

Abstract
Increased cell apoptosis in chondrocytes is a feature of degenerative cartilage. Astragaloside IV (AST) has been proven to possess an antiarthritic effect by preventing interleukin (IL)-1β-induced cartilage damage. However, the role of AST on chondrocyte apoptosis and its underlying mechanism remains unknown. In the present study, degenerative chondrocytes isolated from patients with osteoarthritis (OA) were subjected to AST and IL-1β treatment. Results indicated that AST protected against chondrocyte apoptosis induced by IL-1β. Western blotting indicated that AST increased the protein expression of LC3-II/I and decreased P62/SQSTM1 expression, which suggested that AST upregulated autophagy activity in chondrocytes. Fluorescent protein GFP-LC3 analysis and transmission electron microscopy observation confirmed that autophagy was promoted by AST. In contrast, after autophagy inhibited by 3-methyladenine, chondrocyte apoptosis was further increased under IL-1β treatment. Ultimately, rapamycin was used as a positive control, whose results confirmed that rapamycin-mediated autophagy also decreased chondrocyte apoptosis induced by IL-1β. In conclusion, these results suggested that AST-mediated autophagy serves an anti-apoptotic role in chondrocytes, which may aid the development of novel therapeutic approaches for OA treatment.
Keywords: astragaloside IV, chondrocyte, interleukin-1β, apoptosis, autophagy
Introduction
Osteoarthritis (OA) is characterized by a gradual degradation of articular cartilage and pain, resulting in significant disability in patients, which consumes a significant amount of healthcare resources (1). Articular cartilage consists of abundant extracellular matrix that is synthesized by resident cells, named chondrocytes. Increased apoptotic chondrocyte death has been detected in OA cartilage, which leads to the matrix degradation and the loss of the lubricant and swelling properties to the joint (2). Interleukin (IL)-1β is considered to be the most important inflammatory cytokine involved in multiple pathological processes of OA. Patients with OA have an elevated level of IL-1β in both synovial fluid and cartilage, and chondrocytes subjected to IL-1β tend to induce apoptosis (3).
Astragalus membranaceus is a widely used in traditional Chinese medicine, particularly for autoimmune diseases (4). The extracts of Astragalus membranaceus (Fisch) contain various active ingredients, including saponins, polysacharides and flavonoids. Astragaloside IV (AST) is a natural saponin purified from Astragalus, which exerts numerous therapeutic effects. A previous study has indicated that AST may protect against IL-1β-induced joint inflammation and cartilage damage in rat adjuvant-induced arthritis (5). However, to date, the role and mechanism of AST on the IL-1β-induced chondrocyte apoptosis remain unclear.
Autophagy is a self-degradation process for balancing sources of energy in response to cell stress (6). Autophagy has been proven to be important for cartilage homeostasis (7). Autophagy is increased in OA chondrocytes and cartilage, particularly during the early stage of the degenerative phase, which acts as an adaptive response to protect chondrocyte from sublethal conditions (8). Sasaki et al (9) reported that autophagy activation could inhibit IL-1β-induced apoptosis in chondrocytes. Furthermore, AST demonstrated beneficial effects against mitochondrial dysfunction through promotion of autophagy in rat vascular smooth muscle cells (10). Thus, the authors hypothesized that AST may serve a protective role in chondrocytes against IL-1β-induced apoptosis, and its potential mechanisms may be responsible to autophagy activation.
Materials and methods
Primary chondrocyte isolation and culture
Primary articular chondrocytes were isolated from 7 patients with OA undergoing total knee replacement surgery. Written informed consent was obtained from all tissue donors and the present study approach was approved by Ethics Committee of Beijing University of Chinese Medicine (Beijing, China) before surgery. The human degenerative chondrocytes were isolated and cultured similarly as described previously (11). In brief, articular cartilage was first separated from harvested joint tissues microscopically according to their macroscopic morphologies. Following washing by PBS buffer, cartilage was dissected and digested with by 0.25% trypsin solution and 0.2% type II collagenase (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) at 37°C for 5 h. Chondrocytes were isolated by a 200 µm filter, and resuspended in Dulbecco's modified Eagle's medium/Ham's F-12 (DMEM/F12) medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10% fetal bovine serum (FBS; Invitrogen; Thermo Fisher Scientific, Inc.). Chondrocytes were monolayer cultured at 37°C under 5% CO2 and 95% oxygen in humidified air; cells between passages 1 and 3 were used for subsequent experiments.
Cell treatment
AST (Beijing Solarbio Science & Technology Co., Ltd., Beijing, China) was dissolved in dimethyl sulfoxide (DMSO) to produce a stock solution, the final concentration of AST used in the current study was 50 µg/ml. Differentiated chondrocytes were subjected to DMEM/F12 containing 1% FBS for 12 h prior to exposure to various treatment conditions. Subsequently, cells were pre-incubated with AST (50 µg/ml) for 2 h and then incubated with 10 ng/ml IL-1β for a further 24 h. In addition, chondrocytes were transfected with adenovirus containing GFP-LC3 to monitor the autophagic level changes. Finally, 3-methyladenine (3MA; 5 mM) and rapamycin (100 nM) were used as a chemical inhibitor or activator for autophagy, which were applied 2 h prior to further treatment. Chondrocytes treated with DMEM/F12 medium with 10% FBS was considered as the control.
Cell Counting kit (CCK)-8 measurement
Chondrocyte viability was detected by CCK-8 (Promega Corporation, Madison, WI, USA). Briefly, chondrocytes were seeded into a 96-well plate with concentration of 1×104 cells/well overnight prior to treatment. Following exposeure to IL-1β or AST, 10 µl CCK-8 solution was added to each well and incubated for 2 h at 37°C. Optical density was determined at 450 nm using a microplate reader (Thermo Fisher Scientific, Inc.). The cellular viability was defined as a percentage of the control group.
Flow cytometry analysis
The apoptotic incidence was evaluated by the Annexin V-phycoerythrin (PE)/propidium iodide (PI) apoptosis detection kit (BD Biosciences, Franklin Lakes, NJ, USA) according to the manufacturer's instructions. Briefly, treated chondrocytes were collected and washed by PBS buffer, then incubated with 5 µl Annexin V-PE and 5 µl PI for 30 min at room temperature in the dark. Finally, Ending buffer was added, and cell apoptotic rate was analyzed using a flow cytometer (BD Biosciences).
Western blot analysis
Whole cells were lysed using radioimmunoprecipitation assay lysis buffer (Beyotime Institute of Biotechnology, Haimen, China) on ice. The protein concentration was measured using a bicinchoninic acid protein assay kit (Beyotime Institute of Biotechnology). Equal amounts of protein (40 µg) from each sample were subjected to 10–15% SDS-PAGE and transferred to polyvinylidene difluoride membranes. The membranes were blocked with 5% bovine serum albumin and then incubated with primary antibody against cleaved caspase-3 (dilution, 1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA; cat no. 9661), cleaved caspase-9 (dilution, 1:1,000; Cell Signaling Technology, Inc.; cat no. 9501), cleaved poly (ADP-ribose) polymerase (PARP; dilution, 1:1,000; Cell Signaling Technology, Inc.; cat no. 5625), LC-3 (dilution, 1:500; Abcam, Cambridge, UK; cat no. ab48394), P62/SQSTM1 (dilution, 1:500; Abcam; cat no. ab91526) and β-actin (dilution, 1:5,000; Beyotime Institute of Biotechnology; cat no. AF0003) at 4°C overnight. The next day, after washing with TBS with 1% Tween-20 (TBST), membranes were incubated with goat anti-mouse or rabbit HRP-conjugated secondary antibodies (dilution, 1:1,000; Beyotime Institute of Biotechnology; cat nos. A0216 and A0208) for 1 h at 37°C. Finally, the membranes were visualized by an enhanced chemiluminescence detection kit (EMD Millipore, Billerica, MA, USA). The protein expression levels were detected by Quantity One software (version, 4.62; Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Caspase activity measurement
Cell apoptosis was also assayed by a caspase-3/9 activity assay kit (Beyotime Institute of Biotechnology; cat no. C1115). In brief, whole cell protein was lysed and extracted as aforementioned. Then, 100 µg protein from each sample was incubated with 50 µl reaction buffer and 5 µl Ac-DEVD-pNA (4 mM) substrate at 37°C for 2 h. The absorbance at 405 nm was determined using a microplate reader (Thermo Fisher Scientific, Inc.).
Confocal microscopic analysis
To visualize the autophagosome, cells were transfected with adenovirus containing GFP-LC3 (Hanbio Biotechnology Co., Ltd., Shanghai, China). Briefly, NP cells were incubated in complete medium with the adenovirus at a multiplicity of infection of 50 for 6 h, then replace fresh medium for another 24 h. The transfected cells were used for subsequent experiments. Autophagy was evaluated by analyzing the number of green fluorescent puncta of autophagosomes under laser confocal microscopy.
Transmission electron microscopy
The treated cells were collected and fixed with 2.5% glutaraldehyde overnight, subsequently fixed with 1% osmium tetroxide for 1 h. The fixed specimens were dehydrated and embedded in tetraoctadecylammonium bromide (TAAB) Epon resin (TAAB Laboratories Equipment Ltd., Aldermaston, UK). The samples were cut into 60 nm ultrathin sections and stained with uranyl acetate and lead citrate. A transmission electron micoroscope (Hitachi-7500, Hitachi, Ltd., Tokyo, Japan) was used for observing the cellular ultrastructure at an accelerating voltage of 100 kV.
Statistical analysis
Quantitative data are presented as the mean ± standard deviation. Statistical analyses were performed using Student's t test or one-way analysis of variance by SPSS software (version, 19.0; IBM SPSS, Armonk, NY, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
AST maintains chondrocyte viability under IL-1β treatment
CCK-8 analysis was performed to evaluate the effect of AST on chondrocyte viability under IL-1β treatment. Results demonstrated that the cell viability was significantly decreased to (39.3±5.3%) following stimulation with 10 ng/ml IL-1β. However, pretreatment with AST (50 µg/ml) dramatically increased the cell viability to (67.7±8.1%). Otherwise, the viabilities of the chondrocytes were not altered significantly by AST or 0.1% DMSO alone (Fig. 1). Therefore, AST demonstrated a protective effect on maintaining chondrocyte viability under IL-1β treatment.
Figure 1.
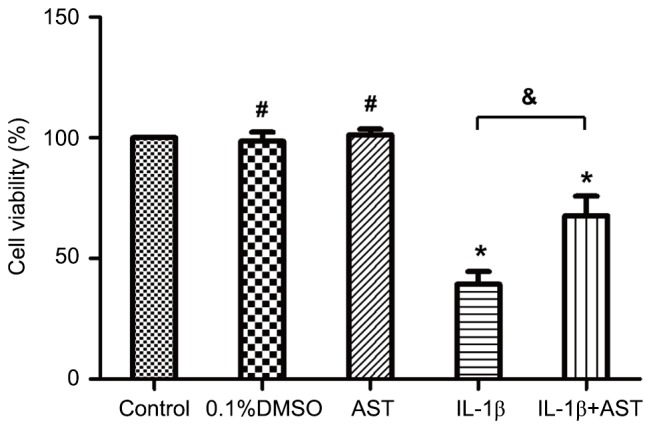
AST maintains chondrocyte viability under IL-1β treatment. Cell viability was detected by Cell Counting kit-8 assay. Chondrocytes were treated with 0.01% DMSO, 10 ng/ml IL-1β or 50 µg/ml AST. Chondrocytes treated with complete medium was considered as a control. The cellular viability was defined as a percentage of the control group. Data are presented as mean ± standard deviation from three independent experiments. *P<0.05 vs. control group; #P>0.05 vs. control group; &P<0.05 as indicated. AST, Astragaloside IV; IL, interleukin; DMSO, dimethylsulfoxide.
AST protects against IL-1β-induced chondrocyte apoptosis
Annexin V-fluorescein isothiocyanate/propidium iodide double staining by flow cytometry analysis was performed to test the effect of AST on IL-1β-induced cell apoptosis. Results indicated that the apoptotic ratio in the control group was (4.3±1.6%), and IL-1β treatment led to a significant increase in cell apoptosis to (28.7±4.8%). However, co-treatment of AST and IL-1β presented an anti-apoptotic effect that decreased the apoptotic ratio to (15.3±3.1%; Fig. 2A). Western blotting further confirmed that IL-1β alone strongly increased the protein levels of cleaved caspase-3/9 and cleaved PARP, while AST under treatment with IL-1β inhibited the increase in these three pro-apoptotic protein expressions (Fig. 2B). These results indicated that IL-1β-induced apoptosis of chondrocytes may be suppressed by AST.
Figure 2.

AST protects against IL-1β-induced chondrocyte apoptosis. (A) Cell apoptosis was detected by flow cytometric analysis. Chondrocytes were treated with 10 ng/ml IL-1β or 50 µg/ml AST, and which treated with complete medium was considered as a control. The apoptosis incidence was calculated by the percentage of early apoptotic (Annexin V+/PI-) cells plus the percentage of late apoptotic (Annexin V+/PI+) cells. (B) The protein expressions of cleaved-caspase3/9 and cleaved PARP were measured by western blot analysis. Data are presented as mean ± standard deviation from three independent experiments. *P<0.05 vs. control group; &P<0.05 as indicated. AST, Astragaloside IV; IL, interleukin; PI, propidium iodide; PARP, poly (ADP-ribose) polymerase; FITC, fluorescein isothiocyanate.
AST stimulates autophagic flux in chondrocytes
To investigate whether autophagy was activated in chondrocytes stimulated by AST, western blot analysis was performed to detect the protein levels of LC3 and P62/SQSTM1, which are markers of autophagy. Results demonstrated that the expression of LC3-II/I was downregulated significantly in chondrocytes with treatment of IL-1β, in contrast, expression of P62/SQSTM1 was obviously upregulated. However, AST presented promoting effect on autophagic flux in chondrocytes, which reversed the expression of LC-II/I and P62/SQSTM1 (Fig. 3A). The autophagy activation was further confirmed by monitoring the subcellular localization of fluorescent GFP-LC3 puncta. Confocal microscopic analysis indicated that AST significantly increased the fluorescent puncta in cytoplasm, and punctate green fluorescence could hardly be observed with IL-1β treatment (Fig. 3B). Furthermore, TEM observation demonstrated that the number of double-membrane autophagosomes was much greater under AST treatment (Fig. 3C). All the evidence suggests that AST stimulated autophagy response in the chondrocyte.
Figure 3.

AST stimulates autophagic flux in chondrocytes. Chondrocytes were treated with 10 ng/ml IL-1β or 50 µg/ml AST, and those treated with complete medium was considered as a control. (A) Western blot analysis was performed for the protein expressions of LC3 and P62. The ratio of LC3-II/I was quantified. (B) Chondrocytes were transfected with adenovirus containing GFP-LC3. Confocal microscopy was observed for the formation and distribution of GFP-LC3 punctate. Magnification, ×800. (C) Morphological observation of autophagy under transmission electron microscope. Magnification, ×2000. Data are presented as mean ± standard deviation from three independent experiments. *P<0.05 vs. control group; &P<0.05 as indicated. AST, Astragaloside IV; IL, interleukin.
AST inhibits chondrocyte apoptosis by promoting autophagy
To investigate the interaction between autophagy and apoptosis in chondrocytes under AST treatment, 3MA was used as a chemical inhibitor of autophagy. 3MA treatment significantly attenuated the LC3-II expression that indicated 3MA decreased the autophagy incidence by AST treatment (Fig. 4A). Flow cytometric analysis revealed that the suppressed apoptosis ratio by AST was obvious increased under 3MA treatment (Fig. 4B). Colorimetric assay revealed that, under 3MA treatment, the activities of caspase-3 and caspase-9 increased to 178 and 258%, respectively, compared to the control group (Fig. 4C). Ultimately, to verify the exact mechanism of autophagy in chondrocytes, rapamycin was used to activate autophagy. The flow analysis presented similar results with those treated by AST. Rapamycin also reversed the increased apoptotic rate induced by IL-1β (Fig. 4D). These results suggested that activation of autophagy could downregulate the IL-1β-induced apoptosis in chondrocytes under AST treatment.
Figure 4.

AST inhibits chondrocyte apoptosis by promoting autophagy. 3MA was used to inhibit autophagy activation. (A) Western blot analysis was performed for the protein expressions of LC3-I and LC3-II. (B) Cell apoptosis was detected by flow cytometric analysis. Chondrocytes were treated with IL-1β and AST or 3MA (5 mM), and which treated with complete medium was considered as control. (C) Colorimetric activity assay for caspase-3, and −9. Cell treatment scheme is the same as in B. The activation of caspase-3/−9 was defined as a percentage of the control group. (D) Chondrocytes were treated with 10 ng/ml IL-1β or with 100 nM rapamycin, and then cell apoptosis was detected by flow cytometric analysis. Data are presented as mean ± standard deviation from three independent experiments. *P<0.05 vs. control group; #P<0.05, &P<0.05 as indicated. AST, Astragaloside IV; 3MA, 3-methyladenine; FITC, fluorescein isothiocyanate.
Discussion
To the best of the authors' knowledge, the present study was the first to demonstrate that AST prevents against IL-1β-induced apoptosis of chondrocytes via autophagy activation. IL-1β was proven to reduce cell viability and increase apoptosis in chondrocytes, however, AST significantly attenuated the IL-1β-induced apoptosis and maintain cell viability. Furthermore, AST treatment induced autophagic flux in chondrocytes, and, after inhibiting this autophagy, the suppressed apoptosis ratio by AST was reversed.
Increased expression of proinflammatory cytokines in cartilage, synovial membrane and subchodral bone is related to the pathological process in the OA joint (12). IL-1β is one of the most important cytokines responsible for chondrocyte apoptosis and cartilage matrix degradation. Therefore, IL-1β is considered as the potential target in OA therapy. AST is a characteristic active saponin compound present in Astragalus, which presents an immunomodulating effect on lymphocyte apoptosis (13) and inflammatory cytokine production (14). IL-1β was used to induce chondrocyte apoptosis in the presence or absence of AST in the present study. Results demonstrated that AST possessed the ability to reverse the inhibition of cell proliferation and enhancement of cell apoptosis induced by IL-1β. These findings suggested that AST could be treated as a candidate for the antiarthritic treatment. However, the underlying mechanism of anti-apoptotic effect by AST needs to be further investigated.
The role of autophagy on the pathogenesis of OA is a research hotspot during recent years. During the early stage of OA, autophagy activation has been indicated to protect from limiting catabolic degradation (9); while in late OA, the level of autophagy decreases in chondrocytes, which is associated with the increase of apoptosis of chondrocytes (15). Previous studies have suggested that autophagy serves a prominent influence on chondrocyte survival. In aged mice, the number of autophagic vesicles decreased, accompanied by reduced expression of LC3 and increased expression of PARP, a marker of apoptosis (16). In addition, in neural cells, AST can concomitantly inhibit multiple cell death pathways following oxygen and glucose deprivation (17). Therefore, the authors speculated that the anti-apoptotic role of AST in chondrocytes is also based on autophagy activation. The present results indicated that IL-1β decreased the expression ratio of LC-3II/I and increased the expression of P62, suggesting IL-1β inhibited autophagy in chondrocytes. However, AST treatment may be activating autophagy, which reverses the expression of LC-3II/I and P62. This was further confirmed by fluorescent microscopy and transmission electron microscopy observation. All the evidence indicated that AST had the effect of promoting autophagy in chondrocytes.
The effect of autophagy on apoptosis is varying depending on the cell type and different stimulations (18). To further investigate the role of autophagy by AST on the apoptosis under IL-1β stimulation, chondrocytes were pre-treated with MA to inhibit autophagy. Results showed that 3MA significantly reduced the level of LC3-II/I that was originally boosted by AST. On the contrary, downregulation of autophagy via 3MA inhibited the anti-apoptotic effect of AST in chondrocytes. This result was in accordance with Huang et al (19) who demonstrated that leptin promotes apoptosis via inhibition of autophagy in chondrocytes during osteoarthritis pathogenesis. Ultimately, rapamycin was used as a positive control to investigate the potency of autophagy in chondrocytes. Results demonstrated that rapamycin reversed the increased apoptotic rate induced by IL-1β, which has a similar effect to AST. Thus, these findings indicated that regulation of autophagy activation may be the underlying mechanism of AST to reduce the apoptosis of chondrocytes under IL-1β stimulation.
The current study also has some limitations. First, chondrocytes were monolayer cultured; it is easy to lose cell phenotype during cell propagation (20). Thus, the authors used the passage 1–3 chondrocytes to avoid and decrease this phenomenon. Secondly, though sufficient results were acquired to elucidate the role of autophagy in the regulation of apoptosis in chondrocytes, the underlying intracellular signaling pathway for regulation of autophagy needs to be further investigated.
In conclusion, the present study demonstrated that inhibition of autophagy enhanced IL-1β-induced chondrocyte apoptosis, while induction of autophagy by AST prevented chondrocyte survival. The findings suggested that autophagy serves an important role in human chondrocytes to protect them from inflammatory stress, and its natural agonist, AST may be a novel candidate for the treatment of OA. In a future investigation, the potential mechanism of AST to regulate autophagy in chondrocytes is required.
Acknowledgements
This study was supported by a grant from the National Natural Science Foundation of China (grant no. 81473800).
References
- 1.Xie F, Kovic B, Jin X, He X, Wang M, Silvestre C. Economic and humanistic burden of osteoarthritis: A systematic review of large sample studies. Pharmacoeconomics. 2016;34:1087–1100. doi: 10.1007/s40273-016-0424-x. [DOI] [PubMed] [Google Scholar]
- 2.Zamli Z, Sharif M. Chondrocyte apoptosis: A cause or consequence of osteoarthritis? Int J Rheum Dis. 2011;14:159–166. doi: 10.1111/j.1756-185X.2011.01618.x. [DOI] [PubMed] [Google Scholar]
- 3.Wojdasiewicz P, Poniatowski ŁA, Szukiewicz D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm. 2014;2014:561459. doi: 10.1155/2014/561459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He Y, Du M, Gao Y, Liu H, Wang H, Wu X, Wang Z. Astragaloside IV attenuates experimental autoimmune encephalomyelitis of mice by counteracting oxidative stress at multiple levels. PLoS One. 2013;8:e76495. doi: 10.1371/journal.pone.0076495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang B, Chen MZ. Astragaloside IV possesses antiarthritic effect by preventing interleukin 1β-induced joint inflammation and cartilage damage. Arch Pharm Res. 2014;37:793–802. doi: 10.1007/s12272-014-0336-2. [DOI] [PubMed] [Google Scholar]
- 6.Rockel JS, Kapoor M. Autophagy: Controlling cell fate in rheumatic diseases. Nat Rev Rheumatol. 2016;12:517–531. doi: 10.1038/nrrheum.2016.92. [DOI] [PubMed] [Google Scholar]
- 7.Li YS, Zhang FJ, Zeng C, Luo W, Xiao WF, Gao SG, Lei GH. Autophagy in osteoarthritis. Joint Bone Spine. 2016;83:143–148. doi: 10.1016/j.jbspin.2015.06.009. [DOI] [PubMed] [Google Scholar]
- 8.Almonte-Becerril M, Navarro-Garcia F, Gonzalez-Robles A, Vega-Lopez MA, Lavalle C, Kouri JB. Cell death of chondrocytes is a combination between apoptosis and autophagy during the pathogenesis of Osteoarthritis within an experimental model. Apoptosis. 2010;15:631–638. doi: 10.1007/s10495-010-0458-z. [DOI] [PubMed] [Google Scholar]
- 9.Sasaki H, Takayama K, Matsushita T, Ishida K, Kubo S, Matsumoto T, Fujita N, Oka S, Kurosaka M, Kuroda R. Autophagy modulates osteoarthritis-related gene expression in human chondrocytes. Arthritis Rheum. 2012;64:1920–1928. doi: 10.1002/art.34323. [DOI] [PubMed] [Google Scholar]
- 10.Lu Y, Li S, Wu H, Bian Z, Xu J, Gu C, Chen X, Yang D. Beneficial effects of astragaloside IV against angiotensin II-induced mitochondrial dysfunction in rat vascular smooth muscle cells. Int J Mol Med. 2015;36:1223–1232. doi: 10.3892/ijmm.2015.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu J, Zhang C. In vitro isolation and cultivation of human chondrocytes for osteoarthritis renovation. In Vitro Cell Dev Biol Anim. 2014;50:623–629. doi: 10.1007/s11626-014-9742-5. [DOI] [PubMed] [Google Scholar]
- 12.Philp AM, Davis ET, Jones SW. Developing anti-inflammatory therapeutics for patients with osteoarthritis. Rheumatology (Oxford) 2017;56:869–881. doi: 10.1093/rheumatology/kew278. [DOI] [PubMed] [Google Scholar]
- 13.Liu R, Jiang H, Tian Y, Zhao W, Wu X. Astragaloside IV protects against polymicrobial sepsis through inhibiting inflammatory response and apoptosis of lymphocytes. J Surg Res. 2016;200:315–323. doi: 10.1016/j.jss.2015.08.024. [DOI] [PubMed] [Google Scholar]
- 14.Cho WC, Leung KN. In vitro and in vivo immunomodulating and immunorestorative effects of Astragalus membranaceus. J Ethnopharmacol. 2007;113:132–141. doi: 10.1016/j.jep.2007.05.020. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y, Vasheghani F, Li YH, Blati M, Simeone K, Fahmi H, Lussier B, Roughley P, Lagares D, Pelletier JP, et al. Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann Rheum Dis. 2015;74:1432–1440. doi: 10.1136/annrheumdis-2013-204599. [DOI] [PubMed] [Google Scholar]
- 16.Caramés B, Olmer M, Kiosses WB, Lotz MK. The relationship of autophagy defects to cartilage damage during joint aging in a mouse model. Arthritis Rheumatol. 2015;67:1568–1576. doi: 10.1002/art.39073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiu BY, Chang CP, Lin JW, Yu JS, Liu WP, Hsu YC, Lin MT. Beneficial effect of astragalosides on stroke condition using PC12 cells under oxygen glucose deprivation and reperfusion. Cell Mol Neurobiol. 2014;34:825–837. doi: 10.1007/s10571-014-0059-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levine B, Yuan J. Autophagy in cell death: An innocent convict? J Clin Invest. 2005;115:2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang ZM, Du SH, Huang LG, Li JH, Xiao L, Tong P. Leptin promotes apoptosis and inhibits autophagy of chondrocytes through upregulating lysyl oxidase-like 3 during osteoarthritis pathogenesis. Osteoarthritis Cartilage. 2016;24:1246–1253. doi: 10.1016/j.joca.2016.02.009. [DOI] [PubMed] [Google Scholar]
- 20.Ma B, Leijten JC, Wu L, Kip M, van Blitterswijk CA, Post JN, Karperien M. Gene expression profiling of dedifferentiated human articular chondrocytes in monolayer culture. Osteoarthritis Cartilage. 2013;21:599–603. doi: 10.1016/j.joca.2013.01.014. [DOI] [PubMed] [Google Scholar]