

Abstract
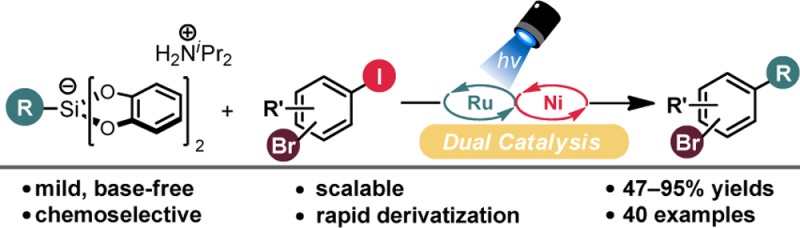
The chemoselective functionalization of polyfunctional aryl linchpins is crucial for rapid diversification. Although well-explored for Csp2 and Csp nucleophiles, the chemoselective introduction of Csp3 groups remains notoriously difficult and is virtually undocumented using Ni catalysts. To fill this methodological gap, a “haloselective” cross-coupling process of arenes bearing two halogens, I and Br, using ammonium alkylbis(catecholato)silicates, has been developed. Utilizing Ni/photoredox dual catalysis, Csp3–Csp2 bonds can be forged selectively at the iodine-bearing carbon of bromo(iodo)arenes. The described high-yielding, base-free strategy accommodates various protic functional groups. Selective electrophile activation enables installation of a second Csp3 center and can be done without the need for purification of the intermediate monoalkylated product.
Keywords: nickel/photoredox dual catalysis, bromo(iodo)arenes, hypervalent silicon, cross-coupling, selective functionalization
Chemoselective processes facilitate rapid divergence in organic synthesis.1 Innate chemical discrimination stemming from myriad factors is often the crux not only of elegant synthetic chemistry but of nature’s precise manipulation of the chemical world. Whereas nature relies on enzymes to effect chemoselective transformations, synthetic chemists increasingly rely on transition metal catalysts to enhance structural complexity selectively and efficiently.2 Indeed, great strides have been made recently, particularly in chemoselective C–C cross-coupling.3 In this context, approaches include selective oxidative addition via differences in C–X bond enthalpies, exploiting the disparate transmetalation rates of nucleophiles, and mechanistic discrimination.3
Although seemingly well-developed in the area of Csp2–Csp2 cross-coupling, the ability to forge Csp3–Csp2 bonds when using bromo(iodo)aryl compounds in a “haloselective” manner remains challenging. The recalcitrance of bench-stable Csp3 nucleophiles (e.g., organoboron or organostannyl species) toward Csp3–Csp2 cross-coupling requires laborious, harsh methods to be employed.4 As such, chemoselective cross-coupling is inherently intractable using these reagents. Alternatively, more reactive nucleophiles (e.g., organozinc species) have been used sporadically with success for chemoselective functionalization via Pd-mediated cross-coupling, although no cohesive study has been performed on this topic.5 Moreover, analogous Ni-mediated processes are virtually undocumented in the literature. More often than not, a multistep approach is used to install the desired alkyl group wherein selectivity is achieved via Csp2–Csp2 or Csp2–Csp cross-coupling (i.e., using Pd-based catalysts) followed by a functional group interconversion (i.e., reduction to an alkane; see Figure 1).6 Although reliable, these multistep processes are inherently less direct and efficient than the former approaches, particularly if installation of an additional Csp3 center is desired. Thus, no general solution for Csp3–Csp2 coupling using bromo(iodo)arenes in a haloselective manner has been reported, despite the fact that such transformations would allow access to useful linchpins for polyfunctional arene construction.
Figure 1.
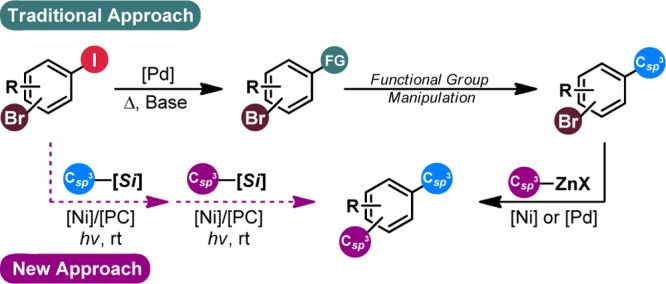
Current strategies versus the envisioned approach for selective electrophilic activation of bromo(iodo)aryl linchpins.
The recent emergence of Ni/photoredox dual catalysis has resulted in a rapid, one-step approach to install alkyl groups and represents an alternative to the two classical Csp3–Csp2 cross-coupling extremes (i.e., reactive nucleophiles or harsh conditions).7,8 In this dual catalytic manifold, a benchtop-stable Csp3 pro-nucleophile is controllably activated by photocatalyst-mediated, single-electron transfer (SET) oxidation to generate the active nucleophile, an alkyl radical. Ultimately, this catalytic system endows chemists with a platform that enables facile, mild Csp3–Csp2 bond formation with the added benefit of broad tolerance toward sensitive moieties. Moreover, numerous feedstocks for pro-nucleophiles have been identified, such as organotrifluoroborates,9a,9b carboxylic acids,9c,9d alkylbis(catecholato)silicates,9e−9g 4-alkyl-1,4-dihydropyridines (DHPs),9h,9i and even activated C–H bonds.9j−9l Consequently, substantial structural diversity in the nucleophilic partner can be readily accessed. Because of the mild conditions and the generally less electron-rich ligands employed, Ni/photoredox dual catalysis presents a broadly applicable solution to the challenge of chemoselective cross-coupling in the bromo(iodo)arene scaffold. Given the improved clinical success of drug molecules possessing higher fractions of Csp3 centers,10 the realization of such a solution would enable the modular synthesis of polyfunctional arenes via the controlled functionalization of each synthetic handle and access to molecules with higher 3D complexity.
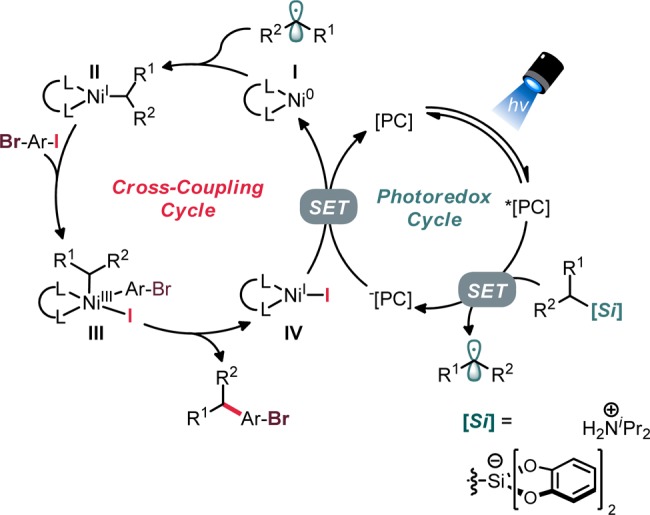
To investigate the tenability of this strategy, ammonium alkylbis(catecholato)silicates were utilized as “plug and play” radical progenitors.9e,9f This choice was made for a number of reasons: (1) the ability to perform dual catalytic cross-coupling free of any additives (thus maximizing functional group tolerance and minimizing complications arising from additives), (2) the amenability to generating stabilized and destabilized radicals (thus allowing assessment of broad applicability), and (3) the known ease with which these hypercoordinate silicon species can be used in cross-coupling with an array of electrophiles (thus assessing electrophile fidelity). Additionally, the low redox potential (E0 ∼ 0.40–0.75 V vs SCE) of ammonium alkylbis(catecholato)silicates and their tunable solubility properties (based on counterion structure) allow flexibility in the identity of the photocatalyst and solvent, respectively.9e,9f Ultimately, haloselective cross-coupling was envisioned to occur via the mechanism shown in Scheme 1.11 Speculatively, selective oxidative addition by the NiI species II would provide discrimination between the iodine and bromine.12−14
Scheme 1. Envisioned Catalytic Cycle for Selective Cross-Coupling of Dihaloarenes Using Ni/Photoredox Dual Catalysis.
Using the previously optimized conditions for Ni/photoredox dual catalytic cross-coupling of alkylsilicates,9e a baseline for innate selectivity and conversion was observed to be 88:12 (3a:4a) at 95% conversion (Table 1, entry 1). Without any optimization, selective functionalization was possible with no detectable overalkylation. However, improvement of the selectivity was still needed. Given that selectivity might hinge on kinetic control of oxidative addition, ligand choice and Ni catalyst loading were examined. When using dtbbpy, Ni loading seemed to have a minimal effect, although lower loadings appeared to be preferable (entries 1–3). Next, a number of bipyridyl and phenanthryl ligands were examined. These two ligand classes have been shown to be privileged in the context of Ni/photoredox dual catalysis but are virtually undocumented in facilitating this type of selective alkyl–aryl coupling. Although substituted bipyridyl ligands failed to improve the ratio (entries 4 and 5), bipyridine itself gave excellent selectivity (entry 6). Selectivity did come at the cost of lowered conversion. Similarly, phenanthroline performed well (entry 9), whereas alteration of the substitution pattern of the phenanthryl core had deleterious effects on conversion (entry 7) or selectivity (entry 8). Using a lower Ni loading, a 2:1 ligand to metal ratio, and slightly increasing the loading of the photocatalyst resulted in excellent conversion and selectivity (entries 10 and 11). Ultimately, both protocols were scaled up and gave good yields of the desired cross-coupled product. Conceivably, these ligand systems are not as efficient at oxidative addition as dtbbpy from either NiI (or Ni0) and thus provide further chemoselective enhancement from innate I/Br selectivity.15
Table 1. Optimization of Haloselective Cross-Couplinga.
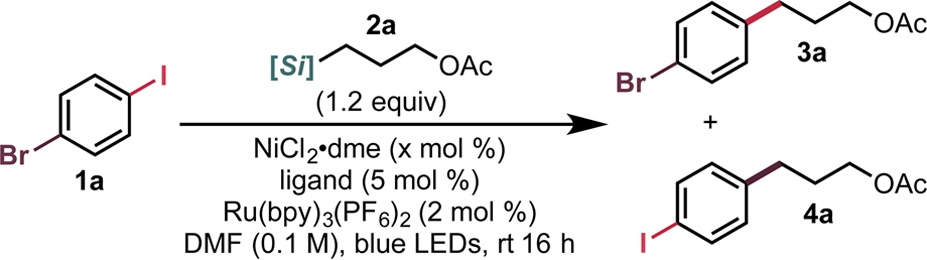
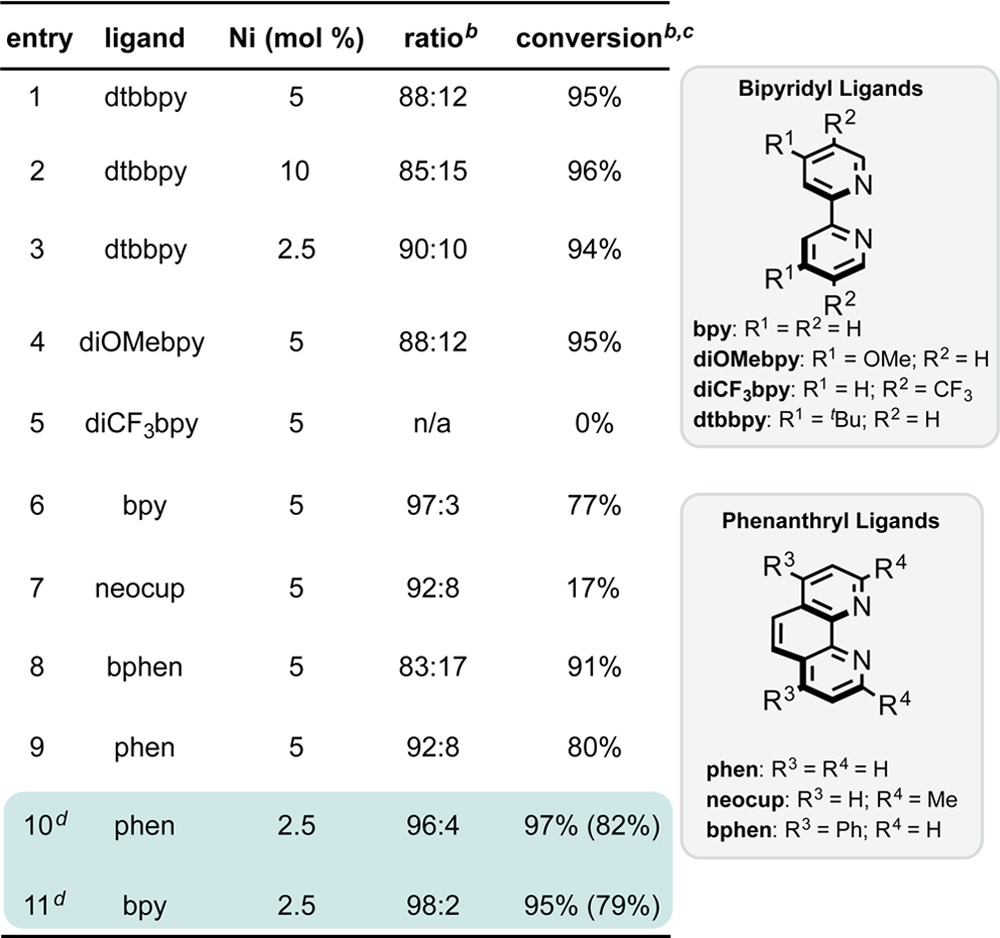
Unless otherwise noted, reactions were run using 0.1 mmol of 1a for 16 h at 27 °C and using a 1:1 Ni/ligand ratio.
Conversions and ratios of 3a to 4a were determined by GC/MS and confirmed by 1H NMR analysis of the crude reaction mixture.
Values in parentheses indicate the isolated yields on a 0.5 mmol scale of 1a.
A 1:2 Ni/ligand ratio and 2.5 mol % of Ru(bpy)3(PF6)2 were used.
With suitable conditions and two viable ligands identified, we next investigated the scope of the process (Table 2). First, the range of bromo(iodo)arenes that could be used was assessed using a representative 1° alkylsilicate, 2a. The reaction proved quite general, accommodating a number of functional groups and giving excellent chemoselectivity in nearly all cases (Table 2). Although in general phen was primarily used, bpy could be used interchangeably with little to no effect on yield or selectivity. Unsubstituted bromo(iodo)arenes underwent haloselective cross-coupling in good yield regardless of the substitution pattern of the two halides (3a–c). In addition, scale up of this process proved facile and did not compromise selectivity. Both electron-donating and electron-withdrawing groups were well-tolerated. Aryl substitution did appear to impact the efficiency and/or selectivity of the described cross-coupling process, with both steric and electronic changes having subtle yet noticeable effects on either product ratio or yield. Indeed, the substrates derived from 2-bromo-4-iodophenol (3h–3k), as well as 3t, best illustrate these sorts of effects. Of note are the polyfunctional arenes 3f, 3j, and 3k, which, in addition to the bromide handle, possess a third site for additional functionalization. We serendipitously discovered that when attempting to isolate 3f, a minor change in the order of the workup (base wash prior to acid wash) led to the facile hydrolysis of the acetoxy group. Thus, the free alcohol, 3f′, could also be obtained under near identical reaction conditions. Heteroaromatics generally proved challenging or low yielding in the described cross-coupling process (possibly because of competitive ligation, poor catalyst efficiency, or catalyst deactivation), although some success was observed with a quinoline-based system (3p). Finally, although a benzylic alcohol failed under the optimized conditions, TBS-protection restored reactivity (3s) and enabled successful cross-coupling, albeit with somewhat diminished chemoselectivity.
Table 2. Selective Cross-Coupling Using Alkylsilicate 2aa.
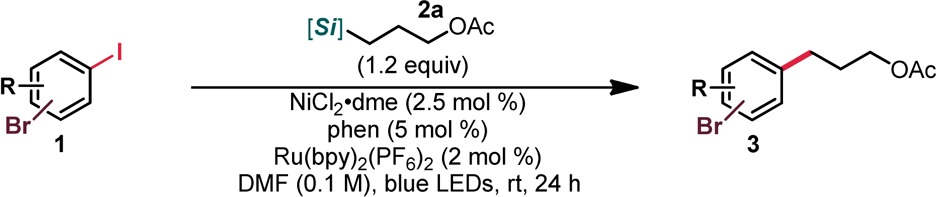
Unless otherwise noted, reactions were performed using 1 (1.0 equiv, 0.5 mmol), 2a (1.2 equiv), NiCl2·dme (2.5 mol %), Ru(bpy)3(PF6)2 (2 mol %), and phen (5 mol %) in DMF (0.1 M) at rt for 24 h, irradiating with blue LEDs; ratios of 3:4 are indicated in parentheses, and these values were obtained before purification. All yields are isolated yields after purification.
Performed using bpy as a ligand.
Value in brackets denotes the yield on a 5 mmol scale using phen as the ligand.
Product obtained as its 1° alcohol rather than acetate by modification of the workup.
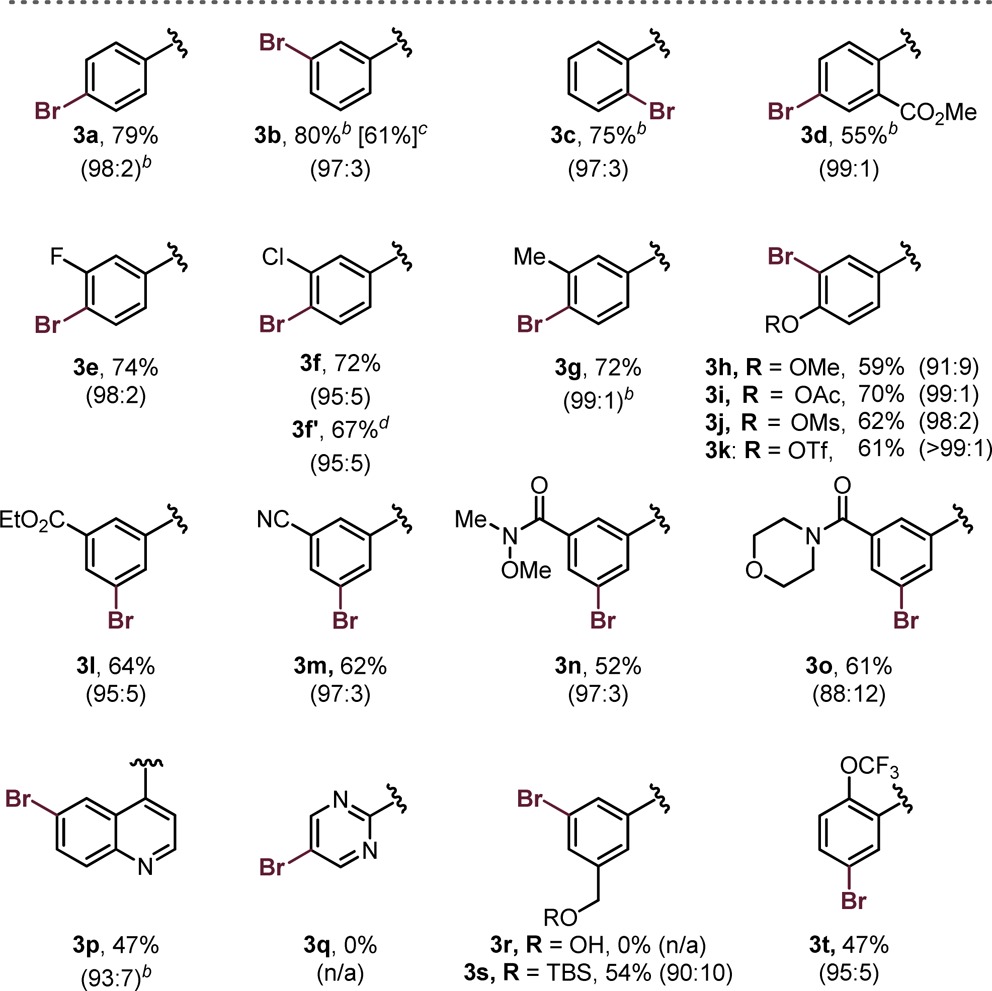
The diversity in radical structure was next assessed (Table 3). Gratifyingly, a variety of alkylsilicates were successfully employed without compromising yield or chemoselectivity. Relatively simple 1° and 2° alkylsilicates performed well in the haloselective cross-coupling process. In addition, the organic dye 4CzIPN could be used in place of the Ru-based photocatalyst at a minimal cost of selectivity and yield (see 3z). Nucleophilic partners that would be typically unattainable as their corresponding organometallics did not pose a problem in this process (3aa–ae, 3ag, 3ah). The reaction is highly tolerant of protic functional groups, a unique aspect of this process that cannot be mimicked with current protocols in haloselective Csp3–Csp2 cross-coupling (e.g., 3ab–ad, 3ae). Notably, the thiolsilicate, used in the synthesis of 3ad, undergoes hydrogen atom transfer (HAT) upon radical generation to furnish a thiyl radical that subsequently couples, as disclosed in our previous report.16 Thus, 3ad was obtained as the corresponding thioether. Not only was this successful, but it also demonstrated that S-centered radicals are also amenable to selective coupling. Finally, stabilized radicals performed quite well in the haloselective cross-coupling (3ah–ai), despite the known reversibility of radical metalation.11
Table 3. Selective Cross-Coupling of 1e with Various Alkylsilicatesa.

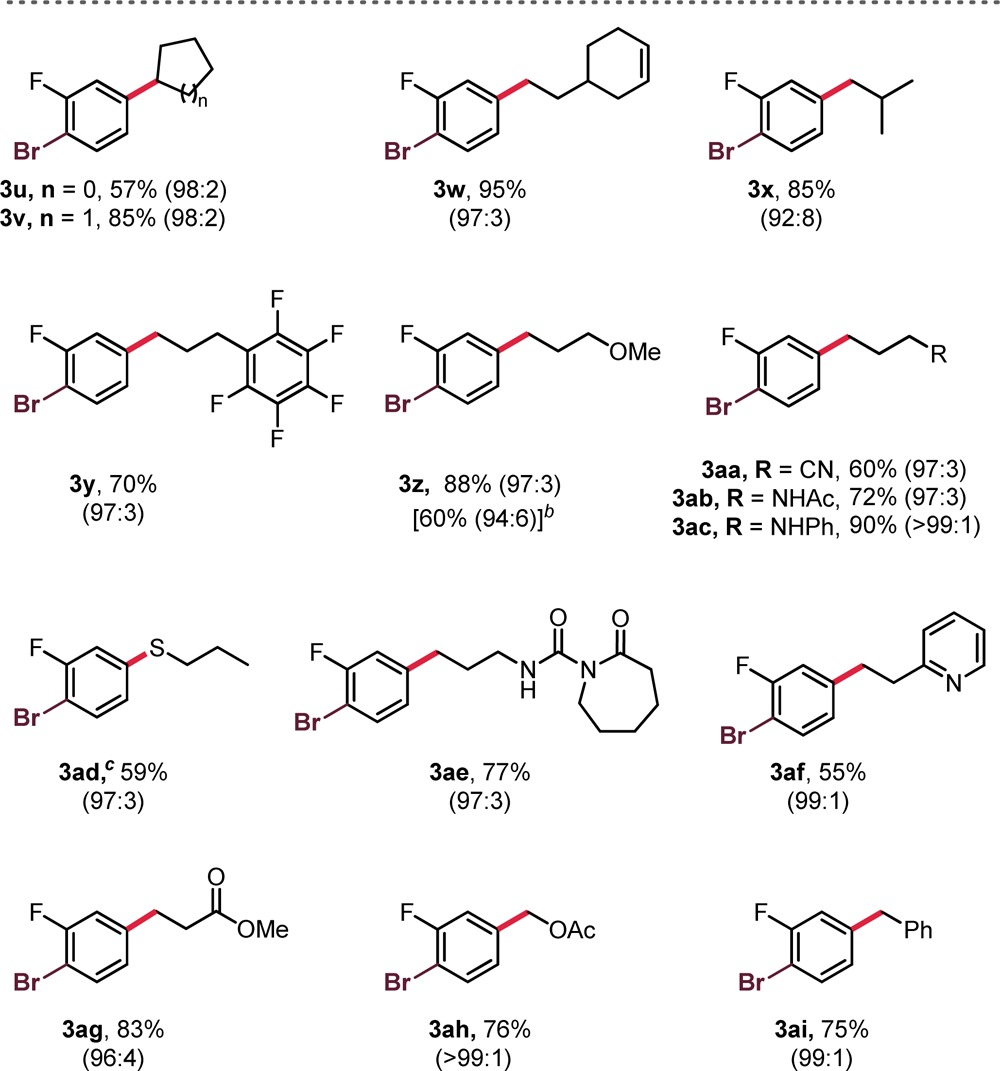
Unless otherwise noted, reactions were performed using 1e (1.0 equiv, 0.5 mmol), 2 (1.2 equiv), NiCl2·dme (2.5 mol %), Ru(bpy)3(PF6)2 (2 mol %), and phen (5 mol %) in DMF (0.1 M) at rt for 24 h, irradiating with blue LEDs; ratios of 3:4 are indicated in parentheses, and these values were obtained before purification. All yields are isolated yields after purification.
Value in brackets denotes the yield when using the organic dye 4CzIPN (5 mol %) as the photocatalyst.
Product obtained as the thioether rather than thiol from (3-mercaptopropyl)silicate.
To showcase the utility of the described haloselective process further, a series of subsequent functionalization reactions were executed using 3aj (Figure 2). In addition to demonstrating the value of the haloselective process, the requisite amount of 3aj for these studies presented the opportunity to push the limits of the scalability of this cross-coupling reaction. The reaction proved to be quite scalable and can be performed on 10 mmol scale (and likely greater) with minimal effect on yield and chemoselectivity. Subsequent installation of Csp2 or Csp centers via Suzuki or Sonogashira coupling processes, respectively, proceeded very smoothly, affording good yields of the corresponding cross-coupled products. Buchwald–Hartwig-type amination similarly was successful, affording the piperidinyl adduct 6aj in 74% yield. Finally, installation of a second Csp3 center via a second Ni/photoredox cross-coupling of an alkylsilicate was successful, giving 8aj in 51% yield. This process was later repeated starting from 1b without intermediate isolation, providing an improved 66% yield of the desired product.
Figure 2.
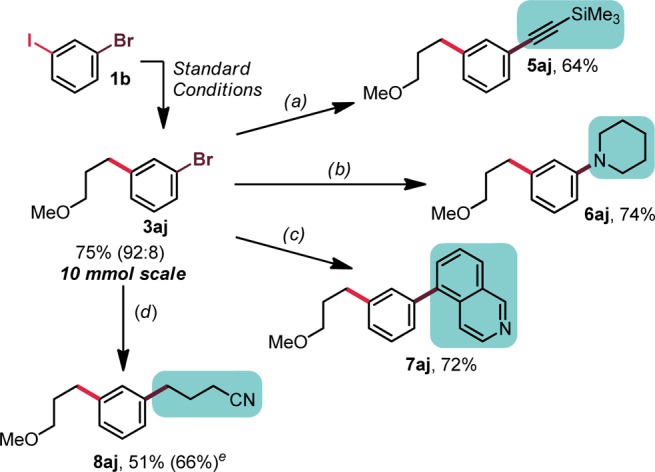
Diversification of 3aj. Conditions: (a) 3aj (0.5 mmol, 1 equiv), TMS-acetylene (3 equiv), Pd(PPh3)2Cl2 (5 mol %), CuI (5 mol %), Et3N (0.2 M), 80 °C, 48 h; (b) 3aj (1 mmol, 1 equiv), piperidine (1.5 equiv), XPhos G2 (2 mol %), Cs2CO3 (2.5 equiv), toluene/t-BuOH (5:1, 0.4 M), 80 °C, 18 h; (c) 3aj (0.5 mmol, 1 equiv), organotrifluoroborate (1.5 equiv), Pd(PPh3)2Cl2 (5 mol %), Cs2CO3 (3 equiv), THF/H2O (2:1, 0.25 M), 80 °C, 18 h; (d) 3aj (1 mmol, 1 equiv), alkylsilicate (1.2 equiv), [Ni(dtbbpy)(H2O)4]Cl2 (5 mol %), and Ru(bpy)3(PF6)2 (2 mol %), DMF (0.1 M), blue LEDs, rt, 24 h. (e) The yield in parentheses indicates the yield of sequential coupling, starting from 1b on 0.5 mmol scale without isolation of 3aj.
Recognizing the power of this method for selective electrophile activation and to build on our successful sequential process with 8aj, a series of tandem Csp3–Csp2 cross-coupling reactions were successfully attempted (Figure 3). Using the now-established conditions for the haloselective process, the first alkyl group was installed at the iodo position in three distinct bromo(iodo)arenes. After a simple aqueous workup, the crude product was subjected to the standard conditions for Ni/photoredox dual catalytic cross-coupling, thus installing a second alkyl group at the bromo position.9e,9f This demonstrates the ability of this process to install alkyl groups regioselectively (compare 3ak to 3am) and further validates the selective electrophile-activation paradigm. In addition, the overall yields were quite good, equating to greater than 75% per chemical step for all four examples.
Figure 3.
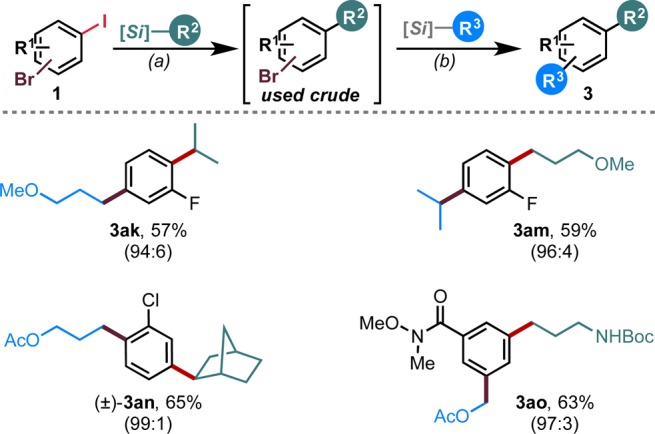
Tandem Csp3–Csp2 cross-coupling. Numbers in parentheses are ratios of 3:4 after the first coupling. Conditions: (a) 1 (1.0 equiv, 0.5 mmol), alkylsilicate (1.2 equiv), NiCl2·dme (2.5 mol %), Ru(bpy)3(PF6)2 (2 mol %), and phen (5 mol %) DMF (0.1 M), blue LEDs, rt, 24 h; (b) alkylsilicate (1.2 equiv), [Ni(dtbbpy)(H2O)4]Cl2 (5 mol %), and Ru(bpy)3(PF6)2 (2 mol %), DMF (0.1 M), blue LEDs, rt, 24 h.
In summary, a mild, user-friendly method for the selective electrophilic activation of bromo(iodo)arenes using ammonium alkylbis(catecholato)silicates is presented. The described “haloselective” reaction tolerates an array of functional groups, including those bearing acidic protons, and is highly scalable. The monofunctionalized products can be readily paired with existing cross-coupling technology, affording a means to fuse classical two-electron cross-coupling with single-electron transformations. Sequential Ni/photoredox cross-coupling of these polyfunctional arenes enables two unique Csp3–Csp2 bonds to be forged selectively. The conditions outlined here can likely be extended to other radical progenitors, further emphasizing the utility of the described process for rapid diversification.
Acknowledgments
The authors are grateful for the financial support provided by NIGMS (R01 GM 113878). C.B.K. is grateful for an NIH NRSA postdoctoral fellowship (F32 GM117634-01). R.J.W. is grateful for an NSF GRFP fellowship. We sincerely thank Mr. David Primer, Dr. James Phelan, and Dr. Simon Lang of the University of Pennsylvania (UPenn) for useful discussions. We thank Mr. Xingpin Li (UPenn) for assistance in the preparation of some alkylsilicates. We thank Dr. Charles W. Ross, III (UPenn) for assistance in obtaining HRMS.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.7b01773.
Experimental procedures and characterization data for all compounds (DOCX)
Author Contributions
† K.L. and R.J.W. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Shenvi R. A.; O’Malley D. P.; Baran P. S. Acc. Chem. Res. 2009, 42, 530–541. 10.1021/ar800182r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Almond-Thynne J.; Blakemore D. C.; Pryde D. C.; Spivey A. C. Chem. Sci. 2017, 8, 40–62. 10.1039/C6SC02118B. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Xu L.; Zhang S.; Li P. Chem. Soc. Rev. 2015, 44, 8848–8858. 10.1039/C5CS00338E. [DOI] [PubMed] [Google Scholar]; c Manabe K.; Yamaguchi M. Catalysts 2014, 4, 307–320. 10.3390/catal4030307. [DOI] [Google Scholar]; c1 Rossi R.; Bellina F.; Lessi M. Adv. Synth. Catal. 2012, 354, 1181–1255. 10.1002/adsc.201100942. [DOI] [Google Scholar]; d Fairlamb I. J. S. Chem. Soc. Rev. 2007, 36, 1036–1045. 10.1039/b611177g. [DOI] [PubMed] [Google Scholar]; e Schröter S.; Stock C.; Bach T. Tetrahedron 2005, 61, 2245–2267. 10.1016/j.tet.2004.11.074. [DOI] [Google Scholar]; f Wang J.-R.; Manabe K. Synthesis 2009, 2009, 1405–1427. 10.1055/s-0029-1216632. [DOI] [Google Scholar]
- For some recent elegant examples of chemoselective cross-coupling, see the following:; a Kalvet I.; Magnin G.; Schoenebeck F. Angew. Chem., Int. Ed. 2017, 56, 1581–1585. 10.1002/anie.201609635. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fyfe J. W. B.; Fazakerley N. J.; Watson A. J. B. Angew. Chem., Int. Ed. 2017, 56, 1249–1253. 10.1002/anie.201610797. [DOI] [PubMed] [Google Scholar]; c Vara B. A.; Jouffroy M.; Molander G. A. Chem. Sci. 2017, 8, 530–535. 10.1039/C6SC03236B. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Yamashita Y.; Tellis J. C.; Molander G. A. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 12026–12029. 10.1073/pnas.1509715112. [DOI] [PMC free article] [PubMed] [Google Scholar]; e He L.-Y.; Schulz-Senft M.; Thiedemann B.; Linshoeft J.; Gates P. J.; Staubitz A. Eur. J. Org. Chem. 2015, 2015, 2498–2502. 10.1002/ejoc.201500138. [DOI] [Google Scholar]; f Demory E.; Devaraj K.; Orthaber A.; Gates P. J.; Pilarski L. T. Angew. Chem., Int. Ed. 2015, 54, 11765–11769. 10.1002/anie.201503152. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Linshoeft J.; Heinrich A. C. J.; Segler S. A. W.; Gates P. J.; Staubitz A. Org. Lett. 2012, 14, 5644–5647. 10.1021/ol302571t. [DOI] [PubMed] [Google Scholar]; h Espino G.; Kurbangalieva A.; Brown J. A. Chem. Commun. 2007, 1742–1744. 10.1039/B701517H. [DOI] [PubMed] [Google Scholar]
- For an example of this approach, see the following:Okochi K.; Jin Y.; Zhang W. Chem. Commun. 2013, 49, 4418–4420. 10.1039/C2CC33078D. [DOI] [PubMed] [Google Scholar]
- Examples of this approach:; a Placzek A. T.; Scanlan T. S. Tetrahedron 2015, 71, 5946–5951. 10.1016/j.tet.2015.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pena M. A.; Perez Sestelo J.; Sarandeses L. A. Synthesis 2005, 2005, 485–492. 10.1055/s-2004-834945. [DOI] [Google Scholar]; c Ebert G. W.; Pfennig D. R.; Suchan S. D.; Donovan T. A. Jr.; Aouad E.; Tehrani S. S.; Gunnersen J. N.; Dong L. J. Org. Chem. 1995, 60, 2361–2364. 10.1021/jo00113a013. [DOI] [Google Scholar]; d Malhotra S.; Seng S. S.; Koenig S. G.; Deese A. J.; Ford K. A. Org. Lett. 2013, 15, 3698–3701. 10.1021/ol401508u. [DOI] [PubMed] [Google Scholar]
- Odedra A.; Datta S.; Liu R.-S. J. Org. Chem. 2007, 72, 3289–3292. 10.1021/jo062573l. [DOI] [PubMed] [Google Scholar]
- Seminal reports:; a Tellis J. C.; Primer D. N.; Molander G. A. Science 2014, 345, 433–436. 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zuo Z.; Ahneman D. T.; Chu L.; Terrett J. A.; Doyle A. G.; MacMillan D. W. C. Science 2014, 345, 437–440. 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reviews on Ni/photoredox cross-coupling:; a Tellis J. C.; Kelly C. B.; Primer D. N.; Jouffroy M.; Patel N. R.; Molander G. A. Acc. Chem. Res. 2016, 49, 1429–1439. 10.1021/acs.accounts.6b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gui Y.-Y.; Sun L.; Lu Z.-P.; Yu D.-G. Org. Chem. Front. 2016, 3, 522–526. 10.1039/C5QO00437C. [DOI] [Google Scholar]; c Skubi K. L.; Blum T. R.; Yoon T. P. Chem. Rev. 2016, 116, 10035–10074. 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Kelly C. B.; Patel N. R.; Primer D. N.; Jouffroy M.; Tellis J. C.; Molander G. A. Nat. Protoc. 2017, 12, 472–476. 10.1038/nprot.2016.176. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Levin M. D.; Kim S.; Toste F. D. ACS Cent. Sci. 2016, 2, 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Shaw M. H.; Twilton J.; MacMillan D. W. C. J. Org. Chem. 2016, 81, 6898–6926. 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Matsui J. K.; Lang S. B.; Heitz D. R.; Molander G. A. ACS Catal. 2017, 7, 2563–2575. 10.1021/acscatal.7b00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The following are selected examples of each radical precursor in this type of cross-coupling. RBF3Ks:; a Primer D. N.; Karakaya I.; Tellis J. C.; Molander G. A. J. Am. Chem. Soc. 2015, 137, 2195–2198. 10.1021/ja512946e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Karakaya I.; Primer D. N.; Molander G. A. Org. Lett. 2015, 17, 3294–3297. 10.1021/acs.orglett.5b01463. [DOI] [PMC free article] [PubMed] [Google Scholar]; Carboxylic acids:; c Noble A.; McCarver S. J.; MacMillan D. W. C. J. Am. Chem. Soc. 2015, 137, 624–627. 10.1021/ja511913h. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Zuo Z.; Cong H.; Li W.; Choi J.; Fu G. C.; MacMillan D. W. C. J. Am. Chem. Soc. 2016, 138, 1832–1835. 10.1021/jacs.5b13211. [DOI] [PMC free article] [PubMed] [Google Scholar]; Alkylsilicates:; e Jouffroy M.; Primer D. N.; Molander G. A. J. Am. Chem. Soc. 2016, 138, 475–478. 10.1021/jacs.5b10963. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Corce V.; Chamoreau L.-M.; Derat E.; Goddard J.-P.; Ollivier C.; Fensterbank L. Angew. Chem., Int. Ed. 2015, 54, 11414–11418. 10.1002/anie.201504963. [DOI] [PubMed] [Google Scholar]; g Patel N. R.; Kelly C. B.; Jouffroy M.; Molander G. A. Org. Lett. 2016, 18, 764–767. 10.1021/acs.orglett.6b00024. [DOI] [PMC free article] [PubMed] [Google Scholar]; DHPs:; h Gutierrez-Bonet A.; Tellis J. C.; Matsui J. K.; Vara B. A.; Molander G. A. ACS Catal. 2016, 6, 8004–8008. 10.1021/acscatal.6b02786. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Nakajima K.; Nojima S.; Nishibayashi Y. Angew. Chem., Int. Ed. 2016, 55, 14106–14110. 10.1002/anie.201606513. [DOI] [PubMed] [Google Scholar]; C–H Activation:; j Shaw M. H.; Shurtleff V. W.; Terrett J. A.; Cuthbertson J. D.; MacMillan D. W. C. Science 2016, 352, 1304–1308. 10.1126/science.aaf6635. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Shields B. J.; Doyle A. G. J. Am. Chem. Soc. 2016, 138, 12719–12722. 10.1021/jacs.6b08397. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Heitz D. R.; Tellis J. C.; Molander G. A. J. Am. Chem. Soc. 2016, 138, 12715–12718. 10.1021/jacs.6b04789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lovering F.; Bikker J.; Humblet C. J. Med. Chem. 2009, 52, 6752–6756. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]; b Schneider P.; Schneider G. Angew. Chem., Int. Ed. 2017, 56, 7971–7974. 10.1002/anie.201702816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez O.; Tellis J. C.; Primer D. N.; Molander G. A.; Kozlowski M. C. J. Am. Chem. Soc. 2015, 137, 4896–4899. 10.1021/ja513079r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Although using phosphine complexes of Ni rather than the bipyrdyl or phenanthryl ligands used here, Kochi has suggested that oxidative addition may proceed via a SET from Ni0 when iodides are used. See the following:Tsou T. T.; Kochi J. K. J. Am. Chem. Soc. 1979, 101, 6319–6332. 10.1021/ja00515a028. [DOI] [Google Scholar]
- Very recently, Schoenebeck and the groups of Sproules and Nelson have engaged in mechanistic studies on oxidative addition via a NiI species. Although the Nelson report expands on the work of Kochi in ref (12), Schoenebeck suggests a NiI oxidative addition with bpy ligands in the context of aryl trifluoromethylthiolation. See the following:; a Kalvet I.; Guo Q.; Tizzard G. J.; Schoenebeck F. ACS Catal. 2017, 7, 2126–2132. 10.1021/acscatal.6b03344. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bajo S.; Laidlaw G.; Kennedy A. R.; Sproules S.; Nelson D. J. Organometallics 2017, 36, 1662–1672. 10.1021/acs.organomet.7b00208. [DOI] [Google Scholar]
- A single example of the Ni/photoredox cross-coupling of 4-bromoiodobenzene with a benzyl radical derived from a DHP was reported; see ref (9i).
- Computational investigations of this innate selectivity are ongoing within our laboratory to explain this peculiar ligand effect further, as well as further understand how Ni oxidation states influence oxidative addition rates.
- Jouffroy M.; Kelly C. B.; Molander G. A. Org. Lett. 2016, 18, 876–879. 10.1021/acs.orglett.6b00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.