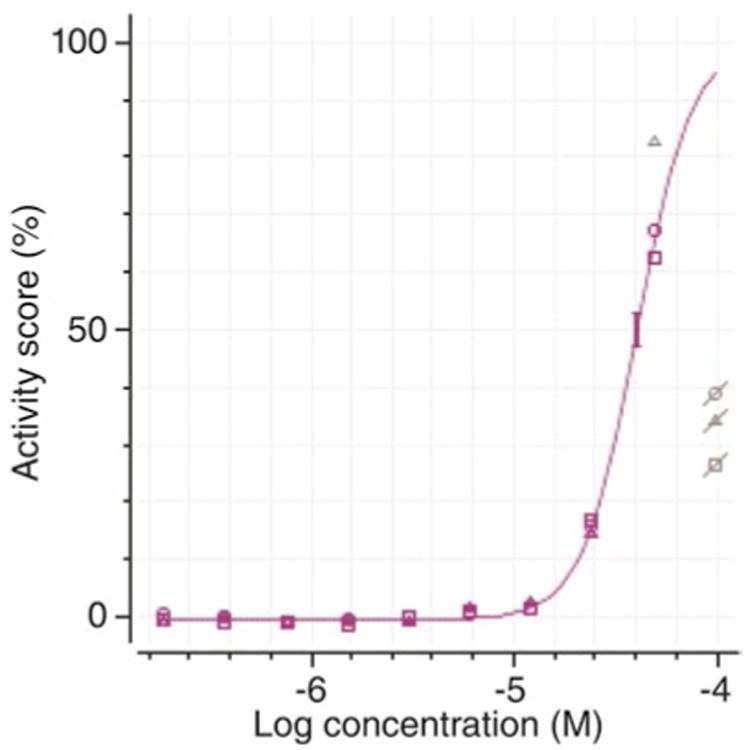
Dose-response curve of a hit compound fitted by the Hill equation. Data points with reduced activity affected by cytotoxicity are masked (gray) and not included for curve fitting
References
- 1.Persidis A. High-throughput screening. Advances in robotics and miniturization continue to accelerate drug lead identification. Nat Biotechnol. 1998;16:488–489. doi: 10.1038/nbt0598-488. [DOI] [PubMed] [Google Scholar]
- 2.Carnero A. High throughput screening in drug discovery. Clin Transl Oncol. 2006;8:482–490. doi: 10.1007/s12094-006-0048-2. [DOI] [PubMed] [Google Scholar]
- 3.Macarron R, Banks MN, Bojanic D, et al. Impact of high-throughput screening in biomedical research. Nat Rev Drug Discov. 2011;10:188–195. doi: 10.1038/nrd3368. [DOI] [PubMed] [Google Scholar]
- 4.Zoete V, Grosdidier A, Michielin O. Docking, virtual high throughput screening and in silico fragment-based drug design. J Cell Mol Med. 2009;13:238–248. doi: 10.1111/j.1582-4934.2008.00665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caliandro R, Belviso DB, Aresta BM, et al. Protein crystallography and fragment- based drug design. Future Med Chem. 2013;5:1121–1140. doi: 10.4155/fmc.13.84. [DOI] [PubMed] [Google Scholar]
- 6.Hoelder S, Clarke PA, Workman P. Discovery of small molecule cancer drugs: successes, challenges and opportunities. Mol Oncol. 2012;6:155–176. doi: 10.1016/j.molonc.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daiger SP, Bowne SJ, Sullivan LS. Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol. 2007;125:151–158. doi: 10.1001/archopht.125.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 9.Gorbatyuk MS, Gorbatyuk OS, LaVail MM, et al. Functional rescue of P23H rhodopsin photoreceptors by gene delivery. Adv Exp Med Biol. 2012;723:191–197. doi: 10.1007/978-1-4614-0631-0_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gorbatyuk MS, Knox T, LaVail MM, et al. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc Natl Acad Sci U S A. 2010;107:5961–5966. doi: 10.1073/pnas.0911991107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aguila M, Bevilacqua D, McCulley C, et al. Hsp90 inhibition protects against inherited retinal degeneration. Hum Mol Genet. 2014;23:2164–2175. doi: 10.1093/hmg/ddt613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fernandez-Sanchez L, Lax P, Esquiva G, et al. Safranal, a saffron constituent, attenuates retinal degeneration in P23H rats. PLoS One. 2012;7:e43074. doi: 10.1371/journal.pone.0043074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vasireddy V, Chavali VR, Joseph VT, et al. Rescue of photoreceptor degeneration by curcumin in transgenic rats with P23H rhodopsin mutation. PLoS One. 2011;6:e21193. doi: 10.1371/journal.pone.0021193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fernandez-Sanchez L, Lax P, Pinilla I, et al. Tauroursodeoxycholic acid prevents retinal degeneration in transgenic P23H rats. Invest Ophthalmol Vis Sci. 2011;52:4998–5008. doi: 10.1167/iovs.11-7496. [DOI] [PubMed] [Google Scholar]
- 15.Rossmiller B, Mao H, Lewin AS. Gene therapy in animal models of autosomal dominant retinitis pigmentosa. Mol Vis. 2012;18:2479–2496. [PMC free article] [PubMed] [Google Scholar]
- 16.Wert KJ, Davis RJ, Sancho-Pelluz J, et al. Gene therapy provides long-term visual function in a pre-clinical model of retinitis pigmentosa. Hum Mol Genet. 2013;22:558–567. doi: 10.1093/hmg/dds466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petrs-Silva H, Linden R. Advances in gene therapy technologies to treat retinitis pigmentosa. Clin Ophthalmol. 2014;8:127–136. doi: 10.2147/OPTH.S38041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaushal S. Effect of rapamycin on the fate of P23H opsin associated with retinitis pigmentosa (an American Ophthalmological Society thesis) Trans Am Ophthalmol Soc. 2006;104:517–529. [PMC free article] [PubMed] [Google Scholar]
- 19.Noorwez SM, Malhotra R, McDowell JH, et al. Retinoids assist the cellular folding of the autosomal dominant retinitis pigmentosa opsin mutant P23H. J Biol Chem. 2004;279:16278–16284. doi: 10.1074/jbc.M312101200. [DOI] [PubMed] [Google Scholar]
- 20.Noorwez SM, Kuksa V, Imanishi Y, et al. Pharmacological chaperone-mediated in vivo folding and stabilization of the P23H-opsin mutant associated with autosomal dominant retinitis pigmentosa. J Biol Chem. 2003;278:14442–14450. doi: 10.1074/jbc.M300087200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Y, Jastrzebska B, Cao P, et al. Inherent instability of the retinitis pigmentosa P23H mutant opsin. J Biol Chem. 2014;289:9288–9303. doi: 10.1074/jbc.M114.551713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sung CH, Schneider BG, Agarwal N, et al. Functional heterogeneity of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991;88:8840–8844. doi: 10.1073/pnas.88.19.8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaushal S, Khorana HG. Structure and function in rhodopsin. 7. Point mutations associated with autosomal dominant retinitis pigmentosa. Biochemistry. 1994;33:6121–6128. doi: 10.1021/bi00186a011. [DOI] [PubMed] [Google Scholar]
- 24.Lin JH, Li H, Yasumura D, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mendes HF, van der Spuy J, Chapple JP, et al. Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol Med. 2005;11:177–185. doi: 10.1016/j.molmed.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 26.Saliba RS, Munro PM, Luthert PJ, et al. The cellular fate of mutant rhodopsin: quality control, degradation and aggresome formation. J Cell Sci. 2002;115:2907–2918. doi: 10.1242/jcs.115.14.2907. [DOI] [PubMed] [Google Scholar]
- 27.Sakami S, Kolesnikov AV, Kefalov VJ, et al. P23H opsin knock-in mice reveal a novel step in retinal rod disc morphogenesis. Hum Mol Genet. 2014;23:1723–1741. doi: 10.1093/hmg/ddt561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakami S, Maeda T, Bereta G, et al. Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J Biol Chem. 2011;286:10551–10567. doi: 10.1074/jbc.M110.209759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haeri M, Knox BE. Rhodopsin mutant P23H destabilizes rod photoreceptor disk membranes. PLoS One. 2012;7:e30101. doi: 10.1371/journal.pone.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 31.Auld DS, Southall NT, Jadhav A, et al. Characterization of chemical libraries for luciferase inhibitory activity. J Med Chem. 2008;51:2372–2386. doi: 10.1021/jm701302v. [DOI] [PubMed] [Google Scholar]