

Abstract
Cytokines IL-4 and IL-13 play important roles in polarization of macrophages/dendritic cells to an M2 phenotype, which is important for recovery from acute kidney injury. Both IL-4 and IL-13 activate JAK3/STAT6 signaling. In mice with diphtheria toxin receptor expression in proximal tubules (selective injury model), a relatively selective JAK3 inhibitor, tofacitinib, led to more severe kidney injury, delayed recovery from acute kidney injury, increased inflammatory M1 phenotype markers and decreased reparative M2 phenotype markers of macrophages/dendritic cells, and development of more severe renal fibrosis after diphtheria toxin administration. Similarly, there was delayed recovery and increased tubulointerstitial fibrosis in these diphtheria toxin-treated mice following tamoxifen-induced deletion of both IL-4 and IL-13, with increased levels of M1 and decreased levels of M2 markers in the macrophages/dendritic cells. Furthermore, deletion of IL-4 and IL-13 led to a decrease of tissue reparative M2a phenotype markers but had no effect on anti-inflammatory M2c phenotype markers. Deletion of IL-4 and IL-13 also inhibited recovery from ischemia-reperfusion injury in association with increased M1 and decreased M2 markers and promoted subsequent tubulointerstitial fibrosis. Thus, IL-4 and IL-13 are required to effectively polarize macrophages/dendritic cells to an M2a phenotype and to promote recovery from acute kidney injury.
Keywords: ischemia-reperfusion, macrophage polarization, STAT6, tofacitinib
Acute kidney injury (AKI) is defined as an abrupt decrease in renal function. The reported incidence of AKI varies from 5% in all hospitalized patients to 30% to 50% in intensive care units.1 Incomplete recovery from AKI leads to chronic kidney injury. The majority of interventional trials in AKI have failed in humans. Therefore, novel therapeutic approaches are needed to prevent or treat AKI.
Increasing evidence indicates that infiltration of immune cells, particularly macrophages, plays an important role in the initiation and propagation of the kidney injury. However, the roles of infiltrating macrophages in the pathogenesis of acute or chronic renal injury are still understood incompletely. Although early studies suggested that macrophages might be detrimental, beneficial, or have no effect in acute renal injury,2–6 recent work has indicated that macrophage infiltration is detrimental in the early injury phase but beneficial in the repair phase after ischemia injury.7–10 The study by Lee et al.7 showed that macrophages infiltrating at early time points after ischemia-reperfusion AKI exhibited primarily a proinflammtory M1 phenotype, but switched to a reparative M2 phenotype at later time points. Macrophage depletion at early time points led to less severe kidney injury, but macrophage deletion at later time points led to impaired recovery, underscoring the importance of polarization of macrophages in recovery from ischemia-reperfusion AKI.7 Recent studies by us8,10 and others7,11,12 have indicated that macrophage colony-stimulating factor 1 is a mediator of macrophage polarization and proliferation and plays a key role in recovery from AKI. Infusion of M2 macrophages polarized ex vivo has been reported to protect against adriamycin-induced nephropathy in association with induction of regulatory T cells.13
Although macrophages were previously characterized as having “M1” (inflammatory) and “M2” (tissue reparative) phenotypes, there is increasing evidence that what was previously characterized as M2 macrophages actually represents a spectrum of phenotypes. Three main subtypes M2a, M2b, and M2c have been described, although it is recognized that even this further differentiation provides somewhat artificial distinctions.14–16 Macrophages/dendritic cells in each of these subcategories produce cytokines and chemokines that overlap with other M2 subtypes as well as cytokines more specific for the individual subtype. In this regard, the M2a subtype is more associated with a wound healing and tissue reparative phenotype, and the M2c subtype was more associated with a regulatory, anti-inflammatory, as well as profibrotic phenotype.17 Interleukin (IL)-4 and IL-13 induce macrophages to polarize to a more M2a-like phenotype whereas IL-10 and transforming growth factor ß induce a more M2c-like phenotype.17 In the current study, we determined the effect of deletion of IL-4 and IL-13 upon recovery from AKI and subsequent development of renal fibrosis.
RESULTS
We have previously described a model of selective proximal tubular injury resulting from diphtheria toxin (DT) administration to mice expressing the human diphtheria toxin receptor (DTR) in the proximal tubule.8 Administration of DT resulted in renal dysfunction, as manifested by increased blood urea nitrogen (BUN), which peaked at 5 to 6 days after DT administration (Figure 1a). IL-4 and IL-13 bind to the same receptor complexes containing IL-4Rα and activate Janus kinase 3 (JAK3), leading to increased phosphorylation of signal transducer and activator of transcription 6 (pSTAT6).18 Following DT administration, both STAT6 expression and activity (pSTAT6) increased in the kidney, and STAT6 expression and activation were inhibited when macrophages/dendritic cells were ablated with clodronate (Figure 1b), suggesting that macrophages/dendritic cells are the major source of STAT6 activation. Quantitative analysis determined that renal STAT6 and pSTAT6 levels increased to similar extent after DT injection (Supplementary Figure S1), suggesting that increased pSTAT6 levels are primarily due to increased STAT6 expression. Administration of the relatively selective JAK3 inhibitor, tofacitinib (CP-690550), decreased kidney expression of pSTAT6 following DT-induced injury (Figure 1c). Administration of tofacitinib significantly delayed recovery from DT-induced AKI, as indicated by persistent elevation of BUN and serum creatinine (Figure 1a) and delayed structural recovery (Figure 1d). Twelve days following injury, there was also increased evidence of tubulointerstitial fibrosis (Figure 1d). In addition, 6 days after DT administration, a time that our previous studies had indicated significant increases in macrophage “M2” marker expression and few “M1” macrophages,8 tofacitinib administration led to increases in “M1” markers, inducible nitric oxide synthase and CC chemokine ligand 3 (Figure 1e) and decreases in “M2” markers, arginase and CD206/mannose receptor (Figure 1f). Huen et al.9 have reported that STAT5 is activated after ischemia-reperfusion AKI. Therefore, we investigated whether STAT5 was also activated in our DTR-AKI model. As indicated in Supplementary Figure S2, pSTAT5 levels increased after DT injection; however, pSTAT5 levels were not affected by treatment with CP-690550, suggesting that CP-690550–mediated worsening of kidney injury in DTR model is primarily due to its inhibition of JAK3/STAT6 activity.
Figure 1. Janus kinase 3 (JAK3) pathway promoted recovery from diphtheria toxin (DT)-mediated acute kidney injury in DT receptor mice.
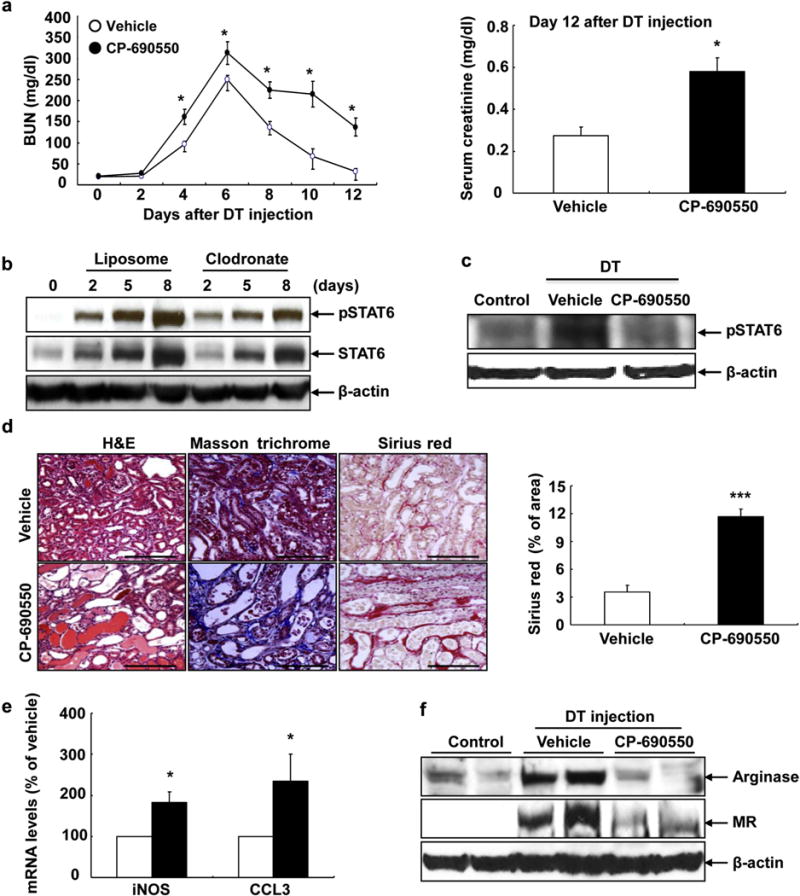
(a) Levels of blood urea nitrogen (BUN) remained higher during recovery from DT-mediated acute kidney injury (AKI) with treatment of tofacitinib (CP-690550), a relatively selective JAK3 inhibitor. At 12 days after DT injection, serum creatinine was higher in mice with CP-690550 treatment. *P < 0.05 versus vehicle-treated group, n = 6. (b) Increases in total signal transducer and activator of transcription 6 (STAT6) and phosphorylation of STAT6 (pSTAT6) levels after DT-mediated AKI were attenuated by macrophage depletion with clodronate. (c) Activation of STAT6 six days following DT-mediated AKI was attenuated by treatment with CP-690550. (d) Twelve days following DT-mediated AKI, kidney injury (hematoxylin and eosin [H&E] staining) and fibrosis (Masson trichrome staining and Sirius red staining and quantification) were apparent in mice with CP-690550 administration. ***P < 0.001 versus vehicle-treated group, n = 4. (e,f) JAK3 inhibition with CP-690550 led to increased mRNA levels of inducible nitric oxide synthase (iNOS) and CC chemokine ligand 3 (CCL3) and (e) markers of M1 phenotypic macrophages/dendritic cells, but decreased protein levels of arginase and mannose receptor (MR), (f) markers of M2 phenotypic macrophages/dendritic cells at 6 days after DT injection. Bar = 120 μm in all.
In addition to inhibiting JAK3 activity, CP-690550 has also been reported to inhibit JAK1 activity.19 To investigate further the role of this signaling pathway in recovery from AKI, we generated mice with global deletion of both IL-4 and IL-13 by crossing IL-4/IL-13f/f mice with a tamoxifen-inducible ubiquitin Cre (Cg-Tg[UBC-Cre/ERT2] 1Ejb/J). These mice were then crossed to the proximal tubular DTR transgenic mice. Expression of both IL-4 and IL-13 decreased in total kidney homogenate after tamoxifen administration (Figure 2a). The residual levels of IL-4 and IL-13 in DTR; IL-4/IL-13 knockout (KO) mice may be due to incomplete Cre recombination. We are also aware that our model is an inducible and global deletion model, and any effect seen in the kidney may also have been affected by decreased levels of IL-4 and IL-13 levels in cells in the circulation. Similar to what we observed with tofacitinib treatment, there was delayed recovery of BUN and serum creatinine from DT-induced AKI in the DTR; IL-4/IL-13 KO mice (Figure 2b). Six days after DT administration, there was also increased renal expression of the proximal tubular injury marker, kidney injury molecule 1 (Figure 2a). Immunostaining showed that pSTAT6 was intensely expressed in cells in the interstitium in DTR mice, and there was decreased expression in DTR; IL-4/IL-13 KO mice 6 days after DT injection (Figure 2c).
Figure 2. Renal interleukin (IL)-4/IL-13 was involved in recovery from diphtheria toxin (DT)-mediated acute kidney injury.
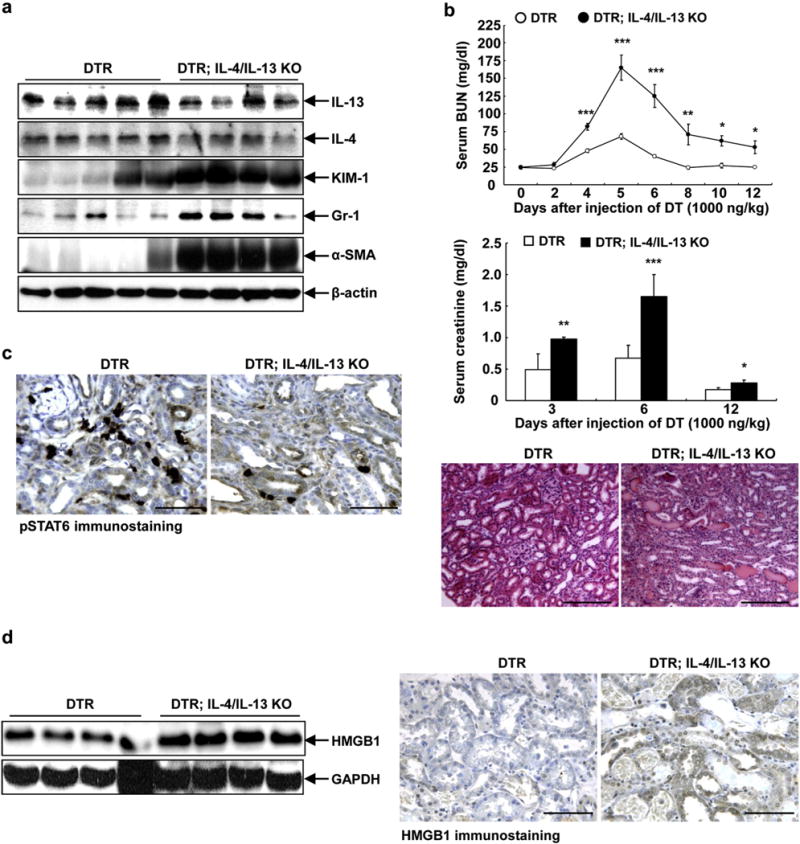
(a) Renal protein levels of both IL-4 and IL-13 were reduced in DT receptor (DTR); IL-4/IL-13 knockout (KO) mice 6 days after DT administration, in association with increased expression levels of kidney injury molecule 1 (KIM-1; a kidney injury marker), Gr-1 (marker of neutrophils), and α-smooth muscle actin (SMA) (marker of myofibroblasts). (b) Levels of blood urea nitrogen (BUN) and serum creatinine remained higher during recovery from DT-mediated acute kidney injury in DTR; IL-4/IL-13 KO mice. *P < 0.05, **P < 0.01, ***P < 0.001 versus corresponding DTR mice, n = 4. Representative histological photomicrographs determined that renal injury was still evident in DTR; IL-4/IL-13 KO mice 12 days after DT injection. Bar =180 μm. (c) Six days after DT administration, phosphorylation of signal transducer and activator of transcription 6 (pSTAT6) was intensely expressed in interstitial cells in wild-type mice, but its expression was much less in DTR; IL-4/IL-13 KO mice. Bar = 64 μm. (d) Both immunoblotting and immunostaining showed that after DT injection for 6 days IL-4/IL-13 ablation led to increases in high mobility group box 1 (HMGB1) expression, a marker of secondary necrosis. Bar = 100 μm. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Our previous studies indicated that DT-induced AKI in proximal tubular DTR mice was primarily due to epithelial cell apoptosis with minimal necrosis, but secondary necrosis occurred if there was inadequate clearance of apoptotic cells secondary to depletion of M2 macrophages.8,10 At 6 days following DT injection, there was significantly increased expression of high mobility group box 1, a marker of secondary necrosis (Figure 2d).
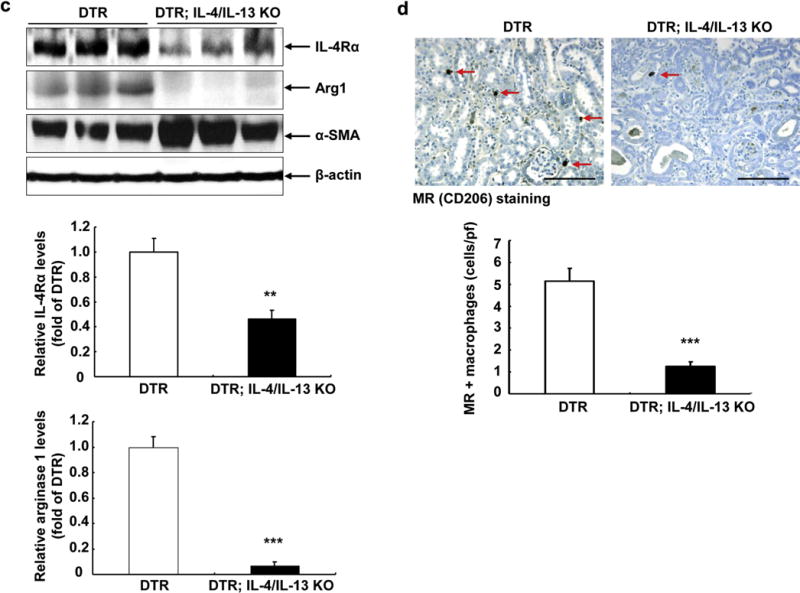
Deletion of IL-4/IL-13 led to increased renal resident macrophages/dendritic cells when compared with wild-type mice at 6 days after DT administration (Figure 3a). However, the percentage of CD206 (mannose receptor)-positive M2 macrophages among total macrophages was markedly decreased when compared with DTR mice (Figure 3b). Flow cytometry also demonstrated increased renal neutrophil infiltration in DTR; IL-4/IL-13 KO mice (Figure 3c). Immunoblotting showed increased expression of Gr-1 (a marker of neutrophils) and α-smooth muscle actin (a marker of myofibroblasts) in DTR; IL-4/IL-13 KO mice compared with DTR mice at 6 days after DT administration (Figure 2a).
Figure 3. Interleukin (IL)-4/IL-13 was essential for macrophage/dendritic cell polarization in response to diphtheria toxin–mediated acute kidney injury.

(a) Flow cytometric analysis gating with F4/80 indicated that there were more renal macrophages (F4/80+ CD11b+ CD11c−) in DTR; IL-4/IL-13 knockout (KO) mice than in diphtheria toxin receptor (DTR) mice at 6 days after diphtheria toxin injection. *P < 0.05 versus DTR mice, n = 6. (b) Further cytometric analysis showed that CD206+ macrophages (difference between F4/80+CD11b+ CD206+ cells and F4/80+ CD11c+ CD206+ cells) accounted for a much smaller percentage of total macrophage in DTR; IL-4/IL-13 KO mice. **P < 0.01 versus DTR mice, n = 8. (c) IL-4/IL-13 KO also led to increased renal neutrophil infiltration at 6 days after DT injection. **P < 0.01 versus DTR mice, n = 6. (d) Isolated renal macrophages/dendritic cells from DTR; IL-4/IL-13 KO mice had lower mRNA levels of IL-4Rα and CD209 (a marker of M2a),=but had comparable mRNA levels of B7-H4 and CD150 (markers of M2c) compared with that in DTR mice. *P < 0.05 versus DTR mice, n = 5. MR, mannose receptor.
To characterize further the polarization of renal resident macrophages/dendritic cells in DTR; IL-4/IL-13 KO mice, we isolated renal macrophages/dendritic cells from kidneys 6 days after DT injection and measured mRNA of M2 subtype markers. As indicated in Figure 3d, deletion of IL-4/IL-13 led to deceased mRNA levels of IL-4Rα, a marker of M2 macrophages. Furthermore, IL-4/IL-13 deletion also led to decreased mRNA levels of CD209, a marker of M2a, but had no effect on the mRNA levels of either B7-H4 or CD150, markers of M2c, which is consistent with previous studies that found that IL-4/IL-13 polarize macrophages to an M2a phenotype (Figure 3d).14
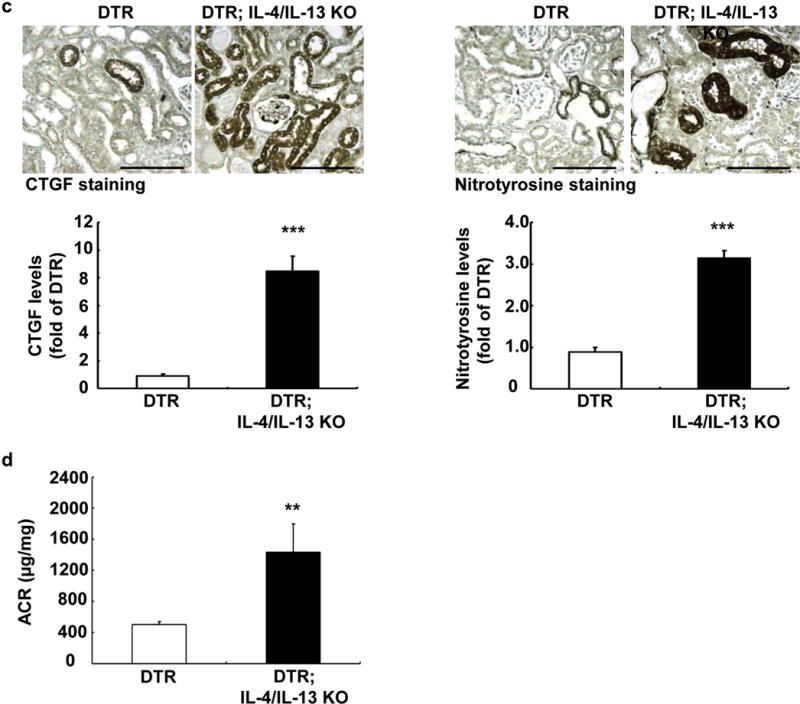
Incomplete recovery from AKI may lead to development of chronic kidney injury. To investigate whether delayed recovery from DTR AKI in DTR; IL-4/IL-13 KO would lead to the development of fibrosis, the mice were killed after 3 weeks of DT injection. There was increased tubular expression of the macrophage-homing chemokine, CX3CL-1 (fractalkine) in DTR; IL-4/IL-13 KO mice 3 weeks following DT administration (Figure 4a), in association with an increase in F4/80+ cells in both renal cortex and medulla (Figure 4b). Immunoblotting and immunohistochemistry demonstrated that DTR; IL-4/IL-13 KO kidneys had decreased expression of the “M2” markers, arginase and IL-4Rα (Figure 4c). In addition, there were fewer mannose receptor-positive interstitial cells detectable by immunostaining (Figure 4d). Furthermore, 3 weeks after DT administration, there was markedly increased tubulointerstitial fibrosis, as indicated by Masson trichrome and picrosirius red staining and increased collagen I detected by immunostaining (Figure 5a). In addition, both immunoblotting and immunostaining demonstrated increased expression of the myofibroblast marker α-smooth muscle actin (Figures 4c and 5b). There was also increased expression of the profibrotic factor, connective tissue growth factor, as well as increased nitrotyrosine, a marker of oxidative stress (Figure 5c). Finally, IL-4/IL-13 deletion led to increased urinary albuminuria (Figure 5d), which is indicative of persistent kidney injury.
Figure 4. Interleukin (IL)-4/IL-13 promoted macrophage/dendritic cell M2 polarization at 3 weeks after diphtheria toxin (DT)-mediated acute kidney injury.

(a) There was increased renal epithelial expression of CX3CL-1 (fractalkine), a macrophage-homing chemokine, in DT receptor (DTR); IL-4/IL-13 knockout (KO) mouse kidney at 3 weeks after DT administration. Bar = 100 μm. (b) Macrophage density was much higher in both renal cortex and medulla in DTR; IL-4/IL-13 KO mice 3 weeks after DT administration. ***P < 0.001 versus DTR mice, n = 4. Bar = 100 μm. (c) IL-4/IL-13 deletion led to decreased renal expression levels of IL-4Rα and arginase 1 (Arg1), markers of M2 phenotypic macrophages, but increased α-smooth muscle actin (SMA) levels at 3 weeks after DT injection. **P < 0.01, ***P < 0.001 versus DTR mice, n = 3. (d) Immunostaining indicated decreased renal CD206-positive macrophages in DTR; IL-4/IL-13 KO mice at 3 weeks after DT injection. Arrows: CD206-positive macrophages. ***P < 0.001 versus DTR mice, n = 4. Bar = 70 μm. MR, mannose receptor; pf, per field.
Figure 5. Interleukin (IL)-4/IL-13 deletion exacerbated the development of chronic kidney injury after diphtheria toxin-mediated AKI.

Both DTR; IL-4/IL-13 knockout (KO) and diphtheria toxin receptor (DTR) mice were injected with diphtheria toxin and killed 3 weeks later. (a) IL-4/IL-13 deletion led to increased renal fibrosis as indicated by increased collagen I immunostaining and Masson trichrome and Sirius red stain. ***P < 0.001 versus DTR mice, n = 4. (b,c) IL-4/IL-13 KO deletion led to increased renal expression of α-smooth muscle actin (SMA) and connective tissue growth factor (CTGF), as well as increased oxidative stress, as indicated by increased nitrotyrosine staining. **P < 0.01, ***P < 0.001 versus DTR mice, n = 3 for α-SMA and n = 4 for CTGF and nitrotyrosine. (d) IL-4/IL-13 KO deletion led to increased urinary albuminuria. **P < 0.01 versus DTR mice, n = 4. Bar = 120 μm in all. ACR, albumin-creatinine ratio.
We also determined whether IL-4/IL-13 deletion inhibited recovery from ischemia-reperfusion injury. As indicated in Figure 6a, there was a significant delay in recovery of renal function, as indicated by elevated BUN and serum creatinine levels. In previous studies, we and others had determined that by 72 hours following ischemia-reperfusion injury, there was a predominant phenotypic shift in renal resident macrophages/dendritic cells from expression of proinflammatory, “M1” phenotype markers to expression of reparative, “M2 phenotype markers.1,8 In contrast, with IL-4/IL-13 deletion, there was persistently elevated expression of M1 markers, IL-23, tumor necrosis factor α, and CC chemokine ligand 3, and decreased expression of IL-4Rα and mannose receptor (Figure 6b). Similar to DT-induced injury, there was markedly increased tubulointerstitial fibrosis in mice with IL-4/IL-13 deletion 3 weeks after ischemia-reperfusion injury, as indicated by quantitation of Picro-sirius red staining and Masson trichrome staining (Figure 7).
Figure 6. Interleukin (IL)-4/IL-13 was essential for recovery and macrophage/dendritic cell polarization in response to ischemia-reperfusion (I/R)-induced acute kidney injury.
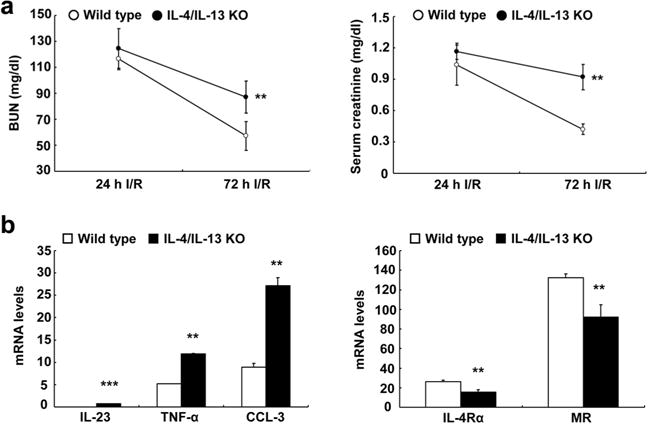
Both IL-4/IL-13 knockout (KO) and wild-type mice were uninephrectomized, immediately followed by unilateral I/R injury and killed 3 days later. (a) IL-4/IL-13 deletion led to slower recovery from I/R-induced acute kidney injury, as indicated by higher blood urea nitrogen (BUN) and serum creatinine 3 days after injury (**P < 0.01, n = 5). (b) IL-4/IL-13 deletion led to increased mRNA levels of markers of M1 phenotypic macrophages, including IL-23, tumor necrosis factor (TNF)-α, and CC chemokine ligand (CCL)-3, but decreased mRNA levels of CD206 and IL-4Rα, markers of M2 phenotypic macrophages, in isolated kidney macrophages from mice 3 days after I/R injury. **P < 0.01, ***P < 0.001, n = 7. MR, mannose receptor.
Figure 7. Interleukin (IL)-4/IL-13 deletion led to development of chronic kidney injury after ischemia-reperfusion-induced acute kidney injury.
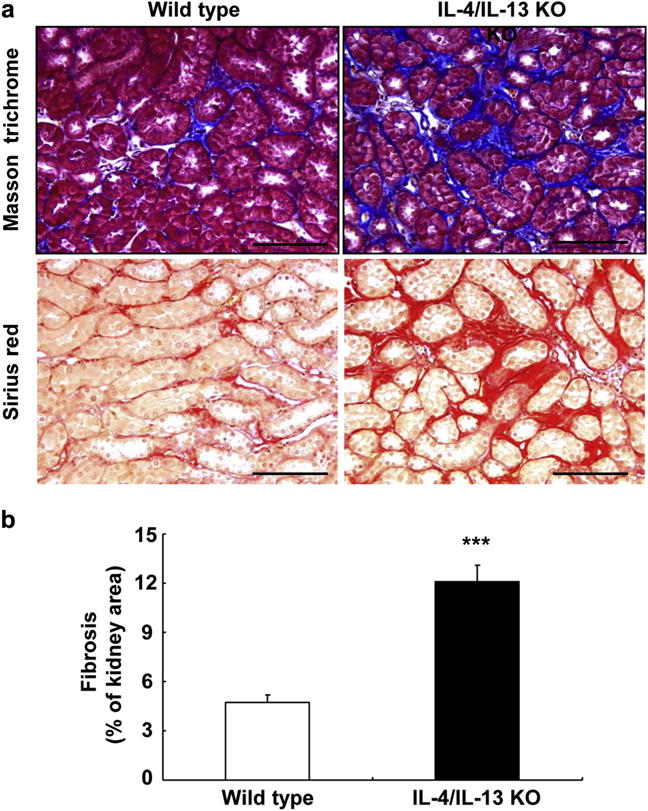
Both IL-4/IL-13 knockout (KO) and wild-type were uninephrectomized, immediately followed by unilateral ischemia-reperfusion injury and killed 4 weeks later. IL-4/IL-13 deletion led to increased renal fibrosis. (a) Masson trichrome and Picro-Sirius red staining. (b) Quantitative Picro-sirius red staining. ***P < 0.001 versus wild-type mice, n = 4. Bar = 100 μm in all.
DISCUSSION
There is increasing evidence that renal resident macrophages/dendritic cells that are polarized to a tissue reparative/anti-inflammatory phenotype are important contributors to the repair of kidney parenchyma following acute injury. In vitro and in vivo studies indicate that IL-4 and IL-13 induce renal resident macrophages/dendritic cell polarization into a wound-healing, tissue-reparative phenotype (“M2a”) whereas IL-10 promotes polarization to a regulatory, anti-inflammatory phenotype (“M2c”). Lipopolysaccharide has been shown to induce polarization to an “M2b” phenotype, which is involved in immunoregulation.14,16 Potential sources of IL-4 and IL-13 include Th2 T cells, basophils, mast cells, and granulocytes, whereas IL-10 may be derived from Tregs or induced by prostaglandins, glucocorticoids, apoptotic cells, and G protein-coupled receptor ligands. However, as mentioned, these M2 subtype constructs are somewhat artificial, because, for example, all 3 subtypes of M2 macrophages can produce IL-10.
We determined the effect of either genetic or pharmacologic inhibition of the IL-4/IL-13 signaling pathway in mediation of recovery from AKI. We used a model of selective targeting of DT to the proximal tubule. Because cell death in response to DT occurs predominantly by apoptosis, there is minimal infiltration of inflammatory macrophages in response to the injury.8 As we have shown in our previous studies, in this model, there is predominantly proliferation and differentiation of resident macrophages/dendritic cells to a reparative “M2-like” phenotype.8 With inhibition of IL-4/IL-13 signaling, there was delayed functional and structural recovery from the acute tubular injury and a subsequent increase in tubulointerstitial fibrosis, consistent with a previous report by Yokota et al.20 that deletion of IL-4 or STAT6 led to markedly worse renal function and tubular injury in ischemia-reperfusion AKI. This decreased potential for recovery from AKI was accompanied by a late increase in renal resident macrophages/dendritic cells with an inflammatory phenotype and a reduction in macrophages expressing M2 markers. In addition, there were decreased “M2a” macrophages without any decrease in expression of markers for the “M2c” phenotype. It is possible that the decreased percentage of M2-activated macrophages after DT injection in the IL-4/IL-13 KO mice was due to increased additional influx of M1 macrophages or persistence of M1-activated macrophages or both driven by the increase in neutrophils. We also determined that neutrophil number increased dramatically in IL-4/IL-13 KO mice in the DTR-AKI model. Whether this increase is a direct effect of loss of IL-4/IL-13 on neutrophil signaling/homing/activation or an indirect effect through the change in macrophage signaling needs to be further investigated.
In the current study, we did not distinguish whether the major effects on renal resident macrophage/dendritic cell polarization either were due to IL-4 or IL-13 or were the result of both cytokines. Both cytokines signal through membrane receptor dimers. IL-13 signals exclusively through a heterodimer composed of IL-4Rα and IL-13Rα1, whereas IL-4 can signal through either this IL-4Rα/IL-13Rα1 hetero-dimer or through an IL-4Rα/γc heterodimer.18,21 Intracellular signaling activated by both cytokines and mediated by either receptor subtype results in JAK3/STAT6 activation. However, studies conducted with knockout mice, neutralizing antibodies, and IL-13 antagonists suggest that in other tissues, IL-4 may serve more as an immune regulator and IL-13 as an effector of reparative functions, as well as fibrosis.22–25 In this regard, IL-13 activation in scleroderma has been shown to stimulate transforming growth factor ß and promote fibrogenesis.22
There are undoubtedly multiple other factors in addition to the IL-4/1L-13 axis that mediate renal resident macrophage/dendritic cell polarization following AKI-induced tubular injury. In addition to IL-10, other studies have implicated colony-stimulating factor 1, granulocyte macrophage–colony-stimulating factor 1 and Wnt7b in either transition to, or expansion of, the regulatory/reparative macrophage phenotype post-AKI.8,9,11,12,26 The current studies now also indicate an important role for IL-4/IL-13-polarized renal resident macrophages/dendritic cells in recovery from AKI and prevention of development of subsequent tubulointerstitial fibrosis.
MATERIALS AND METHODS
Animals
All animal experiments were performed in accordance with the guidelines and with the approval of the Institutional Animal Care and Use Committee of Vanderbilt University. Generation of transgenic mice expressing DTR in proximal tubule (or hHB-EGFTg/+) was reported previously.8 IL-4/IL-13f/f mice on BALB/c backgrounds (stock no.: 015859) and inducible ubiquitin ligase Cre mice on 129SvEv backgrounds (stock no.: 007179) were purchased from Jackson Laboratory (Bar Harbor, ME). Both hHB-EGFTg/+ mice and inducible ubiquitin ligase Cre mice were crossed with IL-4/IL-13f/f mice to generate hHB-EGFTg/+: IL-4/IL-13f/+ and inducible ubiquitin ligase Cre: IL-4/IL-13f/+ mice, which were further crossed with original IL-4/IL-13f/f mice to generate hHB-EGFTg/+: IL-4/IL-13f/f and inducible ubiquitin ligase Cre: IL-4/IL-13f/f mice, respectively. The resultant hHB-EGFTg/+: IL-4/IL-13f/f and inducible ubiquitin ligase Cre: IL-4/IL-13f/f mice were crossed to generate hHB-EGFTg/+: IL-4/IL-13f/f mice (IL-4/IL-13f/f, wild-type control) and inducible ubiquitin ligase Cre: IL-4/IL-13f/f: hHB-EGFTg/+ (inducible IL-4/IL-13 KO mice). Only age-matched male littermates were used for the experiments. Both wild-type and inducible ubiquitin ligase Cre: IL-4/IL-13f/f mice were given tamoxifen in oil (160 mg/kg/day, i.p.) for 5 consecutive days. Two weeks later, they were used for experiments.
AKI models
DT-mediated AKI was induced by a single i.p. injection of DT (200 ng/kg; Sigma-Aldrich, St. Louis, MO). Ischemia-reperfusion-induced AKI was carried out as previously described.8 Briefly, the animal was uninephrectomized, immediately followed by unilateral ischemia-reperfusion with renal pedicle clamping for 31 minutes. Depletion of monocytes/macrophages/dendritic cells was achieved by i.p. administration of clodronate (Encapsula NanoSciences, Nashville, TN) at a dose of 40 mg/kg at day 0, and 20 mg/kg every 3 days thereafter. As control, animals were treated with liposome carrier alone.
Evaluation of kidney function and histology
BUN was monitored using a Urea Assay Kit (BioAssay Systems, Hayward, CA). Serum creatinine was measured by high-performance liquid chromatography. Urinary albumin and creatinine excretion was determined using Albuwell M kits (Exocell, Philadelphia, PA).27
Antibodies
Rat antimouse F4/80 (MCA497R, a marker of macrophages) and Ly-6G (Gr-1, MCA2387, a marker of neutrophils) were purchased from AbD Serotec (now Bio-Rad Laboratories, Hercules, CA); mouse anti–mannose receptor (CD206, MAB25341) and kidney injury molecule 1 (a marker of renal tubular injury, MAB1817) were from R&D Systems (Minneapolis, MN); affinity-purified rabbit antihuman IL-4Rα (NBP1-00884) was from Novus Biologicals (Littleton, CO); goat antiarginase 1 (ab60176) and rabbit anti-CX3CL1 (ab25088) were from Abcam (Cambridge, UK); goat antihuman connective tissue growth factor (SC-14939), IL-4 (SC-1260), IL-13 (SC-1292), pSTAT6, and rabbit antinitrotyrosine (SC-55256) were from Santa Cruz Biotechnology (Santa Cruz, CA); rabbit antimurine collagen type I (600-401-103-01) was from Rockland Immunochemicals (Limerick, PA); mouse anti-α-smooth muscle actin (A5228, a marker of myofibroblasts) was from Sigma (A5228); and pSTAT5 and rabbit anti–high mobility group box 1 (a marker of secondary necrosis) were from Cell Signaling Technologies (Danvers, MA).
RNA isolation and quantitative reverse transcriptase polymerase chain reaction
Total RNA from kidney or isolated renal macrophages/dendritic cells was isolated using TRIzol reagents (Invitrogen, Thermo Fisher Scientific, Waltham, MA). Quantitative reverse transcriptase polymerase chain reaction was performed using TaqMan real-time polymerase chain reaction (7900HT; Applied Biosystems, Thermo Fisher Scientific).28 The Master Mix and all gene probes were also purchased from Applied Biosystems. The probes used in the experiments included mouse S18 (Mm02601778), inducible nitric oxide synthase (Mm00440502), CC chemokine ligand 3 (Mm00441258), mannose receptor (Mrc1, Mm01329362), IL-23 (Mm00518984), IL-4Rα (Mm01275139), tumor necrosis factor α (Mm99999068), CD209 (Cd209a, a marker of M2a), Mm00460067), B7-H4 (Vtcn1, a marker of M2c, Mm00628552), and CD150 (Slamf1, a marker of M2c, Mm00443316).
Immunohistochemistry staining and quantitative image analysis
The animals were anesthetized with Nembutal (70 mg/kg, i.p.) and given heparin (1000 units/kg, i.p.) to minimize coagulation. After 1 kidney was removed for immunoblotting and quantitative reverse transcriptase polymerase chain reaction, the animal was perfused with 3.7% formaldehyde, 10 mmol/l sodium m-periodate, 40 mmol/l phosphate buffer, and 1% acetic acid through the aortic trunk cannulated by means of the left ventricle.29 The fixed kidney sections were dehydrated through a graded series of ethanols, embedded in paraffin, sectioned (4 μm), and mounted on glass slides. Immunostaining and quantitative image analysis were carried out as in previous reports.30
Immunoblotting
Kidney samples were homogenized with buffer containing 10 mmol/l tris(hydroxymethyl)-aminomethane·HCl (pH 7.4), 50 mmol/l NaCl, 2 mmol/l ethyleneglycol-bis-(β-aminoethylether)-N,N,N′,N′-tetraacetic acid, 2 mmol/l ethylenediamine tetraacetic acid, 0.5% Nonidet P-40, 0.1% sodium dodecylsulfate, 100 μmol/l Na3VO4, 100 mmol/l NaF, 0.5% sodium deoxycholate, 10 mmol/l sodium pyrophosphate, 1 mmol/l phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, and 10 μg/ml leupeptin. The homogenate was centrifuged at 15,000g for 20 minutes at 4 °C. An aliquot of supernatant was taken for protein measurement with a bicinchoninic acid protein assay kit (ThermoScientific, Thermo Fisher Scientific). Immunoblotting was described in a recent report.31
Isolation of renal monocytes/macrophages
CD11b-expressing cells in kidney single-cell suspensions were enriched using mouse CD11b MACS Microbeads and MACS Columns (Miltenyi Biotec, Auburn, CA) following the manufacturer’s protocol.
Flow cytometry
Flow cytometric analysis was performed as previously described.27 In brief, after perfusion of the animal with cold phosphate-buffered saline, 1 kidney was removed, minced into fragments, and digested in RMPI 1640 containing 2 mg/ml collagenase type D and 100 μ/ml DNase I for 1 hour at 37 °C, with intermittent agitation. Kidney fragments were passed through a 70-μm mesh (Falcon; BD Bio-sciences, San Jose, CA), yielding single cell suspensions. Cells were centrifuged (800g, 10 minutes, 8 °C), resuspended in fluorescence activated cell sorting (FACS) buffer, kept on ice and counted. Then, 107 cells were incubated in 2.5 μg/ml Fc blocking solution, centrifuged again (800g, 10 minutes, 8 °C), and resuspended with FACS buffer. After that, 106 cells were stained for 20 minutes at room temperature with antibodies including fluorescein isothiocyanate rat antimouse CD45 (clone 30-F11), phycoerythrin (PE)/Cy7 antimouse F4/80 (clone BM8), PE antimouse CD11b (clone M1/70), antigen-presenting cell antimouse CD11c (clone N418), Pacific Blue anti-mouse CD3 (clone 17A2) (BD Biosciences), Alexa fluor647 antimouse neutrophils (clone 7/4) (AbD Serotec), and PE antimouse CD206 (mannose receptor, clone C068C2), then washed in sterile phosphate-buffered saline, and resuspended with 7-Aminoactinomycin D to permit exclusion of nonviable cells. Finally the cells were washed and resuspended in FACS buffer. After immunostaining, cells were analyzed immediately on a Facs Canto II cytomer with DIVA software (Becton Dickinson) and offline list mode data analysis was performed using Winlist software from Verity Software House (Topsham, ME).
Micrography
Bright-field images from a Leitz Orthoplan microscope with a DVC digital RGB video camera were digitized and saved as computer files. Contrast and color level adjustment (Adobe Photoshop, San Jose, CA) were performed for the entire image; in other words, no region-or object-specific editing or enhancements were performed.
Statistics
All values are presented as means, with error bars representing ± SE. Fisher exact test, analysis of variance, and Bonferroni t-tests were used for statistical analysis.
Supplementary Material
Figure S1. Both total signal transducer and activator of transcription 6 (STAT6) and phosphorylation of STAT6 (pSTAT6) levels increased to similar extent after diphtheria toxin (DT) injection, suggesting that increased pSTAT6 levels are primarily due to increased total STAT6.
Figure S2. CP-690550 had no effect on signal transducer and activator of transcription 5 activation. Six days after diphtheria toxin (DT) injection, total kidney phosphorylation of signal transducer and activator of transcription 5 (pSTAT5) levels increased, and CP-690550 treatment had no effect on pSTAT5 levels.
Acknowledgments
This work was supported by National Institute of Health grants DK-95785 and DK-51265 (to MZZ and RCH), DK-62794 (to RCH), and by funds from the Department of Veterans Affairs (to RCH).
Footnotes
DISCLOSURE
All the authors declared no competing interests.
SUPPLEMENTARY MATERIAL
Supplementary material is linked to the online version of the paper at www.kidney-international.org.
References
- 1.Devarajan P. Emerging urinary biomarkers in the diagnosis of acute kidney injury. Expert Opin Med Diagn. 2008;2:387–398. doi: 10.1517/17530059.2.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson RJ, Ray CJ, Popoff MR. Evidence for Rho protein regulation of renal tubular epithelial cell function. Kidney Int. 2000;58:1996–2006. doi: 10.1111/j.1523-1755.2000.00372.x. [DOI] [PubMed] [Google Scholar]
- 3.Day YJ, Huang L, Ye H, et al. Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: role of macrophages. Am J Physiol Renal Physiol. 2005;288:F722–F7231. doi: 10.1152/ajprenal.00378.2004. [DOI] [PubMed] [Google Scholar]
- 4.Lu LH, Oh DJ, Dursun B, et al. Increased macrophage infiltration and fractalkine expression in cisplatin-induced acute renal failure in mice. J Pharmacol Exp Ther. 2008;324:111–117. doi: 10.1124/jpet.107.130161. [DOI] [PubMed] [Google Scholar]
- 5.Jang HS, Kim J, Park YK, Park KM. Infiltrated macrophages contribute to recovery after ischemic injury but not to ischemic preconditioning in kidneys. Transplantation. 2008;85:447–455. doi: 10.1097/TP.0b013e318160f0d1. [DOI] [PubMed] [Google Scholar]
- 6.Vinuesa E, Hotter G, Jung M, et al. Macrophage involvement in the kidney repair phase after ischaemia/reperfusion injury. J Pathol. 2008;214:104–113. doi: 10.1002/path.2259. [DOI] [PubMed] [Google Scholar]
- 7.Lee S, Huen S, Nishio H, et al. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol. 2011;22:317–326. doi: 10.1681/ASN.2009060615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang MZ, Yao B, Yang S, et al. CSF-1 signaling mediates recovery from acute kidney injury. J Clin Invest. 2012;122:4519–4532. doi: 10.1172/JCI60363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huen SC, Huynh L, Marlier A, et al. GM-CSF promotes macrophage alternative activation after renal ischemia/reperfusion injury. J Am Soc Nephrol. 2015;26:1334–1345. doi: 10.1681/ASN.2014060612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Chang J, Yao B, et al. Proximal tubule-derived colony stimulating factor-1 mediates polarization of renal macrophages and dendritic cells, and recovery in acute kidney injury. Kidney Int. 2015;88:1274–1282. doi: 10.1038/ki.2015.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alikhan MA, Jones CV, Williams TM, et al. Colony-stimulating factor-1 promotes kidney growth and repair via alteration of macrophage responses. Am J Pathol. 2011;179:1243–1256. doi: 10.1016/j.ajpath.2011.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Menke J, Iwata Y, Rabacal WA, et al. CSF-1 signals directly to renal tubular epithelial cells to mediate repair in mice. J Clin Invest. 2009;119:2330–2342. doi: 10.1172/JCI39087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Wang YP, Zheng G, et al. Ex vivo programmed macrophages ameliorate experimental chronic inflammatory renal disease. Kidney Int. 2007;72:290–299. doi: 10.1038/sj.ki.5002275. [DOI] [PubMed] [Google Scholar]
- 14.Jones CV, Ricardo SD. Macrophages and CSF-1: implications for development and beyond. Organogenesis. 2013;9:249–260. doi: 10.4161/org.25676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benoit M, Desnues B, Mege JL. Macrophage polarization in bacterial infections. J Immunol. 2008;181:3733–3739. doi: 10.4049/jimmunol.181.6.3733. [DOI] [PubMed] [Google Scholar]
- 18.Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annu Rev Immunol. 2013;31:317–343. doi: 10.1146/annurev-immunol-032712-095906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghoreschi K, Jesson MI, Li X, et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550) J Immunol. 2011;186:4234–4243. doi: 10.4049/jimmunol.1003668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yokota N, Burne-Taney M, Racusen L, Rabb H. Contrasting roles for STAT4 and STAT6 signal transduction pathways in murine renal ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2003;285:F319–F325. doi: 10.1152/ajprenal.00432.2002. [DOI] [PubMed] [Google Scholar]
- 21.Malabarba MG, Rui H, Deutsch HH, et al. Interleukin-13 is a potent activator of JAK3 and STAT6 in cells expressing interleukin-2 receptor-gamma and interleukin-4 receptor-alpha. Biochem J. 1996;319:865–872. doi: 10.1042/bj3190865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wynn TA. IL-13 effector functions. Annu Rev Immunol. 2003;21:425–456. doi: 10.1146/annurev.immunol.21.120601.141142. [DOI] [PubMed] [Google Scholar]
- 23.Novak ML, Koh TJ. Macrophage phenotypes during tissue repair. J Leukoc Biol. 2013;93:875–881. doi: 10.1189/jlb.1012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Reilly S. Role of interleukin-13 in fibrosis, particularly systemic sclerosis. Biofactors. 2013;39:593–596. doi: 10.1002/biof.1117. [DOI] [PubMed] [Google Scholar]
- 25.Lech M, Anders HJ. Macrophages and fibrosis: how resident and infiltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair. Biochim Biophys Acta. 2013;1832:989–997. doi: 10.1016/j.bbadis.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 26.Lin SL, Li B, Rao S, et al. Macrophage Wnt7b is critical for kidney repair and regeneration. Proc Natl Acad Sci U S A. 2010;107:4194–4199. doi: 10.1073/pnas.0912228107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang MZ, Yao B, Yang S, et al. Intrarenal dopamine inhibits progression of diabetic nephropathy. Diabetes. 2012;61:2575–2584. doi: 10.2337/db12-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang MZ, Yao B, Wang Y, et al. Inhibition of cyclooxygenase-2 in hematopoietic cells results in salt-sensitive hypertension. J Clin Invest. 2015;125:4281–4294. doi: 10.1172/JCI81550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang MZ, Wang JL, Cheng HF, et al. Cyclooxygenase-2 in rat nephron development. Am J Physiol. 1997;273:F994–F1002. doi: 10.1152/ajprenal.1997.273.6.F994. [DOI] [PubMed] [Google Scholar]
- 30.Zhang MZ, Yao B, Cheng HF, et al. Renal cortical cyclooxygenase 2 expression is differentially regulated by angiotensin II AT(1) and AT(2) receptors. Proc Natl Acad Sci U S A. 2006;103:16045–16050. doi: 10.1073/pnas.0602176103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang MZ, Wang Y, Yao B, et al. Role of epoxyeicosatrienoic acids (EETs) in mediation of dopamine’s effects in the kidney. Am J Physiol Renal Physiol. 2013;305:F1680–F1686. doi: 10.1152/ajprenal.00409.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Both total signal transducer and activator of transcription 6 (STAT6) and phosphorylation of STAT6 (pSTAT6) levels increased to similar extent after diphtheria toxin (DT) injection, suggesting that increased pSTAT6 levels are primarily due to increased total STAT6.
Figure S2. CP-690550 had no effect on signal transducer and activator of transcription 5 activation. Six days after diphtheria toxin (DT) injection, total kidney phosphorylation of signal transducer and activator of transcription 5 (pSTAT5) levels increased, and CP-690550 treatment had no effect on pSTAT5 levels.