

Abstract
The energetic nature of the interactions of DNA base excision repair glycosylases with undamaged and damaged DNA and the nuclear environment are expected to significantly impact the time it takes for these enzymes to search for damaged DNA bases. In particular, the high concentration of monovalent ions, macromolecule crowding, and densely packed DNA chains in the cell nucleus could alter the search mechanisms of these enzymes as compared to findings in dilute buffers typically used in in vitro experiments. Here we utilize an in vitro system where the concerted effects of monovalent ions, macromolecular crowding and high concentrations of bulk DNA chains on the activity of two paradigm human DNA glycosylases can be determined. We find that the energetic nature of the observed binding free energies of human 8-oxoguanine DNA glycosylase (hOGG1) and human uracil DNA glycosylase (hUNG) for undamaged DNA are derived from different sources. While hOGG1 uses primarily non-electrostatic binding interactions with non-specific DNA, hUNG uses a salt-dependent electrostatic binding mode. Both enzymes turn to a non-electrostatic mode in their specific complexes with damaged bases in DNA, which enhances damage site specificity at physiological ion concentrations. Neither enzyme was capable of efficiently locating and removing their respective damaged bases in the combined presence of physiological ions and a bulk DNA chain density approximating that found in the nucleus. However, the addition of an inert crowding agent to mimic macromolecular crowding in the nucleus largely restored their ability to track DNA chains and locate damaged sites. These findings suggest how the concerted action of monovalent ions and crowding could contribute to efficient DNA damage recognition in cells.
Graphical abstract
DNA is subjected to an array of chemical modifications (oxidation, deamination, alkylation) that result in a diversity of damaged bases. Although these DNA lesions are rare in the context of the billions of bases that make up the human genome, they can have devastating consequences if allowed to persist into S phase where their deleterious effects on the genetic code are made permanent by DNA replication errors1, 2. DNA base excision repair glycosylases are charged with the task of searching the genome for damaged bases and initiating repair by cleaving the glycosidic bond connecting the base and sugar3, 4. The efficiency of the search process relies on the fraction of the total search time that the enzyme remains in the vicinity of DNA as opposed to diffusing through regions of the nucleus that do not contain potential damage sites. The localization of glycosylases to the vicinity of DNA chains involves both intrinsic properties of the enzyme (such as electrostatic or non-electrostatic interaction energies)5, 6, 7, 8, as well as properties of the nuclear environment (such as molecular crowding) that tend to favor the formation of DNA-protein complexes9, 10, 11, 12, 13. An important property that affects the search time is the binding lifetime of a DNA glycosylase with undamaged DNA. This lifetime must be sufficiently long to permit recognition of damage when it is encountered, yet short enough to allow dissociation events that facilitate movement of the enzyme to other DNA chains14, 15, 16. Thus, two fundamental questions are the nature of the energetic interactions of DNA glycosylases with undamaged and damaged DNA and the contribution of the nuclear environment to the search time and recognition mechanism. Indeed, the effects of crowding in a human cell are significant with macromolecule concentrations of ~100–300 mg/ml, indicating that 10–40% of the total cellular volume is consumed by large molecules (often called the excluded volume)10, 13, 17.
We have undertaken a detailed analysis of the effects of key solution parameters (i.e. ion concentration and crowding agents) on the specificity of human uracil DNA glycosylase (hUNG) for undamaged and uracil containing DNA8, 9. This work has shown that the positively charged DNA binding site of hUNG promotes weak electrostatic interactions with undamaged DNA chains. These weak interactions facilitate two general types of translocation events along DNA chains: associative translocations where the enzyme remains within the DNA ion cloud and moves relatively short <10bp distances and dissociative translocations where the enzyme diffuses outside of the local ion environment of the DNA, but eventually rebinds to the same DNA chain18, 19, 20, 21, 22. The two mechanisms work cooperatively to maximize the search coverage and minimize the search time18, 20, 22. Although the specific complex between hUNG and uracil-containing DNA retains the non-specific electrostatic binding energy, specific binding is enhanced by an additional non-electrostatic free energy component (ΔGnon = −7.2 kcal/mol; at a 1 M standard state of [KGlu]), which derives from unique interactions with the extrahelical uracil base in the specific complex8, 23. This additional term enhances specificity for uracil sites at physiological ion concentrations8.
In addition to the above intramolecular translocation events, frequent dissociative transfers also provide an opportunity for the enzyme to move away from the original DNA chain and encounter a new DNA chain (intermolecular transfers). In dilute aqueous solutions where diffusion is not hindered, these events serve to enhance global repair in a population of damaged DNA molecules by allowing the enzyme to move rapidly between DNA chains8, 18. However, in the presence of molecular crowding (i.e. 20% PEG8000), the rate of global repair is decreased because the crowding macromolecule increases the time that hUNG spends translocating along individual DNA chains, thereby hindering its rapid movement to other damaged DNA chains9. These key observations concerning the effect of crowded environments on the efficiency of global DNA repair suggested that efficient repair in the crowded cell nucleus is facilitated by the high hUNG copy number in dividing cells (~105 molecules/nucleus)24. Thus on average, individual enzyme molecules can be charged with efficient scanning of fairly small 15kb DNA domains, forgoing the requirement for global coverage by a limited number of enzyme molecules. A remaining question is whether these mechanistic determinants of hUNG transpose to other DNA repair glycosylases?
In humans, oxidative damage to guanine bases is primarily repaired by 8-oxoguanine DNA glycosylase (hOGG1)25. This glycosylase is a classic member of the Helix-hairpin-Helix Gly/Pro/Asp (HhH-GPD) superfamily and differs significantly from hUNG in overall structure, charge distribution across its active site, and its propensity to deform both damaged and undamaged DNA26, 27. The marked differences between these two enzymes raised the distinct possibility that different mechanisms might be used to solve the search and recognition problem. To investigate this issue, we now describe a comparative study of the factors that govern the activity of hUNG and hOGG1 with damaged and undamaged DNA. In particular, we explore the consequences of three key solution variables that are relevant to the cell nucleus: monovalent ions, molecular crowding, and high solution densities of undamaged DNA chains.
MATERIALS AND METHODS
Experimental Conditions
All experiments with hOGG1 were performed in buffer containing 15 mM potassium phosphate pH 7.5, 1 mM EDTA, and 0.1 mg/mL BSA. This buffer contained a total of 30 mM K+ originating from pH adjustment of potassium phosphate and EDTA. Increasing ionic strength measurements were made by supplementing the buffer with potassium acetate (KOAc). All experiments with hUNG were performed in a buffer consisting of 20 mM HEPES pH 7.5, 3 mM EDTA, 1 mM DTT, 0.002% Brij 35. This buffer contained a total of 22 mM K+ originating from pH adjustment of the HEPES and EDTA stock solutions. The catalytic domain of recombinant hUNG, WT hOGG1 and the K249Q mutant hOGG1 were purified as previously described9. All oligonucleotides were purchased from either Integrated DNA Technologies or Eurofin and purified in-house by denaturing PAGE. All DNA sequences are listed in Supplemental Methods. PEG 8K was purchased from Sigma Chemical and was purified by overnight treatment with activated carbon (0.1g/mL) and filtered to remove UV absorbing impurities. Salmon sperm DNA (salDNA) was purchased from ThermoFisher Scientific.
hOGG1 Equilibrium DNA Binding Measurements
Binding of WT and K249Q hOGG1 to a non-specific 5’ fluorescein-labeled 20mer DNA duplexes was measured by fluorescence anisotropy using a SPEX Fluoromax-3 spectrofluorometer at 20 °C (λex = 494 nm, λem = 518 nm) under magic angle conditions (excitation polarizer at 0° and emission polarizer at 55°). All anisotropy values were corrected for the spectral G factors. Experiments were performed in a back-titration mode by diluting a solution of concentrated hOGG1 containing 25 nM labeled DNA with increasing volumes of a solution containing 25 nM labeled DNA only. After each addition, the solution was allowed to equilibrate for 4 min inside the fluorometer and three anisotropy measurements were averaged. The active hOGG1 concentration present in these experiments (50%) was determined by single-turnover burst amplitude measurements in which 1 nM hOGG1 was mixed with 25 nM of a 5′ 32P-labeled 31mer duplex containing a single 8-oxoG site (S0oxoG)28. Prior to data fitting the raw anisotropy values were converted to fraction of labeled DNA bound (FB) using eq 129, where AOBS is the observed anisotropy, AF and AB are the anisotropy corresponding to free and bound labeled DNA and Q = Fbound/Ffree is the ratio of the fluorescence intensities of the bound and free fluorescein label. The correction for fluorescence intensity changes was necessary because the signal decreased about ~20% upon binding of hOGG1.
| (1) |
The bound fraction (FB) as a function of active hOGG1 concentration was fit to eq 2 using the software Prism, where FB0 and FBmax are the minimum and maximum fraction bound (0 and 1, respectively). The full anisotropy change was not attainable for the higher salt titrations due to limits of protein solubility. However, we achieved ≥75% saturation for all experiments and the binding curves extrapolated to the same final anisotropy when this parameter was used as a floating variable in the curve fitting process.
| (2) |
The binding affinity of hOGG1 for specific, 8-oxoguanine-containing DNA (KDS) was determined using the same method. In these experiments a 10 nM solution of specific DNA (DS) containing a single 8-oxoguanine and a 5′-FAM label on the complimentary strand was mixed with catalytically inactive K249Q hOGG1.
To determine whether the 5′-FAM label had a significant effect on the measured KD values, the binding affinities for the unlabeled versions of DN and DS were determined at low and high salt in a competition binding experiment (30 mM K+ and 150 mM K+). In these titration experiments, WT or K249Q hOGG1 were pre-bound to 5′-FAM-labeled DN and DS, respectively, using enzyme concentrations high enough that ~80% of the labeled DNA was bound. Further details of these competition binding experiments are found in the Supplementary Discussion, Supplementary Figure S1, and Table S1. The KD values for the labeled and unlabeled DNAs differed by a factor of two or less, and more importantly, the salt dependences of the KD values for the labeled and unlabeled DNAs were essentially identical (Table S1). Although direct measurement of the binding stoichiometry was hampered by the comparatively weak binding of the nonspecific DNA sequences, a 1:1 binding stoichiometry with the 20mer DNA sequences is supported by (i) structural information on the hOGG1 footprint and its DNA bending activity, which would preclude the presence of more than one enzyme molecule per DNA (Figure 1), and (ii) an anisotropy–based binding experiment carried out using a shorter 15mer yielded identical binding parameters as the 20mer (Supplementary Figure S2).
Figure 1.

Schematics of undamaged and damaged DNA bound to hOGG1. (A) Depiction of electrostatic (dashed lines) and non-electrostatic (solid lines) contacts between undamaged DNA and WT hOGG1 (Protein Data Bank entry 1YQK26). The top strands of all undamaged DNA sequences used in this studied are listed. A fluorescein label (not shown) is attached to the 5’ end of each strand. Full sequences are listed in Supplemental Methods. (B) Schematic of electrostatic (dashed arrows) and the non-electrostatic (solid arrows) interactions between hOGG1 and specific DNA (Protein Data Bank entry 1EMH27. The top strand of the specific DNA sequence used in this study is shown. A 5’ fluorescein label was attached to the complimentary strand (not shown, see Supplemental Methods). Electrostatic interactions are defined by nitrogen and oxygen atoms of the enzyme within 3.3 Å of DNA phosphate oxygens or heteroatoms on the bases, while non-electrostatic interactions are defined as any enzyme carbon atom within 3.9 Å of a DNA carbon. Hydrogen bonds with the cytosine on the complementary strand are not expected to result in ion displacement. The yellow circles represent a potassium ion, which is likely to be bound in place of the Ca2+ ion observed in crystal structures of both the damaged and undamaged DNA complexes.
Salt Dependence of DNA Binding
An evaluation of the electrostatic and non-electrostatic contributions to the binding free energy of both nonspecific and specific complexes formed by hOGG1 was carried out utilizing counter-ion condensation theory30. This theory attributes the driving force of protein-DNA binding to the cratic entropy of mixing gained by displacement of ions from the condensed DNA ion cloud into the more dilute bulk solution. Experimentally, the log of the binding affinity is plotted against the log of the corresponding salt concentration and the data are fit to eq 3 using linear regression, where the slope N is typically attributed to the
| (3) |
number of anions and cations displaced from the DNA and protein upon binding31, 32. In this study, we use N as a qualitative indicator of the dependence of binding on electrostatic interactions. Exact enumeration of the number ions released upon hOGG1 binding is complicated by the severe distortion of the DNA duplex, which negates use of standard B DNA values for the average number of cations bound per phosphate group33. The second term in eq 3 becomes zero when [salt] = 1 M and accordingly, the first term reports on the binding affinity at a 1 M standard state of salt. Thus, the free energy of non-electrostatic interactions that do not involve ion displacement can be obtained from the equation ΔGnon (1 M) = −2.303RTlogKanon. The electrostatic contribution to the observed binding free energy at physiological salt concentration (taken as 150 mM K+) may be obtained from eq 4.
| (4) |
K249Q Activity Assay
To ensure that the K249Q mutation rendered hOGG1 catalytically inactive, a 25 nM solution of a 5′ 32P-labeled 31mer duplex containing a single 8-oxoG site (S0oxoG) was incubated with 3 µM enzyme for 2 hours at 20°C in buffer containing 150 mM K+. These conditions were chosen to ensure that no appreciable amount of DNA was cleaved within the 2 hours it took to complete the specific DNA binding experiments. Samples were quenched with formamide and heated at 95°C for 30 min the presence and absence of 100 mM NaOH. DNA fragments were separated using a denaturing 10% polyacrylamide gel and the gels were dried and exposed overnight to a storage phosphor screen and imaged with a Typhoon 8600 phosphorimager (GE Healthcare). All gel images were quantified by box method using QuantityOne (Bio-Rad). Background correction was made by subtracting the intensity of the gel directly below each band from the intensity of the corresponding band.
Site Transfer Assay
Site transfer assays were performed as previously reported and the general procedure is recapitulated here34, 35, 36. To initiate the reactions, hUNG (20 pM - 190 nM) was mixed with a dual uracil 32P labeled DNA substrate (40 nM) at 37 °C using the buffer noted above supplemented with all combinations of the following solution components (see results) 22 mM potassium from buffer pH adjustment, 150 mM potassium by addition of 128 mM KOAc, 20% PEG 8K, 100 µM salmon sperm DNA (salDNA), and 1 mM salDNA. The 190 nM concentration of hUNG was only used with buffer containing 22 mM K+, 20% PEG 8K, and either 100 µM or 1 mM salDNA due to extreme inhibition of the enzyme in these solution conditions. In the presence of 0.1 or 1 mM salDNA, most hUNG is bound and the free enzyme concentration is low enough to maintain the initial rate, single encounter conditions required for site transfer experiments34, 36. At various times, aliquots of the reaction mixture were quenched with Uracil DNA Glycosylase Inhibitor protein (UGI, New England Biolabs) and the abasic sites were cleaved by heating at 95°C in the presence of 100 mM KOH for 30 min. For experiments using hOGG1, enzyme (1 to 40 nM) was added to 20 nM 32P-labeled S20oxoG in the presence of all solution conditions noted above. Concentrations of hOGG1 >5 nM were only used in experiments containing salDNA for the same reasons outlined above for hUNG. Aliquots of the reaction were quenched with 20 µL formamide loading buffer and heated for 15 min at 95 °C. For both enzymes, the discrete DNA fragments generated by heating were resolved by electrophoresis on a denaturing 10% PAGE gel containing 7 M urea. All gels were dried, exposed overnight to a storage phosphor screen and imaged with a Typhoon 8600 phosphorimager (GE Healthcare). All gel images were quantified using QuantityOne (Bio-Rad) by the box method and background corrected as described above. The observed transfer probabilities (Ptransobs) were determined using equation 5 and the time independent transfer probability (Ptrans) was calculated by linear extrapolation of the Ptransobs values to zero time as previously described34, 36.
| (5) |
The fractional extent of reaction at each time was quantified by phosphorimaging of the gels and the reaction rates for both hUNG and hOGG1 under all tested conditions were determined from the linear slopes of plots of product concentration against time using Prism.
Turnover Rate Determination
The steady-state turnover rates of hUNG and hOGG1 under various solution conditions were determined using the S20U and S20oxoG substrates, respectively. The fraction reaction at each time point was calculated from the ratio of the summed intensities of all product bands to the summed intensity of substrate and product bands. The fraction reaction was plotted as a function of time and the slopes were determined by linear regression fitting using data where the reaction had proceeded to <30%. The slopes were converted to an observed rate constant (V0/[E]) by multiplying by the total DNA concentration and dividing by the enzyme concentration. For rate measurements with hUNG, [S20U] = 40 nM, [hUNG] = 20 pM (no salDNA) to 190 nM (in the presence of salDNA). For hOGG1, [S20oxoG] = 20 nM; [hOGG1] = 1 nM (no salDNA) to 40 nM (in the presence of salDNA). Because of day-to-day variability in the activity of hOGG1, we compared the turnover rates under various conditions using a single stock of enzyme on the same day.
RESULTS
Structural Aspects of Non-specific and Specific hOGG1-DNA Complexes
Crystal structures of complexes of hOGG1 with undamaged and damaged DNA have provided a detailed model of the binding interface, which provides a useful starting place for understanding the effects of solution ions on binding of non-specific and specific DNA (PDB entries 1YQR and 1EMH, respectively; Figure 1A, 1B). The binding interfaces in these structures encompass over 2,200 Å2 of surface area and include a peripheral ‘exo’ binding site that can accommodate an undamaged guanine base in the non-specific complex (Figure 1A) and the catalytic active site, which is specific for 8-oxoG (Figure 1B)26, 27. Unlike most DNA-binding proteins, the DNA binding site of hOGG1 is lacking in charged residues with the possible exception of a single basic residue, H270, which is implicated in the discrimination between guanine and 8-oxoguanine27, 37. Upon binding to an undamaged DNA segment (Figure 1A), hOGG1 induces a pronounced bend (~ 80°) and rotation of the DNA (~20° about the helix axis), which apparently precludes the formation of potential ionic interactions between protein residues and the phosphate backbone of the DNA. This structure also suggests that non-electrostatic binding energy might be gained by displacement of an undamaged guanine residue from the base stack into the ‘exo’ guanine binding site. The distorted DNA helix is apparently stabilized by insertion of an asparagine into the space vacated by the guanine base.
When hOGG1 binds to 8-oxoG-containing DNA, all of the contacts in the non-specific complex are maintained (Figure 1B). However, the reduced bend angle of the DNA helix positions the phosphate backbone in close proximity to two side chains that form unique hydrogen bonds with the phosphate backbone (His270 and Asn150) and also allows positioning of the 8-oxoG base deep into the active site with the formation of additional non-ionic interactions. We note that the severely bent non-specific DNA complex of hOGG1 and DNA differs considerably from the relatively unperturbed B duplex structure of non-specific DNA bound to hUNG, even though hUNG partially extrudes normal thymine bases into a similar exo-site bound conformation during its search for uracils in the context of U/A base pairs14. In contrast, the specific complex of hUNG with damaged DNA shows significant bending similar to that of hOGG123.
Ion Effects on Binding of hOGG1 to Undamaged DNA
We were interested in whether the structural differences between hOGG1 and hUNG led to distinct electrostatic and non-electrostatic contributions to their binding energies for damaged and undamaged DNA. As in a previous study with hUNG,8 we used fluorescence anisotropy to measure the equilibrium association constants between hOGG1 and non-specific DNA (KaN) under a range of potassium ion concentrations (30 mM – 300 mM) using acetate as the counterion. While physiological steady-state potassium levels in human cells have been estimated to be ~150 mM17, we chose to sample a range of concentrations above and below that value to characterize the electrostatic nature of DNA binding. A carboxylate ion like acetate is a relevant counterion given the prevalence of carboxylate anions in the cell and the weak binding of carboxylate anions to ammonium side chains of proteins38. We also explored several DNA sequences to probe whether the G–C content of the DNA affected the binding energetics of hOGG1. The different DNA sequences were designed to contain a random distribution of A–T and G–C base pairs (DN), no G–C base pairs (DTATA), a single G–C base pair (DGC), or entirely G-C base pairs (DGCGC) (Figure 1A). A fluorescein label was attached to the 5′ end of one DNA strand to follow the increase in fluorescence anisotropy that accompanied hOGG1 binding (Figure 2A). The logarithm of the binding affinities of hOGG1 for each duplex was plotted against the logarithm of the salt concentration present in the binding reaction (Figure 2C). The slopes (N) and intercepts of these plots were then used to determine the electrostatic (ΔGelect) and non-electrostatic (ΔGnon) contributions to the binding free energies as previously described6, 8. Binding affinities at all salt concentrations for DN are listed in Table 1.
Figure 2.

Salt dependence of the non-specific and specific DNA binding affinity determined by fluorescence anisotropy measurements at 20°C. (A) Fraction bound of the non-specific DNA (DN = 25 nM) as a function of hOGG1 concentration at varying potassium ion concentrations (30 mM – 300 mM). Binding curves for the additional non-specific DNA oligos are provided in Figure S3. The salt concentrations listed match the relative positions of the corresponding data set. (B) Fraction bound of the specific DNA (DS = 10 nM) as a function of K249Q concentration at various potassium ion concentrations (30 mM – 200 mM). The salt concentrations listed match the relative positions of the corresponding data set. Data presented in both A and B are single trial measurements. The standard deviation in three replicate anisotropy intensity measurements was less than ±10% and that the Ka values and associated errors from independent measurements are reported in Tables 1 and 2. (C) Dependence of non-specific association constant (KaN) on the concentration of potassium acetate for WT hOGG1 binding to DN (black), DTATA (blue), DGC (purple), and DGCGC (red). Error bars are smaller than the size of the data markers. (D) Salt dependence of the non-specific (KaN) (black, left y-axis) and specific association constants (KaS) (red, right y-axis) for K249Q binding to DN and DS. Error bars are smaller than the size of the data markers. For comparison, black and grey dashed lines in C and D show the published salt dependences for non-specific and specific DNA binding by hUNG8.
Table 1.
Binding affinities (KD = 1/Ka) of hOGG1 for nonspecific DNA and specific DNA sequences at various concentrations of potassium acetatea
| [K+] (mM) | 1/Ka (µM)
|
|
|---|---|---|
| DN | DSb | |
| 30 | 0.11 ± 0.01 | 0.060 ± 0.01 |
| 0.18 ± 0.06b | ||
| 50 | 0.12 ± 0.04 | 0.08 ± 0.01 |
| 0.11 ± 0.03b | ||
| 75 | 0.16 ± 0.02 | 0.15 ± 0.02 |
| 0.26 ± 0.04b | ||
| 100 | 0.15 ± 0.02 | 0.9 ± 0.4 |
| 0.7 ± 0.02b | ||
| 150 | 0.39 ± 0.03 | 1.3 ± 0.1 |
| 1.1 ± 0.2b | ||
| 200 | 0.64 ± 0.05 | 2.6 ± 0.2 |
| 1.4 ± 0.3b | ||
| 300 | 0.9 ± 0.1 | --c |
Errors were estimated from three independent experiments for 50 mM K+, 150 mM K+, and 300 mM K+. The other errors are obtained from the standard deviation of the data from the best non-linear fit.
These values were determined using K249Q hOGG1.
Not determined.
Regardless of the DNA sequence, the salt dependence of the association constants for hOGG1-DNA complexes was small compared to analogous studies with hUNG, indicating a smaller electrostatic component to binding of hOGG1 [NhOGG1 = −1.0 to −2.0; NhUNG = −3.2) (Table 2)8. Within this series of DNA sequences, the binding of DN (35% G/C content) was about five to six times less sensitive to a ten-fold increase in salt concentration than the other DNA sequences (Supplementary Figure S3 and Table S2). The different slopes for the various duplex sequences (Table 2) suggests subtle differences in the electrostatic components of the binding free energies that may in turn stem from differences in the structures of these complexes. The sequence dependent salt effects on binding may arise from differences in the heterogeneous ion density around the free duplexes and/or differential reorganization of the ion cloud arising from structural differences between hOGG1 and these non-specific DNA sequences. For these reasons it is unwarranted to use simple polyelectrolyte theory to calculate the number of ions that are released to bulk solution upon complex formation as we did previously with hUNG binding to non-specific DNA6, 8, 30.
Table 2.
Analysis of the salt dependences of non-specific and specific binding of hOGG1.a
| Slope (N)b | Kanon (M−1)c | |
|---|---|---|
| KaN | −1.0 ± 0.2 | 4.0 ± 0.1 × 105 |
| −1.3 ± 0.3d | 8.8 ± 0.6 × 104d | |
| KaTATA | −1.9 ± 0.4 | 2.6 ± 0.2 × 104 |
| KaGC | −2.0 ± 0.5 | 2.5 ± 0.3 × 104 |
| KaGCGC | −1.8 ± 0.4 | 5.4 ± 0.4 ×104 |
| KaS | −2.1 ± 0.3d | 1.5 ± 0.1 × 104d |
All experiments were conducted using WT hOGG1 unless otherwise noted and the error is derived from the linear fit.
The slope (N) obtained from nonlinear regression fitting to eq 3.
Kanon is the extrapolated value of the Ka to the condition of 1 M [K+].
These values were determined using K249Q hOGG1.
Estimation of the non-electrostatic contributions to the binding free energies for these duplexes (ΔGnon) may be obtained from extrapolation of the linear salt dependences to a standard state of 1M salt6, 8, 30, 39. Under these conditions the second term in eq 3 is zero and the equation reduces to log Ka = log Kanon (with ΔGnon = −RT ln Kanon) (Tables 2 and 3). Using this formalism, DNA sequences with G/C content in the range 35 to 100% had non-electrostatic contributions to their binding free energies that were about −0.4 to −1.6 kcal/mol more favorable as compared to sequences containing a single G/C or no G/C base pairs (Table 3).
Table 3.
Electrostatic (ΔGelect) and non-electrostatic (ΔGnon) contributions to the binding free energy (ΔGbind) for non-specific (DN, DTATA, DGC, DGCGC) and specific (DS) hOGG1 complexes in the presence of 150 mM K+.a
| DN | DTATA | DGC | DGCGC | DS | |
|---|---|---|---|---|---|
| ΔGbindb (kcal mol−1) | −8.6 ± 0.4 | −8.2 ± 0.3 | −8.4 ± 0.3 | −8.5 ± 0.3 | −7.9 ± 0.4e |
| −8.0 ± 0.2e | |||||
| ΔGelectc (kcal mol−1) | −1.1 ± 0.6 | −2.3 ± 0.4 | −2.5 ± 0.7 | −2.2 ± 0.5 | −2.3 ± 0.8 |
| −1.4 ± 0.6e | |||||
| ΔGnond (kcal mol−1) | −7.5 ± 0.2 | −5.9 ± 0.1 | −5.9 ± 0.4 | −6.3 ± 0.2 | −5.6 ± 0.4 |
| −6.6 ± 0.4e |
All values are derived from experiments using KOAc.
Calculated ΔGbind = −RT ln Ka, using Ka at 150 mM [K+].
ΔGelect = ΔGbind - ΔGnon. ΔGelect pertains to the condition of 150 mM [K+].
ΔGnon = −RT ln Ka, using the measured Ka at 1M [K+].
These values were determined using K249Q
The electrostatic contribution to the binding free energy of hOGG1 with non-specific DNA (ΔGelect) at a physiological salt concentration of 150 mM was obtained by subtracting ΔGnon from the observed binding free energy. As detailed in Table 3, the values for the various sequences fell in the range ΔGelect = −1.1 to −2.5 kcal/mol. The enhanced electrostatic terms for the low or no-G/C sequences allows these sequences to bind hOGG1 with nearly the same affinity as the two G/C-rich sequences under physiological salt concentrations, despite the more favorable ΔGnon binding terms for the G/C-rich sequences. Thus even though the energetic basis for the binding of these duplex sequences differs, the observed affinity with hOGG1 is very similar under physiological conditions (see ΔGbind in Table 3).
The major conclusions from the above data are that binding of hOGG1 to non-specific DNA is (i) less driven by electrostatic effects than for hUNG, (ii) subject to sequence dependent differences in the non-electrostatic and electrostatic binding terms, and (iii) largely independent of sequence context at physiological salt concentrations.
Ion Effects on Binding of hOGG1 to DNA containing 8-oxoguanine
A quantitative evaluation of the energetic contributions of these additional backbone interactions in the specific complex was performed by measuring the binding affinity for 8-oxoG-containing DNA (DS) at a range of salt concentrations (30 mM – 200 mM K+) (Figure 2B). These experiments required the use of a catalytically inactive form of hOGG1 containing a K249Q mutation (Supplementary Figure S4)27, 40, 41. To ensure that this active site mutation did not disrupt the ability of hOGG1 to bind to DNA, we confirmed that the salt dependence of non-specific binding for K249Q was similar to that of WT hOGG1 (Supplementary Figure S4B, Table 1). While the salt sensitivity of K249Q and WT hOGG1 were essentially identical, K249Q was found to bind slightly more weakly to nonspecific DNA. Accordingly, the binding affinity K249Q for undamaged DNA is used as the basis for comparison with the specific complex to avoid small differences arising from the mutation.
The salt dependence of the binding affinity for specific DNA (N = −2.1 ± 0.3) was similar to what was observed for undamaged DNA (N = −1.3 ± 0.3). The slightly steeper salt dependence for the specific complex may arise from two additional contacts made with a phosphate group neighboring the 8-oxoG site (Figure 2C, 2D), which could result in the displacement of an additional ion. The apparent lack of thermodynamic discrimination between undamaged and 8-oxoguanine-containing DNA at physiological salt is consistent with the structural similarities between the non-specific and specific complexes (see Discussion). All of the binding affinities determined using K249Q are reported in Table 1.
Environmental Effects on DNA Translocation by hOGG1
A number of environmental factors in the cell nucleus could modulate the efficiency by which hOGG1 uses translocation along a single DNA chain to locate 8-oxoG residues. Three major factors to consider are the physiological salt composition8, molecular crowding by nuclear proteins9, and the local density of DNA chains. Although it is not possible to completely mimic the nuclear environment in a test tube, the response of hOGG1 and hUNG to these variables provides a useful basis set for understanding future measurements in cells.
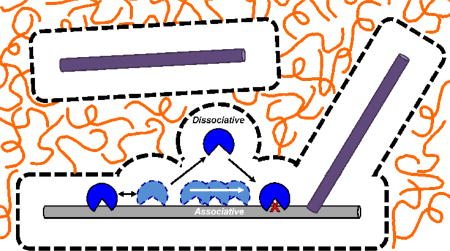
hOGG1 DNA translocation experiments were carried out with 90mer substrates containing two 8-oxoG residues positioned 10bp and 20bp apart on the same DNA strand (S10oxoG and S20oxoG)35. As previously described, translocation between two damage sites within the context of a single DNA chain can occur by either an associative or dissociative pathway20, 21, 22, 34, 35. The associative pathway involves movement of a loosely bound enzyme along the surface of the DNA and the dissociative pathway involves short-range dissociation and diffusion outside of the DNA ion cloud before reassociation with the DNA chain (Figure 3A). The overall probability of an enzyme molecule transferring between damage sites (Ptrans) is the sum of the probabilities of transferring by each pathway. The enzyme molecules that do not translocate successfully are lost to bulk solution after reacting at only a single site. As described in previous studies34, 36, Ptrans is calculated by determining the relative amounts of DNA product fragments that result from base excision at a single site only (producing fragments AB or BC), as compared both sites in a single encounter event (producing fragments A and C) (Supplementary Figure S5).
Figure 3.

Site transfer probabilities and kinetic activity of hOGG1 in the presence of variable cosolutes. (A) The multistep search and repair pathway of DNA glycosylases involves associative and dissociative translocation along the DNA chain (see text). (B) Phosphorimages of the products derived from reaction of hOGG1 with two 90mer substrates (S20oxoG and S10oxoG) with two oxoG residues positioned 10 and 20bp apart. Solution conditions and co-solutes are indicated under each lane of the gel images. (C) The transfer probabilities between oxoG sites in S10oxoG and S20oxoG in the presence of variable cosolutes using 30 mM (red) and 150 mM salt (black) (T = 37°C). (D) Effects of salt, 20% PEG 8K, and salDNA on the observed rate of 8-oxoG excision from S20oxoG. Error is derived from 3 independent trials of each condition.
The 32P end-labeled DNA fragments resulting from base excision are separated on a denaturing polyacrylamide gel and quantified by phosphorimaging (Figure 3B). Productive intramolecular site transfer results in more A and C products relative to the AB and BC products, which can be discerned by visual comparison of the band intensities at low fractional extents of reaction (Figure 3B). We have shown that for both hOGG1 and hUNG, associative transfers dominate when damage sites are closely spaced (<10 bp separation) and that dissociative transfers dominate for spacings much greater than 20 bp34, 35. Therefore, to probe the effects of the cellular environment on the two modes of translocation, we used sequences that are primarily repaired through associative transfer (S10oxoG) or dissociative transfers (S20oxoG).
We first investigated the effects of molecular crowding and the presence of bulk non-specific DNA using a buffer containing 30 mM K+ (Figure 3C, red bars). Crowding was introduced with the use of 20% (w/v) polyethylene glycol (PEG) 8K, which we have previously used to introduce excluded volume and viscosity effects on DNA translocation9. For the first time, we now also include two high concentrations of non-specific DNA that bracket the expected density of 30bp DNA binding sites in the cell nucleus. These concentrations are based on an average nuclear volume of 4200 µm3 and the size of the human diploid genome (~7 billion bp)36 and were achieved by the addition of salmon sperm DNA (salDNA) at 0.10 mM and 1 mM [bp]. The possible effect of chromatinized DNA is best explored in the context of experiments performed in human cells.
At a low 30 mM salt concentration introduction of the crowding agent doubled the translocation efficiency of hOGG1 over a site spacing of 20bp (Ptransbuffer = 0.26 ± 0.04; PtransPEG = 0.50 ± 0.03). Interestingly, the same increase was not observed for a 10 bp spacing (Ptransbuffer = 0.46 ± 0.01; PtransPEG = 0.45 ± 0.01) (Figure 3C, red bars). The basis for this result is not known but may arise from the possibility that translocation of hOGG1 over such short distances is inhibited by the time it takes for the bent DNA to relax its normal structure, which may force the enzyme to dissociate and rebind before reaching the second site35. The further addition of 0.1 and 1 mM salDNA to the crowded solution containing S20oxoG reduced Ptrans by about 50 and 100%, respectively. The concentration dependence of bulk DNA indicates that the local density of exposed DNA chains in a heterogeneous nuclear environment could dramatically impact the efficiency of translocation on single DNA chains. In contrast, addition of 1 mM salDNA to a crowded reaction solution containing S10oxoG only decreased Ptrans by 40% (Figure 3C, red bars). These effects suggest that crowding agents and bulk DNA chain density have reduced effects on short-range translocation where the associative pathway dominates. All low salt Ptrans values are reported in Supplementary Table S3.
As previously reported, increasing the salt concentration to 150 mM completely abolished site transfer by hOGG1 for the substrates with 10 and 20 bp site spacings (compare black and red bars in Figure 3C)35, but addition of the crowder nearly restored the translocation efficiencies to the levels observed in the absence of salt (Figure 3C). The beneficial effect of the crowder on DNA translocation in the presence of high salt is most likely attributed to an increased lifetime of hOGG1 on the DNA chain9. In other words, the crowder serves to reflect hOGG1 back to the DNA chain, whereas in its absence, hOGG1 dissociation is made irreversible by the rapid condensation of salt ions around the enzyme and DNA. The addition of 0.1 and 1 mM salDNA reduced translocation across the 10 and 20bp site spacings in a concentration dependent manner like that observed for the low salt conditions. However, residual translocation was still observed for the short 10bp spacing even at 1 mM salDNA (Ptrans ~ 0.15)(Figure 3C, black bar). We conclude that short-range transfers are possible under conditions of physiological concentrations of salt, crowding and dense concentrations of decoy DNA chains. All high salt Ptrans values are reported in Supplementary Table S4.
Environmental Effects on Steady-State Turnover of hOGG1
The effect of the same environmental factors on the steady-state rate of 8-oxoG excision from S20oxoG was investigated (Figure 3D). Under low salt conditions, the addition of 20% PEG8K reduced the enzyme turnover by about one-order of magnitude, and the further addition of salDNA had no appreciable effect on the rate (Figure 3D). The adverse effect of the crowder on steady-state turnover is attributed to (i) its trapping of hOGG1 on abasic product sites, from which the enzyme releases very slowly28, 42, and (ii) the increased time spent translocating on DNA chains, rather than cycling through the population of DNA molecules by rapid 3D diffusive steps. This leads to reduced turnover of the 8-oxoG sites in the population of DNA molecules (a similar effect has been previously reported with hUNG)9.
The addition of 150 mM salt had little effect on the rate of hOGG1 turnover as compared to low salt, regardless of the presence of crowder or bulk DNA (compare red and black bars in Figure 3D)(Supplementary Table S5, Figure S6). This finding indicates that the rate-limiting step leading to product release is salt-independent. We hypothesize that the overall rate-limiting step for turnover involves resolution of the covalent Schiff base linkage between hOGG1 and the abasic sugar product or a slow conformational change prior to enzyme release from the product43. Neither of these steps would be expected to be strongly dependent on bulk salt concentration.
Environmental Effects on DNA Translocation of hUNG
We performed the same experiments described above with human uracil DNA glycosylase (hUNG) to determine if the effects of salt, molecular crowding, and bulk DNA density are common between the two enzymes. We have previously characterized hUNG with respect to the variables of high salt and molecular crowding, but the effects of bulk decoy DNA have not yet been addressed8, 9. This question is interesting because hUNG binds to non-specific DNA using primarily electrostatic interactions and uses non-electrostatic interactions to enhance its specificity for specific DNA at physiological salt concentrations8.
The site spacings chosen for measuring DNA translocation by hUNG were five (S5U) and twenty base pairs (S20U) in order to satisfy the condition of testing site spacings where uracil excision occurred by primarily associative or dissociative transfers (Figure 4A)34. The low salt condition employed 22 mM KOAc to be consistent with previous measurements8, 9, 34. Translocation by hUNG at low salt was greatly enhanced by the presence of 20% PEG 8K for both site spacings (red bars, Figure 4B)(Supplementary Table S6). The addition 0.1 and 1 mM salDNA to the crowded solution modestly reduced Ptrans for both substrates. Comparing the trends in Ptrans for the 20bp spacing between hOGG1 and hUNG reveals similar trends with respect to the effects of the crowding agent and salDNA (compare red bars in Figure 3C and 4B).
Figure 4.

Site transfer probabilities and kinetic activity of hUNG in the presence of variable cosolutes. (A) Phosphorimages of the products derived from reaction of hUNG with two 90mer substrates (S5U and S20U) with two uracil residues spaced 5 (black) and 20bp (red) apart. Solution conditions and co-solutes are indicated under each lane of the gel images. The Ptrans values for S5U reflect directional bias as indicated by the low population of the single excision AB fragment (see text and Supplemental Information). (B) The transfer probabilities between uracil sites in S5U and S20U in the presence of various co-solutes at 22 (red) and 150 mM salt (black) (T = 37°C). The Ptrans values for S5U at 150 mM salt represent unidirectional site transfer from site 2 to site 1 (Ptrans2→1). (C) Effects of salt, 20% PEG 8K, and salDNA on the observed rate of 8-oxoG excision from S20U. Ptrans and reaction rate data for 22 mM K+ in the presence of 20% PEG 8K has been published and is shown here for comparison9. Error is derived from 3 independent trials of each condition.
The introduction of 150 mM salt reduced or eliminated DNA translocation over the 5 and 20bp spacings in the absence of crowding (compare red and black bars in Figure 4B)(Supplementary Table S7)8. As seen with the low salt condition, the addition of crowder to the high salt solution increased Ptrans by at least 8-fold for the S20U substrate (black bars, in Figure 4B). The further introduction of salDNA to the crowded solution reduced, but did not eliminate transfers over the 20bp spacing. Once again, these effects of crowder and salDNA broadly parallel those of hOGG1, even though the enzymes have different driving forces for interacting with non-specific DNA.
Although similar effects of salt, crowding and bulk DNA were observed for the 5bp spacing, the translocation events were biased in the direction of site 2 → 1 in the presence of high, but not low salt (Figure 4B). Such an effect leads to an atypical product fragment distribution as seen in Figure 4A and described previously (see also Supplementary Information and Supplementary Figure S7)9, 44. Although we cannot ascertain the detailed basis for this effect, we have speculated that it arises from an asymmetric interaction of the enzyme with the DNA that is manifested under select conditions44. The Ptrans2→1 values that we report in Figure 4B reflect this directional bias.
Environmental Effects on Steady-State Turnover of hUNG
As with hOGG1, the effect of solution environment on the steady-state rate of uracil excision was determined. Under low salt conditions, the introduction of 20% PEG 8K caused an ~10-fold reduction in turnover of SU (red bars, Figure 4C). This reduction was previously attributed to the crowding agent trapping hUNG on non-specific DNA, thereby increasing the amount of time the enzyme spends translocating on DNA chains at the expense of diffusing rapidly through bulk solution to new chains9. Addition of 0.1 or 1 mM salDNA to the crowded low salt solution resulted in turnover rates that were suppressed by ~105-fold relative to buffer alone (Figure 4C, Supplementary Table S8, and Supplementary Figure S8). This rate reduction is attributed to competitive binding of the vast excess of non-specific DNA sites, which is driven by electrostatic interactions with hUNG at low salt concentration. Remarkably, the slow turnover in the presence of salDNA is restored by a factor of 104 to 105 by the addition of high salt (black bars, Figure 4C). These substantial salutary effects of high salt on the steady-state turnover of hUNG in the presence of crowding and decoy DNA are accounted for by reduced electrostatic binding to non-specific DNA sites and the enhanced uracil site specificity at high salt provided by the non-electrostatic binding energy for specific sites8.
DISCUSSION
The general question of whether DNA glycosylases from distinct fold families use different energetic and kinetic mechanisms to locate rare damaged DNA bases in a densely crowded nuclear environment is largely unexplored. The present study utilizes a simple in vitro model that consists of an inert crowding agent, physiological concentrations of potassium acetate and high concentrations of bystander DNA chains to mimic the nuclear environment17. The common and unique aspects of the mechanisms utilized by hUNG and hOGG1 are discussed below and summarized in Figure 5. In Figure 5A, the observed “salt effect” (X150/XLS) is indicated for each solution condition we have explored. The salt effect is defined as the value of a given measurement (KD, Ptrans or V0/[E]) at 150 mM salt (X150) to the same measurement at low salt (XLS). We chose to compare our low salt standard conditions to the condition of 150 mM potassium ions because this condition closely matches the cation concentration of a human cell nucleus17. A comparison of the salt effect in dilute buffered solution as compared to when 20% PEG8K, 1 mM bulk DNA, or both are present is shown in Figure 5A and Figure 5B presents a reasonable model for the combined effects of salt, crowding and bulk DNA chains on enzyme-DNA binding and chain translocation (see below).
Figure 5.

Summary of the effects of salt, molecular crowding, and bulk DNA on each measured thermodynamic and kinetic parameter. (A) The salt effect (X150/XLS) is indicated for each solution condition we have explored. The salt effect is defined as the value of a given measurement (KD, Ptrans or V0/[E]) at 150 mM salt (X150) divided by the same measurement at low salt (XLS). This panel compares the salt effect in dilute buffered solution as compared to when 20% PEG8K, 1 mM bulk DNA, or both are present. Salt effects falling below the dashed line are reduced by high ionic strength and those above are enhanced. hUNG and hOGG1 respond differently to the introduction of salt with respect to non-specific DNA binding (KDN) and kinetic activity in the presence of crowder and salDNA. (B) General model for the effects of molecular crowders (orange lines) and bulk DNA (purple bars) on DNA translocation at a physiological salt concentration. The image is drawn to scale using a DNA duplex of with a 2 nm diameter as a scale reference. Dashed lines depict the depletion layer (RgPEG = 4.2 nm) where the PEG 8K polymer is excluded around the protein and DNA macromolecules60. Enzyme association with a substrate DNA chain (light gray) is limited by the bulk viscosity which hinders translational diffusion through the crowded solution, but is also is facilitated when the depletion layers of the enzyme and DNA overlap (see reference 9). Once this happens, the enzyme diffuses as if in a low viscosity environment. The depletion layer and polymer cage are very effective at enhancing DNA translocation by keeping dissociated enzyme molecules in the vicinity of the substrate DNA chain. Two effects of bulk DNA chains (dark gray) are also depicted. In the first scenario, the bulk DNA chain is enclosed in its own depletion layer and separated from the substrate DNA chain at a distance determined by the bulk DNA chain concentration. Such bulk DNA chains would be hindered from trapping translocating enzyme molecules compared to the absence of crowding. In the second scenario, the depletion layers of the bulk and substrate DNA have overlapped, which is facilitated by the reduced volume of complexation and shielding of the repulsive phosphate charge at high salt51, 53.
Salt Effects on DNA Binding in the Absence of Crowding or Bulk DNA
One of the significant differences between hOGG1 and hUNG is their response to physiological concentrations of monovalent ions, which can be attributed to their different structural modes of interacting with undamaged DNA sequences (Figure 1 and references 25 and 32). The binding of uracil DNA glycosylase to undamaged DNA has a large salt effect (Figure 5A), leading to weak binding at 150 mM K+ (ΔGbind150 = −5.5 kcal/mol). In contrast, binding of hOGG1 is comparatively salt resistant (ΔGbind150 = −8.6 kcal/mol, Figure 5A). Thus, hOGG1 binds to nonspecific DNA about −3.1 kcal/mol more tightly than hUNG at 150 mM K+. The energetic basis for these differences has been assigned to the greater electrostatic term in the binding free energy of hUNG and the larger non-electrostatic term for hOGG1 (Table 3). Structurally, these energetic differences can be attributed to the greater number of electrostatic interactions in the hUNG-DNA complex and the nonpolar interactions in the hOGG1 complex that arise from the DNA distortion and docking of guanine into the exo-site (Figure 1).
Relating Salt Effects to the Efficiency of Damage Recognition
The respective weak and stronger interactions hUNG and hOGG1 with non-specific DNA and their different electrostatic attributes are suited to the catalytic properties of these two enzymes and the nature of the DNA damage that these enzymes must recognize. Uracil DNA glycosylase is a highly active enzyme capable of flipping and excising uracil from the DNA duplex on a millisecond timescale14, 15, 24. Thus, if the lifetime of hUNG on non-specific DNA sites were significantly longer than milliseconds, its search and repair efficiency would be diminished. The millisecond timescale for hUNG binding and catalysis are also consistent with the rapid sub-millisecond intrinsic breathing dynamics of base pairs containing uracil or thymidine14, 45. These motions allow the enzyme to inspect T/A and U/G base pairs by taking advantage of their frequent excursions from the base stack without severely distorting the DNA 3, 14, 45. Distortion of an undamaged DNA duplex by hUNG would likely lead to greater non-electrostatic interactions (as observed with hOGG1) and a non-productive increase in hUNG’s bound lifetime. Thus, a physiological salt concentration facilitates the hUNG reaction by decreasing its lifetime on non-specific DNA and intrinsic base pair breathing precludes the requirement for DNA distortion until a uracil base is detected and flipped into the enzyme active site3. Consistent with the above proposal, the hUNG specific DNA complex is severely distorted and displays a +1 kcal/mol smaller electrostatic binding term and a −5 kcal/mol more favorable non-electrostatic binding term than the non-specific complex8.
In contrast, hOGG1 excises 8-oxoG on a time scale of seconds28, 42, 43, which requires a greater lifetime on non-specific DNA to allow damage recognition to occur efficiently. The lower rates of base pair breathing for of G/C and 8-oxoG/C pairs as compared to those of uracil and thymidine45, 46, 47, and the requirement for a longer extrahelical lifetime to allow excision, may have driven the evolution of an active base flipping mechanism for hOGG1. This requirement for damage recognition would give rise to hOGG1’s primarily non-electrostatic DNA binding mode for both undamaged and damaged DNA (see KD, Figure 5A). Thus unlike hUNG, hOGG1 is not imbued with salt dependent DNA binding specificity. It is important to point out that most specificity for damaged bases occurs in the transition state for base excision by both hUNG and hOGG143, 48. For instance, the transition state specificity for uracil as opposed to thymine or cytosine has been estimated to be at least 105-fold for hUNG3, 48.
Salt Effects on DNA Translocation in the Absence of Crowding or Bulk DNA
Physiological salt concentration also diminishes the translocation efficiency (Ptrans) of hUNG and hOGG1 in the absence of crowding and bulk DNA chains (Figures 3C, 4B, 5A). For both enzymes, Ptrans is reduced to zero for the substrates with a 10 to 20bp site spacing. For hUNG, the effects of salt on Ptrans can be rationalized in much the same way as the antagonistic effects of salt on non-specific DNA binding. That is, when hUNG makes a dissociative step away from the DNA and escapes the DNA ion cloud, the presence of high bulk salt takes away the entropic advantage of rebinding the same DNA chain. Thus, more enzyme molecules are lost to bulk solution when attempting to transfer between the sites. However, at a shorter five base pair spacing hUNG can still transfer in the presence of 150 mM K+, which is attributed to the increased use of the associative pathway that is more salt resistant34. The antagonistic effect of salt on Ptrans of hOGG1 may also arise from similar entropic effects even though its non-specific DNA binding is far less sensitive to salt concentration than hUNG. For hOGG1, the 3 to 5-fold decrease in its non-specific binding affinity as the salt is increased from 30 to 150 mM is apparently sufficient to ablate effective translocation between sites. In addition, the relative efficiency of transfer of hOGG1 and hUNG between sites will be affected by the average number of microscopic dissociative steps that are taken by each enzyme during the transfer. In other words, each dissociative step introduces a probability where the enzyme may reassociate with the same DNA chain or escape to bulk solution. Even a relatively small salt effect on several dissociative steps would increase the escape probability multiplicatively, leading to a significant reduction in successful site transfers8, 21.
Salt Effects on Turnover in the Absence of Crowding or Bulk DNA
A physiological concentration of salt can also increase or decrease the catalytic turnover of an enzyme if there are salt sensitive rate-limiting steps along the reaction coordinate8. If salt sensitive binding steps are involved, an increased salt concentration will reduce the entropic driving force for ion release in the transition state for binding6, 8, and facilitate the same transition state in the reverse direction (by microscopic reversibility). For the turnover studies performed here in the absence of crowding or bulk DNA, the kinetic effects of salt are modest and reflect the net effect on the binding steps and product release (see V0/[E], Figure 5A). Below we return to discuss the major effect of salt, which becomes profoundly important when crowding and bulk DNA are introduced.
Impacts of Inert Macromolecule Crowding on the Salt Effect
The addition of 20% PEG8K to the aqueous buffer substantially enhances the translocation efficiency for both hOGG1 and hUNG (see Ptrans, Figure 3C and 4B). The effect is especially impactful at physiological salt concentration using the longer site spacing of 20bp, where Ptrans is increased from essentially zero to around 0.25–0.3 for both enzymes. This appears to be a general effect of crowding that derives from increased sequestration of the enzymes in the vicinity of the DNA chain by the polymer cage. Thus, the partitioning of dissociated enzyme molecules to bulk solution is disfavored, which dramatically attenuates the effect of high salt for both enzymes. The other effect of crowding is to reduce the observed rate of reaction by about 10-fold for both enzymes when low salt conditions are used (Figure 3D and 4C). For hUNG, we have previously attributed the cause of this effect to the increased time spent translocating on long (but not short) DNA chains before and after uracil excision9. The increased time spent translocating on DNA chains reduces opportunities for rapid 3D diffusion steps, which is the most efficient mechanism for rapid sampling all DNA chains in the population18, 19, 20, 21, 22, 34, 49. For hUNG, the inhibitory effect of crowding on the rate is strikingly relieved when a physiological salt concentration is used (Figure 4C, see V0/[E] Figure 5A). This effect is reasonably attributed to an increase in the dissociation frequency from both non-specific DNA and the product complex, which provides more opportunities for escape of the enzyme from the polymer cage around the DNA8, 9. For hOGG1, the same salutary effect of increased ions on the rate is not observed (Figure 5A), which is reasonably attributed to its weaker electrostatic binding component to non-specific DNA and the very slow nature of product release, which likely involves salt independent resolution of the covalent Schiff base linkage with the abasic sugar aldehyde43, 50.
Bulk DNA Effects on Translocation and Turnover
In the absence of crowding, the general effect of a high density of bulk DNA chains on hOGG1 and hUNG activity is to reduce Ptrans and enzyme turnover regardless of the monovalent cation concentration (Figure 5A), which is attributed to bulk DNA trapping of dissociated enzymes molecules in the process of translocating between two sites and equilibrium competitive inhibition, respectively. Trapping of translocating enzymes by bulk DNA chains is entirely plausible because at a 1 mM salDNA concentration a single bp of salDNA would be located an average of ~10 nm from a substrate DNA molecule. This separation is close to the mean distance for diffusion of these enzymes from the substrate DNA chain during a single productive dissociative transfer step (~7 nm), suggesting a high probability of being captured by the DNA trap (Figure 5B)34, 35.
The antagonistic effect of bulk DNA chains on Ptrans is greatly attenuated in the presence of crowding, regardless of the monovalent ion concentration (Figure 3C, 4B, 5A). This effect of crowding may arise from the individual substrate and bulk DNA chains being sequestered in their own polymer cages (Figure 5B), which would tend to diminish trapping by bulk DNA chains near the site of a translocating enzyme independent of salt effects. The data also indicate that Ptrans decreases as the bulk DNA concentration is increased when crowding is held constant. This effect may arise from merging of the bulk and substrate DNA polymer cages and the tendency of DNA chains to interact at high DNA concentrations and in the presence of Coulombic shielding effects provided by of high salt concentrations (Figure 5B)51. Thus, the combined effects of crowding and high salt may allow intermolecular DNA complexes to form between bulk and substrate DNA chains and populate the microscopic region where the enzyme is translocating. This would lead to the observed reduction in Ptrans (Figure 3C and 4B)52, 53.
The introduction of high salt results in a profound increase in the exceedingly slow turnover rate of hUNG under conditions of crowding and high bulk DNA concentration (Figure 5A). No such effect of salt was observed with hOGG1 (Figure. 3D). The beneficial kinetic effect of 150 mM salt on turnover of hUNG but not hOGG1 under these conditions is reasonably attributed to the larger electrostatic contribution to non-specific DNA binding by hUNG, the expected enhanced specificity for uracil sites under such conditions, and the previously reported accelerating effects of salt on the product release step (Table 3). For hOGG1, the same stimulatory effect of high monovalent ions would not be expected because of its smaller non-electrostatic binding free energy and its rate-limiting product release step is not stimulated by salt ions. In summary, monovalent cations are required for hUNG to maintain high turnover in the presence of high concentrations of bulk DNA chains, whereas hOGG1 avoids this problem by avoiding strong electrostatic interactions with non-specific DNA.
Glycosylase Activity in a Cellular Environment
It is abundantly clear that monovalent ions, molecular crowding, and high-densities of bulk DNA have profound effects on the activities of hUNG and hOGG1. It is also clear that the individual effects of these solution components, which can either enhance or antagonize a given activity parameter, can be significantly altered when all three components are present simultaneously. Remarkably, we find that the basic features of the damage search and repair process that have been previously determined at low salt concentrations or in the presence of a crowding agent only9, 34, 35, persist under conditions that more closely mimic the nuclear environment. These features include short-range associative translocations over ~10bp distances and frequent dissociative transfers that allow rapid 3D diffusion to new positions on the same DNA chain or to new DNA chains when the chain density is high. A repair mechanism involving short-range, rapid 1D searches interspersed with frequent diffusive events leading to reencounter with the DNA is well suited for the limited accessibility of damage sites in the context of a nucleosome54.
A new finding in the current work is that the caging effect of macromolecular crowding agents and the electrostatic effects of monovalent cations can act cooperatively to retain an efficient search mechanism in the presence of high concentrations of bulk DNA chains. Thus, the caging effect of the crowder counteracts the antagonistic effect of high monovalent ion concentrations on DNA translocation, and high monovalent ion concentrations promote dissociation of enzyme molecules from non-specific DNA chains providing more opportunities for escape from the polymer cage. For hUNG, these effects lead to retention of an efficient search mechanism even in the presence of high concentrations of bulk DNA. Despite the different energetic basis for the interaction of hOGG1 with DNA, this enzyme also follows the same general trends. One unique aspect of hOGG1 that derives from its non-electrostatic binding mechanism is its slow turnover, which is exacerbated in a crowded environment irrespective of the salt concentration (Figure 3D). The low turnover rate of hOGG1 is attributed to steps after glycosidic bond cleavage (i.e. slow lyase activity and/or product release)55, 56, but is greatly accelerated by the next enzyme in the base excision repair pathway, AP endonuclease 1 (APE1)56, 57. Based on the expectation that crowding agents act by shifting binding equilibria towards the formation of lower volume protein complexes as opposed to the free species12, 58, 59, we speculate that APE1-facilitated release of hOGG1 from its product complex will be promoted in a crowded nuclear environment and resolve the intrinsic problem of slow turnover.
Supplementary Material
Abbreviations
- hUNG
human uracil DNA glycosylase
- hOGG1
8-oxoguanine DNA glycosylase
- 8-oxoG
8-oxoguanine
- CC
Counterion Condensation
- ΔGbind
binding free energy at physiological salt (150 mM K+)
- ΔGelect
electrostatic component of the binding free energy
- ΔGnon
non-electrostatic component of the binding free energy
- DN
20mer non-specific DNA with randomized G-C and A-T base pairs
- DTATA
20mer non-specific DNA with only A-T base pairs
- DGC
20mer non-specific DNA containing a central G-C base pair
- DGCGC
20mer non-specific DNA containing only G-C base pairs
- DS
20mer specific DNA containing an oxoG-C pair
- salDNA
salmon sperm DNA
- PEG
polyethylene glycol
- S20U
90mer-DNA with two U-A base pairs separated by 20 bp
- S5U
90mer-DNA with two U-A base pairs separated by 5 bp
- S20oxoG
90mer-DNA with two oxoG-C base pairs separated by 20 bp
- S10oxoG
90mer-DNA with two oxoG-C base pairs separated by 10 bp
Footnotes
This work was supported by the National Institute of Health Grants T32 GM8403-23, RO1 GM056834, and National Science Foundation CHE1307275 (to J.T.S). Shannen Cravens was supported in part by NIH F31 GM114994-01A1.
ASSOCIATED CONTENT
Supporting Information Available. Supplemental methods, eight supporting tables, and eight supporting figures. Dissociation constants for unlabeled DNAs determined by competition binding (Table S1); nonspecific binding affinities for DTATA, DGC, and DGCGC determined under various salt conditions (Table S2); Ptrans determined at 30 mM K+ with combinations of 20% PEG 8K and salDNA for hOGG1 (Table S3); Ptrans determined at 150 mM K+ with combinations of 20% PEG 8K and salDNA for hOGG1 (Table S4); reaction rates for hOGG1 with S20oxoG in the presence of salt, 20% PEG 8K, and salDNA (Table S5); Ptrans determined at 22 mM K+ with combinations of 20% PEG 8K and salDNA for hUNG (Table S6); Ptrans determined at 150 mM K+ with combinations of 20% PEG 8K and salDNA for hUNG (Table S7); reaction rates for hUNG with S20oxoG in the presence of salt, 20% PEG 8K, and salDNA (Table S8); Competition binding measurements for unlabeled DNAs DN′ and DS′ (Fig. S1); binding curves for a 15bp and 20bp DNA (Fig. S2); full nonspecific binding curves for DTATA, DGC, DGCGC at various salt conditions (Fig. S3); nonspecific binding of K249 with DN and activity assay with S0oxoG (Fig. S4); time-dependent Ptransobs example traces for hUNG and hOGG1 (Fig. S5); example gels of hOGG1 reacting with S20oxoG (Fig. S6); evidence of biased transfer of hUNG along S5U at 150 mM K+ (Fig. S7); example gels of hUNG reacting with S20U (Fig. S8). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hoeijmakers JH. DNA damage, aging, and cancer. N. Engl. J. Med. 2009;361:1475–1485. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- 2.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 3.Stivers JT, Jiang YL. A mechanistic perspective on the chemistry of DNA repair glycosylases. Chem. Rev. 2003;103:2729–2759. doi: 10.1021/cr010219b. [DOI] [PubMed] [Google Scholar]
- 4.Seeberg E, Eide L, Bjørås M. The base excision repair pathway. Trends Biochem. Sci. 1995;20:391–397. doi: 10.1016/s0968-0004(00)89086-6. [DOI] [PubMed] [Google Scholar]
- 5.Alsallaq R, Zhou HX. Electrostatic rate enhancement and transient complex of protein-protein association. Proteins. 2008;71:320–335. doi: 10.1002/prot.21679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Privalov PL, Dragan AI, Crane-Robinson C. Interpreting protein/DNA interactions: Distinguishing specific from non-specific and electrostatic from non-electrostatic components. Nucleic Acids Res. 2011;39:2483–2491. doi: 10.1093/nar/gkq984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou H. Disparate ionic-strength dependencies of on and off rates in protein-protein association. Biopolymers. 2001;59:427–433. doi: 10.1002/1097-0282(200111)59:6<427::AID-BIP1047>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 8.Cravens SL, Hobson M, Stivers JT. Electrostatic properties of complexes along a DNA glycosylase damage search pathway. Biochemistry. 2014;53:7680–7692. doi: 10.1021/bi501011m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cravens SL, Schonhoft JD, Rowland MM, Rodriguez AA, Anderson BG, Stivers JT. Molecular crowding enhances facilitated diffusion of two human DNA glycosylases. Nucleic Acids Res. 2015;43:4087–4097. doi: 10.1093/nar/gkv301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ellis RJ. Macromolecular crowding: Obvious but underappreciated. Trends Biochem. Sci. 2001;26:597–604. doi: 10.1016/s0968-0004(01)01938-7. [DOI] [PubMed] [Google Scholar]
- 11.Minton AP. The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media. J. Biol. Chem. 2001;276:10577–10580. doi: 10.1074/jbc.R100005200. [DOI] [PubMed] [Google Scholar]
- 12.Phillip Y, Sherman E, Haran G, Schreiber G. Common crowding agents have only a small effect on protein-protein interactions. Biophys. J. 2009;97:875–885. doi: 10.1016/j.bpj.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou HX, Rivas G, Minton AP. Macromolecular crowding and confinement: Biochemical, biophysical, and potential physiological consequences. Annu. Rev. Biophys. 2008;37:375–397. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parker JB, Bianchet MA, Krosky DJ, Friedman JI, Amzel LM, Stivers JT. Enzymatic capture of an extrahelical thymine in the search for uracil in DNA. Nature. 2007;449:433–437. doi: 10.1038/nature06131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krosky DJ, Song F, Stivers JT. The origins of high-affinity enzyme binding to an extrahelical DNA base. Biochemistry. 2005;44:5949–5959. doi: 10.1021/bi050084u. [DOI] [PubMed] [Google Scholar]
- 16.Veksler A, Kolomeisky AB. Speed-selectivity paradox in the protein search for targets on DNA: Is it real or not? J Phys Chem B. 2013;117:12695–12701. doi: 10.1021/jp311466f. [DOI] [PubMed] [Google Scholar]
- 17.Theillet FX, Binolfi A, Frembgen-Kesner T, Hingorani K, Sarkar M, Kyne C, Li C, Crowley PB, Gierasch L, Pielak GJ, Elcock AH, Gershenson A, Selenko P. Physicochemical properties of cells and their effects on intrinsically disordered proteins (IDPs) Chem. Rev. 2014;114:6661–6714. doi: 10.1021/cr400695p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gowers DM, Wilson GG, Halford SE. Measurement of the contributions of 1D and 3D pathways to the translocation of a protein along DNA. Proc. Natl. Acad. Sci. U. S. A. 2005;102:15883–15888. doi: 10.1073/pnas.0505378102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Halford SE, Marko JF. How do site-specific DNA-binding proteins find their targets? Nucleic Acids Res. 2004;32:3040–3052. doi: 10.1093/nar/gkh624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leonid Mirny, Michael Slutsky, Zeba Wunderlich, Anahita Tafvizi, Jason Leith, Andrej Kosmrlj. How a protein searches for its site on DNA: The mechanism of facilitated diffusion. Journal of Physics A: Mathematical and Theoretical. 2009;42:434013. [Google Scholar]
- 21.Stanford NP, Szczelkun MD, Marko JF, Halford SE. One-and three-dimensional pathways for proteins to reach specific DNA sites. EMBO J. 2000;19:6546–6557. doi: 10.1093/emboj/19.23.6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.von Hippel PH, Berg OG. Facilitated target location in biological systems. J. Biol. Chem. 1989;264:675–678. [PubMed] [Google Scholar]
- 23.Parikh SS, Walcher G, Jones GD, Slupphaug G, Krokan HE, Blackburn GM, Tainer JA. Uracil-DNA glycosylase-DNA substrate and product structures: Conformational strain promotes catalytic efficiency by coupled stereoelectronic effects. Proceedings of the National Academy of Sciences. 2000;97:5083–5088. doi: 10.1073/pnas.97.10.5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedman JI, Stivers JT. Detection of damaged DNA bases by DNA glycosylase enzymes. Biochemistry. 2010;49:4957–4967. doi: 10.1021/bi100593a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.David SS, O’Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Banerjee A, Yang W, Karplus M, Verdine GL. Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damaged DNA. Nature. 2005;434:612–618. doi: 10.1038/nature03458. [DOI] [PubMed] [Google Scholar]
- 27.Bruner SD, Norman DP, Verdine GL. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature. 2000;403:859–866. doi: 10.1038/35002510. [DOI] [PubMed] [Google Scholar]
- 28.Leipold MD, Workman H, Muller JG, Burrows CJ, David SS. Recognition and removal of oxidized guanines in duplex DNA by the base excision repair enzymes hOGG1, yOGG1, and yOGG2. Biochemistry. 2003;42:11373–11381. doi: 10.1021/bi034951b. [DOI] [PubMed] [Google Scholar]
- 29.Roehrl MHA, Wang JY, Wagner G. A general framework for development and data analysis of competitive high-throughput screens for small-molecule inhibitors of Protein-Protein interactions by fluorescence polarization. Biochemistry. 2004;43:16056–16066. doi: 10.1021/bi048233g. [DOI] [PubMed] [Google Scholar]
- 30.Manning GS. The molecular theory of polyelectrolyte solutions with applications to the electrostatic properties of polynucleotides. Q. Rev. Biophys. 1978;11:179–246. doi: 10.1017/s0033583500002031. [DOI] [PubMed] [Google Scholar]
- 31.Record MT, Zhang W, Jr, Anderson CF. Analysis of effects of salts and uncharged solutes on protein and nucleic acid equilibria and processes: A practical guide to recognizing and interpreting polyelectrolyte effects, hofmeister effects, and osmotic effects of salts. Adv. Protein Chem. 1998;51:281–353. doi: 10.1016/s0065-3233(08)60655-5. [DOI] [PubMed] [Google Scholar]
- 32.Record MT, Lohman TM, Haseth Pd. Ion effects on ligand-nucleic acid interactions. J. Mol. Biol. 1976;107:145–158. doi: 10.1016/s0022-2836(76)80023-x. [DOI] [PubMed] [Google Scholar]
- 33.Olmsted MC, Bond JP, Anderson CF, Record MT. Grand canonical monte carlo molecular and thermodynamic predictions of ion effects on binding of an oligocation (L8+) to the center of DNA oligomers. Biophys. J. 1995;68:634–647. doi: 10.1016/S0006-3495(95)80224-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schonhoft JD, Stivers JT. Timing facilitated site transfer of an enzyme on DNA. Nat. Chem. Biol. 2012;8:205–210. doi: 10.1038/nchembio.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rowland MM, Schonhoft JD, McKibbin PL, David SS, Stivers JT. Microscopic mechanism of DNA damage searching by hOGG1. Nucleic Acids Research. 2014;42:9295–9303. doi: 10.1093/nar/gku621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Porecha RH, Stivers JT. Uracil DNA glycosylase uses DNA hopping and short-range sliding to trap extrahelical uracils. Proceedings of the National Academy of Sciences. 2008;105:10791–10796. doi: 10.1073/pnas.0801612105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blainey PC, van Oijen AM, Banerjee A, Verdine GL, Xie XS. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proceedings of the National Academy of Sciences. 2006;103:5752–5757. doi: 10.1073/pnas.0509723103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y, Cremer PS. Interactions between macromolecules and ions: The hofmeister series. Curr. Opin. Chem. Biol. 2006;10:658–663. doi: 10.1016/j.cbpa.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 39.Jen-Jacobson L, Jacobson LA. Chapter 2 Role of Water and Effects of Small Ions in Site-Specific Protein-DNA Interactions. The Royal Society of Chemistry. 2008:13–46. [Google Scholar]
- 40.Nash HM, Lu R, Lane WS, Verdine GL. The critical active-site amine of the human 8-oxoguanine DNA glycosylase, hOgg1: Direct identification, ablation and chemical reconstitution. Chem. Biol. 1997;4:693–702. doi: 10.1016/s1074-5521(97)90225-8. [DOI] [PubMed] [Google Scholar]
- 41.Chen L, Haushalter KA, Lieber CM, Verdine GL. Direct visualization of a DNA glycosylase searching for damage. Chem. Biol. 2002;9:345–350. doi: 10.1016/s1074-5521(02)00120-5. [DOI] [PubMed] [Google Scholar]
- 42.Dherin C, Radicella JP, Dizdaroglu M, Boiteux S. Excision of oxidatively damaged DNA bases by the human alpha-hOgg1 protein and the polymorphic alpha-hOgg1(Ser326Cys) protein which is frequently found in human populations. Nucleic Acids Res. 1999;27:4001–4007. doi: 10.1093/nar/27.20.4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuznetsov NA, Kuznetsova AA, Vorobjev YN, Krasnoperov LN, Fedorova OS. Thermodynamics of the DNA damage repair steps of human 8-oxoguanine DNA glycosylase. PLoS ONE. 2014;9:e98495. doi: 10.1371/journal.pone.0098495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schonhoft JD, Stivers JT. DNA translocation by human uracil DNA glycosylase: The case of single-stranded DNA and clustered uracils. Biochemistry. 2013;52:2536–2544. doi: 10.1021/bi301562n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parker JB, Stivers JT. Dynamics of uracil and 5-fluorouracil in DNA. Biochemistry. 2011;50:612–617. doi: 10.1021/bi101536k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crenshaw CM, Wade JE, Arthanari H, Frueh D, Lane BF, Nunez ME. Hidden in plain sight: Subtle effects of the 8-oxoguanine lesion on the structure, dynamics, and thermodynamics of a 15-base pair oligodeoxynucleotide duplex. Biochemistry. 2011;50:8463–8477. doi: 10.1021/bi201007t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Every AE, Russu IM. Opening dynamics of 8-oxoguanine in DNA. J. Mol. Recognit. 2013;26:175–180. doi: 10.1002/jmr.2262. [DOI] [PubMed] [Google Scholar]
- 48.Stivers JT, Pankiewicz KW, Watanabe KA. Kinetic mechanism of damage site recognition and uracil flipping by escherichia coli uracil DNA glycosylase. Biochemistry. 1999;38:952–963. doi: 10.1021/bi9818669. [DOI] [PubMed] [Google Scholar]
- 49.Berg OG, Winter RB, von Hippel PH. Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. models and theory. Biochemistry. 1981;20:6929–6948. doi: 10.1021/bi00527a028. [DOI] [PubMed] [Google Scholar]
- 50.Fromme JC, Bruner SD, Yang W, Karplus M, Verdine GL. Product-assisted catalysis in base-excision DNA repair. Nat. Struct. Biol. 2003;10:204–211. doi: 10.1038/nsb902. [DOI] [PubMed] [Google Scholar]
- 51.Bai Y, Das R, Millett IS, Herschlag D, Doniach S. Probing counterion modulated repulsion and attraction between nucleic acid duplexes in solution. Proc. Natl. Acad. Sci. U. S. A. 2005;102:1035–1040. doi: 10.1073/pnas.0404448102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kombrabail MH, Krishnamoorthy G. Fluorescence dynamics of DNA condensed by the molecular crowding agent poly(ethylene glycol) J. Fluoresc. 2005;15:741–747. doi: 10.1007/s10895-005-2982-8. [DOI] [PubMed] [Google Scholar]
- 53.Vasilevskaya VV, Khokhlov AR, Matsuzawa Y, Yoshikawa K. Collapse of single DNA molecule in poly(ethylene glycol) solutions. J. Chem. Phys. 1995;102:6595–6602. [Google Scholar]
- 54.Ye Y, Stahley MR, Xu J, Friedman JI, Sun Y, McKnight JN, Gray JJ, Bowman GD, Stivers JT. Enzymatic excision of uracil residues in nucleosomes depends on the local DNA structure and dynamics. Biochemistry (N. Y. ) 2012;51:6028–6038. doi: 10.1021/bi3006412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sidorenko VS, Nevinsky GA, Zharkov DO. Mechanism of interaction between human 8-oxoguanine-DNA glycosylase and AP endonuclease. DNA Repair (Amst) 2007;6:317–328. doi: 10.1016/j.dnarep.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 56.Vidal AE, Hickson ID, Boiteux S, Radicella JP. Mechanism of stimulation of the DNA glycosylase activity of hOGG1 by the major human AP endonuclease: Bypass of the AP lyase activity step. Nucleic Acids Res. 2001;29:1285–1292. doi: 10.1093/nar/29.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008;18:27–47. doi: 10.1038/cr.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kozer N, Schreiber G. Effect of crowding on protein-protein association rates: Fundamental differences between low and high mass crowding agents. J. Mol. Biol. 2004;336:763–774. doi: 10.1016/j.jmb.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 59.Kozer N, Kuttner YY, Haran G, Schreiber G. Protein-protein association in polymer solutions: From dilute to semidilute to concentrated. Biophys. J. 2007;92:2139–2149. doi: 10.1529/biophysj.106.097717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Gennes P. Scaling Concepts in Polymer Physics. Cornell University Press; Ithaca, N.Y.: 1979. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.