

Abstract
There is growing recognition that glial proinflammatory activation importantly contributes to the rewarding and reinforcing effects of a variety of drugs of abuse, including cocaine, methamphetamine, opioids, and alcohol. It has recently been proposed that glia are recognizing, and becoming activated by, such drugs as a CNS immunological response to these agents being xenobiotics; that is, substances foreign to the brain. Activation of glia, primarily microglia, by various drugs of abuse occurs via toll like receptor 4 (TLR4). The detection of such xenobiotics by TLR4 results in the release of glial neuroexcitatory and neurotoxic substances. These glial products of TLR4 activation enhance neuronal excitability within brain reward circuitry, thereby enhancing their rewarding and reinforcing effects. Indeed, selective pharmacological blockade of TLR4 activation, such as with the non-opioid TLR4 antagonist (+)-naltrexone, suppresses a number of indices of drug reward/reinforcement. These include: conditioned place preference, self-administration, drug-primed reinstatement, incubation of craving, and elevations of nucleus accumbens shell dopamine. Notably, TLR4 blockade fails to alter self-administration of food, indicative of a selective effect on drugs of abuse. Genetic disruption of TLR4 signaling recapitulates the effects of pharmacological TLR4 blockade, providing converging lines of evidence of a central importance of TLR4. Taken together, multiple lines of evidence converge to raise TLR4 as a promising therapeutic target for drug abuse.
Keywords: (+)-naloxone, (+)-naltrexone, cocaine, opioid, morphine, drug reward, drug reinforcement, reinstatement, psychostimulants, alcohol
Historically, neurons have been a primary focus of studies of the mechanistic underpinnings of drug abuse. While tremendous strides have been made by this approach, new players in drug abuse have emerged in recent years. These new players are non-neuronal cells called glia; namely, microglia and astrocytes [2]. This mini-review will summarize the history that led to glia becoming recognized as important contributors to drug abuse. In addition, background will be presented as to how a key glial activation receptor, Toll-like receptor 4 (TLR4), came to the forefront as a candidate non-neuronal therapeutic target for drug abuse. Alcohol, opioids, and psychostimulants will be used as exemplars as to how a focus on TLR4 can be productively exploited for understanding the importance of glial proinflammatory reactivity to drug abuse. Finally, a discussion of paths forward will be explored as they relate to how best to build these recent findings to optimize drug development.
How did a focus on TLR4 arise?
The discovery of a role of glial reactivity in drug abuse was preceded by about 2 decades by the recognition of its role in the creation and maintenance of pathological pain states, including neuropathic pain [3]. Transition of spinal glia into a reactive state in response to bodily injury was documented to drive pathological pain states via the activation of the oxidation sensitive transcription factor nuclear factor kappa B (NFκB) and the release of a host of neuroexcitatory products from spinal glia, including the proinflammatory cytokine interleukin-1beta (IL-1β), nitric oxide, superoxide, etc. [3]. As a result of trauma-induced glial reactivity, direct excitation of neurons occurs along with enhancement of neurotransmitter release from presynaptic terminals, downregulation of glutamate transporters, upregulation of NMDA and AMPA receptor numbers and function, downregulation of GABA, and a variety of associated changes all in the direction of enhanced neuronal excitability. As is already evident, an amplification of abuse potential could be readily envisioned were such a pattern of changes to occur in response to drugs of abuse within brain regions mediating reward/reinforcement.
Following acceptance of the importance of glial reactivity in neuropathic pain, exploration of a potential role of glial reactivity in opioid tolerance followed, based on known parallelisms between these two phenomena [4]. With the confirmation that glial reactivity and the same cast of neuroinflammatory products indeed contributed to opioid tolerance [5, 6], the net was cast more broadly leading to the discovery that suppressing glial activation by a variety of drugs led to an enhancement of opioid analgesia and suppression of signs of opioid dependence such as tolerance and withdrawal [7, 8].
Early work established that chronic morphine increased the glial cell marker, glial fibrillary acidic protein, in the ventral tegmental area, an effect that was inhibited by naltrexone [9]. It was later discovered that opioid effects on glial activation were non-stereoselective [7, 10]. That is, both (+)- and (−)-morphine activated glia [10, 11], and both (+)- and (−)-naloxone and naltrexone inhibited activation of glia by morphine [10], and trauma [12]. These observations were critical as this mandated that whatever the (as yet unknown) receptor was on glia, it could not be classical opioid receptors, which are strictly (−)-isomer selective. This demonstration of non-stereoselectivity was pivotal in that it began the search for an explanation of effects on glia beyond classical opioid receptors. This, in turn, led to the discovery that opioids activate glia via TLR4 and its critical co-receptor myeloid differentiation factor 2 (MD2) [10, 13]. Evidence has since accrued suggesting that alcohol and cocaine produce glial activation through TLR4/MD2 mechanisms as well [14, 15].
The discovery that TLR4/MD2 recognized, and became activated by, diverse drugs of abuse led to the Xenobiotic hypothesis [11, 16]. This hypothesis rests on the fact that the blood brain barrier ensures that few substances from the blood can enter the brain. Notable exceptions to this are many drugs of abuse such as opioids, cocaine and alcohol that all readily cross from blood to brain. As these are “foreign” from an immunological perspective, they can be considered as Xenobiotics; that is, foreign chemical substances found within an organism that is not normally produced by, or expected to be present with, that organism. Foreign substances within the brain are prime for recognition by the brain’s immune system, namely glia, via receptors such as TLR4/MD2 that are specifically tuned to detect foreign and dangerous challenges (Figure 1). Importantly, evidence from classical immunology suggests that MD2 engagement with TLR4 is fundamental to producing an active signaling complex when activated by classical pathogen associated molecular patterns (PAMPs) such as LPS [17]. Available data for xenobiotics indicates a similar TLR4/MD2 complex requirement.
Figure 1.
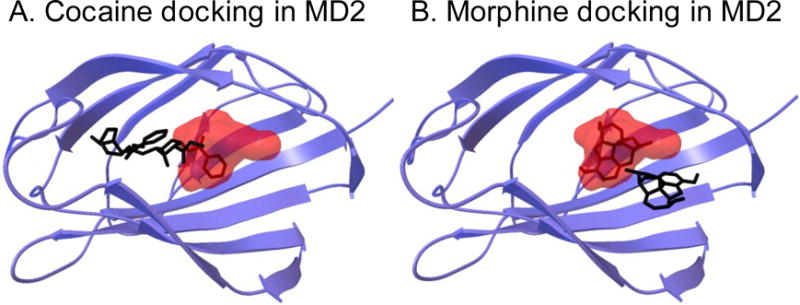
Computer modeling of cocaine and morphine interactions with MD2, whose heterodimerization with TLR4 initiates TLR4 signaling. In silico docking simulation used the high resolution crystalline structure of the dimer of human TLR4 and MD2, and the software suite AutoDock 4. All dockings were executed with Lamarkian genetic algorithms (for details see [43]). Lipopolysaccharide (LPS) is a classical TLR4 agonist known to preferentially dock in a specific binding pocket of MD2. This docking location of LPS is illustrated as the red cloud. Docking of cocaine (chemical structure embedded within the red cloud, Panel A) and morphine (chemical structure embedded within the red cloud, Panel B) were discovered to overlap the docking location of LPS within MD2. Pre-docking of (+)-naltrexone disrupted the preferred binding sites of cocaine and morphine, leading to their docking to be displaced to regions outside of the LPS binding pocket (black chemical structures outside the red cloud, Panel A for cocaine, Panel B for morphine). For visualization on the hypothesized heterodimerization tertiary structure of the TLR4/MD2 complex see [1].
While glial/immune recognition of xenobiotics via TLR4 makes inherent sense, how was the link to inhibition by naloxone and naltrexone discovered? This first arose over 30 years ago through the recognition that naloxone and naltrexone could block effects of lipopolysaccharide (LPS, component of endotoxin from the cell walls of gram negative bacteria) [18, 19], now considered as a prototypic TLR4 agonist. Interestingly, recognition that naloxone/naltrexone blocked LPS effects pre-dated the 1998 discovery of TLR4 by close to two decades [20, 21]. Hence a sizable literature on blockade of LPS effects by naloxone and naltrexone makes no mention of TLR4, as it had not yet been discovered. The evidence of naloxone/naltrexone blockade of LPS effects is now quite far-reaching including excitatory post-synaptic potentials [22], seizures [23, 24], microglial activation [25–27], proinflammatory cytokines [25, 28], nitric oxide and superoxide [29, 30], neurotoxicity and neurodegeneration [31–34], hepatitis [35], septic shock [36, 37], hormone release [38], fever [39], pain [40], reduction in morphine analgesia [41, 42], and so on. Even when (+)-isomers of naloxone and naltrexone are employed to prevent possible influence of opioid receptors, there is suppression of LPS-induced proinflammatory responses [34, 43], LPS-induced excitotoxic death of dopamine neurons [33, 34]; suppression of neuropathic pain [12, 44]; potentiation of opioid analgesia [10, 45]); and decreased opioid-induced withdrawal, tolerance, hyperalgesia and constipation [2, 7, 10, 40]. Highlighted below are several lines of evidence using pharmacological and genetic approaches to demonstrate the role of TLR4/MD2 complex on microglia in regulating the responsiveness to drugs of abuse from different drug classes.
Role of TLR4 in drugs of abuse
The neurobiology of the acute and chronic effects of drugs of abuse has been heavily investigated over the past several decades. It has become clear that drugs of abuse from different drug classes interact with the mesocorticolimbic dopamine system contributing to their acute reinforcing effects [46]. Chronic administration of drugs of abuse produces perturbations within this and other circuitries, culminating in withdrawal-induced anhedonia, increased impulsivity and an enduring susceptibility to relapse [47]. The focus of these studies has largely pertained to the ability of drugs of abuse to interact with neuronal sites of action such as alcohol’s interaction with GABA and glutamate receptor systems, opioid drugs’ stimulation of opioid receptors, and psychostimulant-induced alterations of the function of dopamine transporters. As described above, there has been increased interest in exploring non-neuronal mechanisms of drug actions. Research has begun to identify how non-neuronal mechanisms contribute to the acute effects of abused drugs and how these mechanisms may contribute to the dysregulation of neuronal systems that contribute to the lasting effects. Evidence from several drug classes suggests that microglia expressing the TLR4/MD2 complex may provide a potential mechanism by which non-neuronal cells could identify drugs of abuse. From an immunological perspective, drugs of abuse may be identified as “foreign” and initiate an innate immune response in the brain reflected through the release of proinflammatory substances (e.g. cytokines, chemokines, etc) and an upregulation of cell surface markers (e.g. CD11b). Despite the increased appreciation that several drugs of abuse initiate these immune responses, there are remaining questions of whether this mechanism provides a common mechanism for other classes and types of abused and non-abused drugs. Importantly, additional questions also relate to how immune signaling in the brain synergize with known neuronal mechanisms of drug action and the resulting behaviors that are associated with the development of drug abuse. Here, we will highlight recent findings describing how three commonly abused drugs (e.g. alcohol, opioids, and psychostimulants) interact to initiate an innate immune response and the potential behavioral manifestations of this response.
Alcohol
Alcohol potently activates a series of events that can be linked to the activation of microglia and immune signaling. Acute alcohol administration increases NFκB-DNA binding in the brain and initiates an increase in the transcription of NFκB target genes including the proinflammatory cytokines, IL-1β, IL-6, and TNFα [48, 49]. These acute alcohol actions are initiated by alcohol interactions at TLR4 [50]. Chronic alcohol and the resulting continued activation of the TLR4/MD2-induced NFκB are thought to contribute to alcohol-induced neurodegeneration. Although there is little work explicitly exploring the functional contribution of the TLR4/MD2 activation by alcohol using behavioral models, a few studies suggest that TLR4/MD2 activation may play a role in alcohol sensitivity measures and reward. Administration of the non-selective phosphodiesterase inhibitor and global microglia inhibitor, ibudilast, reduces alcohol drinking in several established high alcohol-consuming rodent lines [51]. The C3H/HeJ mouse strain possesses a TLR4 point mutation that can disrupt TLR4 signaling. Interestingly, these mice show very low alcohol preference and consumption using a two-bottle choice paradigm of alcohol drinking [52]. Similarly, TLR4 and MyD88 knockout animals have reduced alcohol-induced sedation and motor impairments compared to control animals [53]. An equivalent reduction in these effects is also observed using the pharmacological TLR4 inhibitor, (+)-naloxone. These findings suggest that both acute and chronic alcohol activate the microglial TLR4/MD2 complex and that this activation may contribute to the subjective effects of alcohol administration.
Opioids
Opioids, such as morphine and remifentanil, have been experientially demonstrated to bind to MD2 [13, 14, 45, 54, 55] and activate TLR4 signaling [56]. Some of the first evidence for a role of glial cells in opiate reward came from studies in which astrocyte-conditioned medium containing a complement of pro-inflammatory cytokines and chemokines was injected into the nucleus accumbens [57]. This enhances the development of conditioned place preference to morphine. Recently, it has been established that opioid interactions with the TLR4/MD2 complex (Figure 1) also potently contribute to opioid actions within the brain’s reward system to potentially influence the development of substance abuse [45]. Thus, TLR4 knockout animals show impaired development of conditioned place preference to morphine and oxycodone. Likewise, the pharmacological TLR4 inhibitor, (+)-naloxone, not only impairs the development of morphine conditioned place preference and self-administration of remifentanil, but also abolishes the ability of morphine to produce dopamine elevations in the nucleus accumbens. These findings reflect the importance of opioid actions at the TLR4/MD2 complex for the acute reinforcing effects of opioids. Other recent evidence illustrates the importance of the TLR4/MD2 complex in the development of tolerance and withdrawal [58, 59]. Lastly, chronic antagonism of the TLR4/MD2 complex during withdrawal impairs the incubation of heroin craving that occurs after prolonged withdrawal [60]. Together, these findings suggest that opioid actions at the microglial TLR4/MD2 complex initiates proinflammatory signaling that powerfully contributes to many behavioral indices contributing to the development of opioid dependence.
This TLR4 stimulatory effect of opioids on microglia may, at first glance, seem contrary to the well-established literature on immunosuppressant effects of morphine and other opioids. Opioids can suppress immune cell functions involved in delayed type hypersensitivity, lymphocyte proliferation, natural killer cell cytotoxicity, antibody production, phagocytic function, immune chemotaxis and recruitment across blood vessels, induce lymphoid organ atrophy, and diminish T-cell CD+/CD8+ ratios [61, 62]. Notably, when one considers immune cell types within the reported immunosuppressive effects, what one finds is that many of the immunosuppressant effects of opioids are on T-cells, neutrophils, and mast cells. That is, a large portion of the literature arises from immune cells other than macrophages, the peripheral immune cell type with the closest functional relationship to microglia. Reports of opioid effects on macrophages are mixed as to direction of effect and the basis of these conflicting reports is unknown [61, 62]. However, morphine has been reported to increase LPS-induced expression of IL-6 and TNFα through the NFkB pathway [61, 62]. Likewise, macrophage nitric oxide, superoxide and proinflammatory cytokine production are enhanced by morphine under both basal and LPS-activated states [61, 62]. Thus, it appears that the effects of opioids on microglia and macrophages may be distinct from other immune cell types that display immunosuppressant effects.
Psychostimulants
There are many indications that psychostimulant drugs such as cocaine and methamphetamine initiate immune signaling and the TLR4/MD2 complex plays a prominent role (Figure 2). One of the first illustrations of increased immune response in the brain and microglial activation was shown with the administration of methamphetamine [63, 64]. Here, high doses of methamphetamine produce enhanced NFκB-DNA binding and induction of proinflammatory cytokine gene expression. Subsequent post-hoc analysis of publicly available databases illustrate that many more proinflammatory genes are upregulated upon psychostimulant administration [65]. It is becoming clear that psychostimulants activate microglia and this powerfully contributes to the behavioral effects of psychostimulant drugs. Thus, microglial inhibitors, ibudilast and sulforaphane, reduce cocaine-induced dopamine release and conditioned place preference [14], and suppress methamphetamine self-administration, locomotion, sensitization, and relapse [66–68]. While there is some indication that these effects are produced through psychostimulant actions at sigma1-receptors [69], there is emerging support for psychostimulant-induced activation of the TLR4/MD2 complex on microglial cells [14]. Recent evidence shows that cocaine [14] and methamphetamine (Wang et al., unpublished observations) each competitively binds to MD2 (Figure 1). Further, psychostimulant administration increases the expression of NFκB in isolated microglia [14, 70], an effect dependent on the activation of the TLR4/MD2 complex [14]. There is also support for a role of the TLR4/MD2 complex in the behavioral effects of cocaine (Figure 2). Pharmacological inhibition of the TLR4/MD2 complex impairs the ability of cocaine to elevate dopamine in the nucleus accumbens, disrupts the development of conditioned place preference and reduces the self-administration of cocaine [14]. Genetic models of disrupted TLR4 function also show reduced cocaine-induced locomotion and impairments in cocaine self-administration [14, 71]. Furthermore, there is support for a role of the TLR4/MD2 complex in cocaine relapse as cocaine-primed reinstatement is diminished by inhibiting immune signaling initiated by the TLR4/MD2 pro-inflammatory pathways (unpublished observations, RKB et al.). Interestingly, inhibition of the TLR4/MD2 complex during withdrawal did not reduce the incubation of craving as was shown for heroin suggesting that there may be differences in the regulation of this phenomenon between drug classes [60]. Nonetheless, these findings cumulatively suggest that microglial activation through the TLR4/MD2 complex is very influential in the behavioral responsiveness of psychostimulants.
Figure 2.
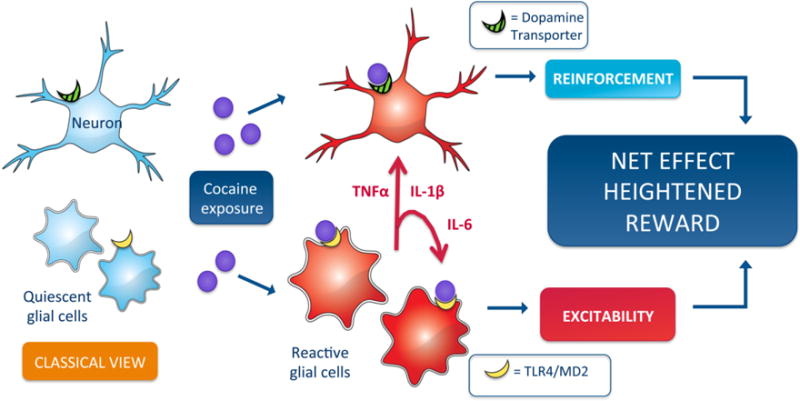
Classically, dopaminergic neurons and glia within the reward neurocircuitry are thought to form parallel relationships with minimal inter-communication. However, a growing body of evidence demonstrates that the tripartite and tetrapartite synapse structures facilitate neuronal-glial connections and enables glial driven behavioral adaptations following drug exposure. For example, cocaine exposure causes dual neuronal dopamine transporter and glial Toll-like receptor 4 actions, creating a complex dopamine and cytokine dependent heightened rewarding response. Viewing cocaine pharmacology from this glial-neuronal vantage point provides opportunities to investigate glial sensitive stimuli as primers for cocaine and other drugs of abuse actions.
Implications for clinical translation
There are currently no approved pharmacotherapies on the market that target the brain’s immune system for the treatment of substance abuse. The ability to generate pharmacotherapies is critical not only to evaluate the xenobiotic hypothesis, but to also identify whether candidate drugs could be used clinically for substance abuse. This is a necessary, but very challenging next step. Specifically, it is difficult to evaluate whether pharmacotherapies should act as global microglial inhibitors, inhibitors of the TLR4/MD2 complex, or inhibit specific proinflammatory markers (i.e. interleukin 1β, interleukin 6, etc). Many of the currently available blood-brain barrier permeable microglia inhibitors are notoriously non-selective and have a plethora of off-target effects. It is not entirely clear whether microglia inhibitors would also impair normal, beneficial anti-inflammatory and pro-inflammatory signals upon other immune challenges. Likewise, inhibition of proinflammatory mediators could also impair normal immune function to all immune challenges and many currently available inhibitors do not penetrate the blood brain barrier. Thus, one may argue that targeting the site of drug action at the TLR4/MD2 complex may be the best approach.
To date, evidence suggests that the blood-brain permeable TLR4 antagonists, (+)-naloxone and (+)-naltrexone, are striking in their breadth of clinically relevant effects on drug abuse. In vitro cell studies show (+)-naltrexone and (+)-naloxone are TRIF-IRF3 axis biased TLR4 antagonists and block TLR4 signaling pathways leading to NO, TNF-α and ROS (Wang et al., unpublished observations). In considering their translation potential, a first question that arises is the selectivity of these compounds for TLR4 versus potential off-target effects. NovaScreen assays of ~70 neurotransmitter and peptide receptors, growth factor/hormone receptors, ion channels, second messengers, and enzymes revealed no effects on these analytes, nor were there binding or functional effects on transporters for serotonin, norepinephrine, or dopamine [45]. While there is evidence for (+)-naloxone and/or (+)-naltrexone as inhibitors of gp91-phox [29] and Filamin A [72], the fact that diverse TLR4 antagonists and TLR4 knockout/mutant strains recapitulate the effects of (+)-naloxone and (+)-naltrexone suggest that TLR4 is likely the predominant target rather than gp91-phox, Filamin A, or other yet to be discovered off-target effects.
Another issue to consider in developing (+)-naltrexone or similar compounds for clinical trial is that (+)-naltrexone has a far lower affinity for TLR4/MD2 than does LPS [54]. While this may be viewed as a negative feature of (+)-naltrexone, the upside is that it would provide a measure of protection under conditions of bacterial infection, as LPS could out compete (+)-naltrexone for TLR4 on immune cells that would normally be activated for host protection in response to LPS. Studies of TLR4/MD2 with (+)-naltrexone indicate that it has comparable affinity to drugs of abuse and endogenous danger signals [13, 14, 45, 54, 55] Equivalent affinities between these compounds would be predicted to bode well for translation of (+)-naltrexone or similar compound for targeted use in the treatment of drug abuse and chronic pain.
A final issue is a practical one for drug development. Namely, what is the appropriate in vitro method for screening compounds to identify drug candidates that target TLR4? We initially used commercially available human embryonic kidney-293 (HEK293) cells that were stably transfected by Invivogen to over-express human TLR4 (hTLR4), MD2, and CD14. This cell line was commercially generated to provide a human relevant (i.e., expressing human TLR4), sensitive measure of endotoxin contamination in test samples, given the potent signaling of LPS through NFκB/AP-1 downstream of TLR4. This HEK293 cell line is engineered to signal TLR4 activation solely via NFκB/AP-1 activation, leading to the production of a reporter protein. However, given the instability in the HEK293-TLR4 cell line that arose over time, we have abandoned it in favor of several other approaches. For example, BV-2 microglia allow the study of effects on NFκB/AP-1 leading to IL-1β, NFkB/IRF3 leading to TNFα, and TRIF/IRF3 leading to nitric oxide and interferon-beta [54] This is important as TLR4 agonists and antagonists can exert biased effects; that is, they can signal or block signaling in select downstream pathways. Hence interrogating TLR4 signaling via HEK293 cells that report only via NFkB/AP-1 provides at best incomplete answers for issues of critical importance to drug development. Indeed, these more recent screens reveal that (+)-naloxone and (+)-naltrexone predominantly block the activation of TLR4 intracellular pathways other than NFkB [54].
Summary
We describe an emerging story in which drugs of abuse produce a neuroinflammatory response in the brain via the activation of a specific ancient receipt on at least microglia, called TLR4. We propose that TLR4 recognizes and is activated by drugs of abuse as xenobiotics. The TLR4/MD2 complex not only plays a central role in the initiation of this response but it is also positioned to create heightened glial reactivity upon chronic drug use that perpetuates innate immune responses potentially contributing to the development of drug dependence. It is essential to seize this enhanced understanding of the ability of drugs of abuse to enhance immune reactivity in the brain by identifying novel strategies to target the TLR4/MD2 complex. The ability of (+)-naloxone and (+)-naltrexone to inhibit the TLR4/MD2 complex has been exploited in several basic science and preclinical animal models of drug abuse. The findings of these studies largely demonstrate the significant contribution of TLR4-induced microglial reactivity on behavioral manifestations of drug dependence. It will be important to consider whether these compounds should be moved forward for testing and approval in clinical settings, or whether new TLR4-based pharmacotherapies are developed and testing for effectiveness. Regardless, it is essential to appreciate and consider the important impact that central immune signaling has on the initiation and development of drug abuse and that this information can be used to guide future treatment approaches.
Acknowledgments
This work was supported in part by NIH R01 DA023132 and DA033358, DoD grant PR110146 and Australian Research Council Research Fellowship (DP110100297).
List of abbreviations
- CD14
cluster determinant 14, a protein of the TLR4 signaling complex
- HEK293-hTLR4
human embryonic kidney-293 cells expressing human toll like receptor 4
- IL-1β
interleukin-1beta
- HMGB1
high mobility group box 1, an endogenous danger signal
- LPS
lipopolysaccharide
- MD2
myeloid differentiation 2
- NAc
nucleus accumbens shell
- NFκB
nuclear factor-kappa B
- TLR4
toll like receptor 4
- VTA
ventral tegmental area
Footnotes
Conflict of interest: LRW is a co-founder and co-chair of the Scientific Advisory Board of Xalud Therapeutics.
References
- 1.Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–5. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 2.Hutchinson MR, Watkins LR. Why is neuroimmunopharmacology crucial for the future of addiction research? Neuropharmacology. 2014;76(Pt B):218–27. doi: 10.1016/j.neuropharm.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grace PM, Hutchinson MR, Maier SF, Watkins LR. Pathological pain and the neuroimmune interface. Nat Rev Immunol. 2014;14:217–31. doi: 10.1038/nri3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mayer DJ, Mao J, Holt J, Price DD. Cellular mechanisms of neuropathic pain, morphine tolerance, and their interactions. Proc Natl Acad Sci U S A. 1999;96:7731–6. doi: 10.1073/pnas.96.14.7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song P, Zhao ZQ. The involvement of glial cells in the development of morphine tolerance. Neurosci Res. 2001;39:281–6. doi: 10.1016/s0168-0102(00)00226-1. [DOI] [PubMed] [Google Scholar]
- 6.Fairbanks CA, Wilcox GL. Spinal plasticity of acute opioid tolerance. J Biomed Sci. 2000;7:200–12. doi: 10.1007/BF02255467. [DOI] [PubMed] [Google Scholar]
- 7.Hutchinson MR, Shavit Y, Grace PM, Rice KC, Maier SF, Watkins LR. Exploring the neuroimmunopharmacology of opioids: an integrative review of mechanisms of central immune signaling and their implications for opioid analgesia. Pharmacol Rev. 2011;63:772–810. doi: 10.1124/pr.110.004135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watkins LR, Hutchinson MR, Ledeboer A, Wieseler-Frank J, Milligan ED, Maier SF. Norman Cousins Lecture. Glia as the “bad guys”: implications for improving clinical pain control and the clinical utility of opioids. Brain Behav Immun. 2007;21:131–46. doi: 10.1016/j.bbi.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beitner-Johnson D, Guitart X, Nestler EJ. Glial fibrillary acidic protein and the mesolimbic dopamine system: regulation by chronic morphine and Lewis-Fischer strain differences in the rat ventral tegmental area. J Neurochem. 1993;61:1766–73. doi: 10.1111/j.1471-4159.1993.tb09814.x. [DOI] [PubMed] [Google Scholar]
- 10.Hutchinson MR, Zhang Y, Shridhar M, Evans JH, Buchanan MM, Zhao TX, et al. Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav Immun. 2010;24:83–95. doi: 10.1016/j.bbi.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hutchinson MR, Lewis SS, Coats BD, Rezvani N, Zhang Y, Wieseler JL, et al. Possible involvement of toll-like receptor 4/myeloid differentiation factor-2 activity of opioid inactive isomers causes spinal proinflammation and related behavioral consequences. Neuroscience. 2010;167:880–93. doi: 10.1016/j.neuroscience.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hutchinson MR, Zhang Y, Brown K, Coats BD, Shridhar M, Sholar PW, et al. Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4) Eur J Neurosci. 2008;28:20–9. doi: 10.1111/j.1460-9568.2008.06321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, Loram LC, Ramos K, de Jesus AJ, Thomas J, Cheng K, et al. Morphine activates neuroinflammation in a manner parallel to endotoxin. Proc Natl Acad Sci U S A. 2012;109:6325–30. doi: 10.1073/pnas.1200130109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Northcutt A, Hutchinson M, Wang X, Baratta MV, Hiranita T, Cochran TA, et al. DAT isn’t all that: cocaine reward and reinforcement requires Toll Like Receptor 4 signaling. Molecular Psychiatry. 2014 doi: 10.1038/mp.2014.177. in review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blanco AM, Valles SL, Pascual M, Guerri C. Involvement of TLR4/type I IL-1 receptor signaling in the induction of inflammatory mediators and cell death induced by ethanol in cultured astrocytes. J Immunol. 2005;175:6893–9. doi: 10.4049/jimmunol.175.10.6893. [DOI] [PubMed] [Google Scholar]
- 16.Nicotra L, Loram LC, Watkins LR, Hutchinson MR. Toll-like receptors in chronic pain. Exp Neurol. 2012;234:316–29. doi: 10.1016/j.expneurol.2011.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meng J, Gong M, Bjorkbacka H, Golenbock DT. Genome-wide expression profiling and mutagenesis studies reveal that lipopolysaccharide responsiveness appears to be absolutely dependent on TLR4 and MD-2 expression and is dependent upon intermolecular ionic interactions. J Immunol. 2011;187:3683–93. doi: 10.4049/jimmunol.1101397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Traber DL, Thomson PD, Blalock JE, Smith EM, Adams T, Jr, Sziebert LA, et al. Action of an opiate receptor blocker on ovine cardiopulmonary responses to endotoxin. Am J Physiol. 1983;245:H189–93. doi: 10.1152/ajpheart.1983.245.2.H189. [DOI] [PubMed] [Google Scholar]
- 19.Sziebert L, Thomson PD, Jinkins J, Rice K, Adams T, Jr, Henriksen N, et al. Effect of naloxone treatment on the cardiopulmonary response to endotoxin in sheep. Adv Shock Res. 1983;10:121–8. [PubMed] [Google Scholar]
- 20.Rock FL, Hardiman G, Timans JC, Kastelein RA, Bazan JF. A family of human receptors structurally related to Drosophila Toll. Proc Natl Acad Sci U S A. 1998;95:588–93. doi: 10.1073/pnas.95.2.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–8. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 22.Bajo M, Madamba SG, Roberto M, Blednov YA, Sagi VN, Roberts E, et al. Innate immune factors modulate ethanol interaction with GABAergic transmission in mouse central amygdala. Brain Behav Immun. 2014 doi: 10.1016/j.bbi.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahmadi A, Sayyah M, Khoshkholgh-Sima B, Choopani S, Kazemi J, Sadegh M, et al. Intra-hippocampal injection of lipopolysaccharide inhibits kindled seizures and retards kindling rate in adult rats. Exp Brain Res. 2013;226:107–20. doi: 10.1007/s00221-013-3415-6. [DOI] [PubMed] [Google Scholar]
- 24.Sayyah M, Javad-Pour M, Ghazi-Khansari M. The bacterial endotoxin lipopolysaccharide enhances seizure susceptibility in mice: involvement of proinflammatory factors: nitric oxide and prostaglandins. Neuroscience. 2003;122:1073–80. doi: 10.1016/j.neuroscience.2003.08.043. [DOI] [PubMed] [Google Scholar]
- 25.Xu YF, Fu LL, Jiang CH, Qin YW, Ni YQ, Fan JW. Naloxone inhibition of lipopolysaccharide-induced activation of retinal microglia is partly mediated via the p38 mitogen activated protein kinase signalling pathway. J Int Med Res. 2012;40:1438–48. doi: 10.1177/147323001204000422. [DOI] [PubMed] [Google Scholar]
- 26.Jiang X, Ni Y, Liu T, Zhang M, Ren H, Xu G. Inhibition of LPS-induced retinal microglia activation by naloxone does not prevent photoreceptor death. Inflammation. 2013;36:42–52. doi: 10.1007/s10753-012-9518-6. [DOI] [PubMed] [Google Scholar]
- 27.Block L, Forshammar J, Westerlund A, Bjorklund U, Lundborg C, Biber B, et al. Naloxone in ultralow concentration restores endomorphin-1-evoked Ca(2)(+) signaling in lipopolysaccharide pretreated astrocytes. Neuroscience. 2012;205:1–9. doi: 10.1016/j.neuroscience.2011.12.058. [DOI] [PubMed] [Google Scholar]
- 28.Wang TY, Su NY, Shih PC, Tsai PS, Huang CJ. Anti-inflammation effects of naloxone involve phosphoinositide 3-kinase delta and gamma. J Surg Res. 2014 doi: 10.1016/j.jss.2014.06.022. [DOI] [PubMed] [Google Scholar]
- 29.Wang Q, Zhou H, Gao H, Chen SH, Chu CH, Wilson B, et al. Naloxone inhibits immune cell function by suppressing superoxide production through a direct interaction with gp91phox subunit of NADPH oxidase. J Neuroinflammation. 2012;9:32. doi: 10.1186/1742-2094-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu SL, Li YH, Shi GY, Chen YH, Huang CW, Hong JS, et al. A novel inhibitory effect of naloxone on macrophage activation and atherosclerosis formation in mice. J Am Coll Cardiol. 2006;48:1871–9. doi: 10.1016/j.jacc.2006.07.036. [DOI] [PubMed] [Google Scholar]
- 31.Shen D, Cao X, Zhao L, Tuo J, Wong WT, Chan CC. Naloxone ameliorates retinal lesions in Ccl2/Cx3cr1 double-deficient mice via modulation of microglia. Invest Ophthalmol Vis Sci. 2011;52:2897–904. doi: 10.1167/iovs.10-6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang P, Lokuta KM, Turner DE, Liu B. Synergistic dopaminergic neurotoxicity of manganese and lipopolysaccharide: differential involvement of microglia and astroglia. J Neurochem. 2010;112:434–43. doi: 10.1111/j.1471-4159.2009.06477.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu B, Jiang JW, Wilson BC, Du L, Yang SN, Wang JY, et al. Systemic infusion of naloxone reduces degeneration of rat substantia nigral dopaminergic neurons induced by intranigral injection of lipopolysaccharide. J Pharmacol Exp Ther. 2000;295:125–32. [PubMed] [Google Scholar]
- 34.Liu B, Du L, Kong LY, Hudson PM, Wilson BC, Chang RC, et al. Reduction by naloxone of lipopolysaccharide-induced neurotoxicity in mouse cortical neuron-glia co-cultures. Neuroscience. 2000;97:749–56. doi: 10.1016/s0306-4522(00)00057-9. [DOI] [PubMed] [Google Scholar]
- 35.Wang CC, Cheng PY, Peng YJ, Wu ES, Wei HP, Yen MH. Naltrexone protects against lipopolysaccharide/D-galactosamine-induced hepatitis in mice. J Pharmacol Sci. 2008;108:239–47. doi: 10.1254/jphs.08096fp. [DOI] [PubMed] [Google Scholar]
- 36.Lin SL, Lee YM, Chang HY, Cheng YW, Yen MH. Effects of naltrexone on lipopolysaccharide-induced sepsis in rats. J Biomed Sci. 2005;12:431–40. doi: 10.1007/s11373-005-0647-x. [DOI] [PubMed] [Google Scholar]
- 37.Greeneltch KM, Haudenschild CC, Keegan AD, Shi Y. The opioid antagonist naltrexone blocks acute endotoxic shock by inhibiting tumor necrosis factor-alpha production. Brain Behav Immun. 2004;18:476–84. doi: 10.1016/j.bbi.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 38.Xiao E, Xia-Zhang L, Ferin M. Inhibitory effects of endotoxin on LH secretion in the ovariectomized monkey are prevented by naloxone but not by an interleukin-1 receptor antagonist. Neuroimmunomodulation. 2000;7:6–15. doi: 10.1159/000026415. [DOI] [PubMed] [Google Scholar]
- 39.Romanovsky AA, Shido O, Ungar AL, Blatteis CM. Peripheral naloxone attenuates lipopolysaccharide fever in guinea pigs by an action outside the blood-brain barrier. Am J Physiol. 1994;266:R1824–31. doi: 10.1152/ajpregu.1994.266.6.R1824. [DOI] [PubMed] [Google Scholar]
- 40.Hutchinson MR, Ramos KM, Loram LC, Wieseler J, Sholar PW, Kearney JJ, et al. Evidence for a role of heat shock protein-90 in toll like receptor 4 mediated pain enhancement in rats. Neuroscience. 2009;164:1821–32. doi: 10.1016/j.neuroscience.2009.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu HE, Sun HS, Cheng CW, Terashvili M, Tseng LF. dextro-Naloxone or levo-naloxone reverses the attenuation of morphine antinociception induced by lipopolysaccharide in the mouse spinal cord via a non-opioid mechanism. Eur J Neurosci. 2006;24:2575–80. doi: 10.1111/j.1460-9568.2006.05144.x. [DOI] [PubMed] [Google Scholar]
- 42.Johnston IN, Westbrook RF. Inhibition of morphine analgesia by LPS: role of opioid and NMDA receptors and spinal glia. Behav Brain Res. 2005;156:75–83. doi: 10.1016/j.bbr.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 43.Liu B, Du L, Hong JS. Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J Pharmacol Exp Ther. 2000;293:607–17. [PubMed] [Google Scholar]
- 44.Lewis SS, Loram LC, Hutchinson MR, Li CM, Zhang Y, Maier SF, et al. (+)-naloxone, an opioid-inactive toll-like receptor 4 signaling inhibitor, reverses multiple models of chronic neuropathic pain in rats. J Pain. 2012;13:498–506. doi: 10.1016/j.jpain.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hutchinson MR, Northcutt AL, Hiranita T, Wang X, Lewis SS, Thomas J, et al. Opioid activation of toll-like receptor 4 contributes to drug reinforcement. J Neurosci. 2012;32:11187–200. doi: 10.1523/JNEUROSCI.0684-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–38. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomas MJ, Kalivas PW, Shaham Y. Neuroplasticity in the mesolimbic dopamine system and cocaine addiction. Br J Pharmacol. 2008;154:327–42. doi: 10.1038/bjp.2008.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crews F, Nixon K, Kim D, Joseph J, Shukitt-Hale B, Qin L, et al. BHT blocks NF-kappaB activation and ethanol-induced brain damage. Alcohol Clin Exp Res. 2006;30:1938–49. doi: 10.1111/j.1530-0277.2006.00239.x. [DOI] [PubMed] [Google Scholar]
- 49.Zou J, Crews F. Induction of innate immune gene expression cascades in brain slice cultures by ethanol: key role of NF-kappaB and proinflammatory cytokines. Alcohol Clin Exp Res. 2010;34:777–89. doi: 10.1111/j.1530-0277.2010.01150.x. [DOI] [PubMed] [Google Scholar]
- 50.Alfonso-Loeches S, Pascual-Lucas M, Blanco AM, Sanchez-Vera I, Guerri C. Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. J Neurosci. 2010;30:8285–95. doi: 10.1523/JNEUROSCI.0976-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bell RL, Lopez MF, Cui C, Egli M, Johnson KW, Franklin KM, et al. Ibudilast reduces alcohol drinking in multiple animal models of alcohol dependence. Addict Biol. 2013 doi: 10.1111/adb.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li XC, Karadsheh MS, Jenkins PM, Stitzel JA. Genetic correlation between the free-choice oral consumption of nicotine and alcohol in C57BL/6JxC3H/HeJ F2 intercross mice. Behav Brain Res. 2005;157:79–90. doi: 10.1016/j.bbr.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 53.Wu Y, Lousberg EL, Moldenhauer LM, Hayball JD, Coller JK, Rice KC, et al. Inhibiting the TLR4-MyD88 signalling cascade by genetic or pharmacological strategies reduces acute alcohol-induced sedation and motor impairment in mice. Br J Pharmacol. 2012;165:1319–29. doi: 10.1111/j.1476-5381.2011.01572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang X, al. e. MS in preparation. 2014
- 55.Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A. 2010;107:11942–7. doi: 10.1073/pnas.1003893107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jacobsen JH, Watkins LR, Hutchinson MR. Discovery of a Novel Site of Opioid Action at the Innate Immune Pattern-Recognition Receptor TLR4 and its Role in Addiction. Int Rev Neurobiol. 2014;118:129–63. doi: 10.1016/B978-0-12-801284-0.00006-3. [DOI] [PubMed] [Google Scholar]
- 57.Narita M, Miyatake M, Narita M, Shibasaki M, Shindo K, Nakamura A, et al. Direct evidence of astrocytic modulation in the development of rewarding effects induced by drugs of abuse. Neuropsychopharmacology. 2006;31:2476–88. doi: 10.1038/sj.npp.1301007. [DOI] [PubMed] [Google Scholar]
- 58.Shen H, Hu X, Szymusiak M, Wang ZJ, Liu Y. Orally administered nanocurcumin to attenuate morphine tolerance: comparison between negatively charged PLGA and partially and fully PEGylated nanoparticles. Mol Pharm. 2013;10:4546–51. doi: 10.1021/mp400358z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hutchinson MR, Lewis SS, Coats BD, Skyba DA, Crysdale NY, Berkelhammer DL, et al. Reduction of opioid withdrawal and potentiation of acute opioid analgesia by systemic AV411 (ibudilast) Brain Behav Immun. 2009;23:240–50. doi: 10.1016/j.bbi.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Theberge FR, Li X, Kambhampati S, Pickens CL, St Laurent R, Bossert JM, et al. Effect of chronic delivery of the Toll-like receptor 4 antagonist (+)-naltrexone on incubation of heroin craving. Biol Psychiatry. 2013;73:729–37. doi: 10.1016/j.biopsych.2012.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schwacha MG. Opiates and the development of post-injury complications: a review. Int J Clin Exp Med. 2008;1:42–9. [PMC free article] [PubMed] [Google Scholar]
- 62.Wang J, Barke RA, Ma J, Charboneau R, Roy S. Opiate abuse, innate immunity, and bacterial infectious diseases. Arch Immunol Ther Exp (Warsz) 2008;56:299–309. doi: 10.1007/s00005-008-0035-0. [DOI] [PubMed] [Google Scholar]
- 63.Asanuma M, Cadet JL. Methamphetamine-induced increase in striatal NF-kappaB DNA-binding activity is attenuated in superoxide dismutase transgenic mice. Brain Res Mol Brain Res. 1998;60:305–9. doi: 10.1016/s0169-328x(98)00188-0. [DOI] [PubMed] [Google Scholar]
- 64.Asanuma M, Miyazaki I, Higashi Y, Tsuji T, Ogawa N. Specific gene expression and possible involvement of inflammation in methamphetamine-induced neurotoxicity. Ann N Y Acad Sci. 2004;1025:69–75. doi: 10.1196/annals.1316.009. [DOI] [PubMed] [Google Scholar]
- 65.Clark KH, Wiley CA, Bradberry CW. Psychostimulant abuse and neuroinflammation: emerging evidence of their interconnection. Neurotox Res. 2013;23:174–88. doi: 10.1007/s12640-012-9334-7. [DOI] [PubMed] [Google Scholar]
- 66.Snider SE, Hendrick ES, Beardsley PM. Glial cell modulators attenuate methamphetamine self-administration in the rat. Eur J Pharmacol. 2013;701:124–30. doi: 10.1016/j.ejphar.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Snider SE, Vunck SA, van den Oord EJ, Adkins DE, McClay JL, Beardsley PM. The glial cell modulators, ibudilast and its amino analog, AV1013, attenuate methamphetamine locomotor activity and its sensitization in mice. Eur J Pharmacol. 2012;679:75–80. doi: 10.1016/j.ejphar.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen H, Wu J, Zhang J, Fujita Y, Ishima T, Iyo M, et al. Protective effects of the antioxidant sulforaphane on behavioral changes and neurotoxicity in mice after the administration of methamphetamine. Psychopharmacology (Berl) 2012;222:37–45. doi: 10.1007/s00213-011-2619-3. [DOI] [PubMed] [Google Scholar]
- 69.Beardsley PM, Hauser KF. Glial modulators as potential treatments of psychostimulant abuse. Adv Pharmacol. 2014;69:1–69. doi: 10.1016/B978-0-12-420118-7.00001-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wires ES, Alvarez D, Dobrowolski C, Wang Y, Morales M, Karn J, et al. Methamphetamine activates nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kappaB) and induces human immunodeficiency virus (HIV) transcription in human microglial cells. J Neurovirol. 2012;18:400–10. doi: 10.1007/s13365-012-0103-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thomsen M, Caine SB. Psychomotor stimulant effects of cocaine in rats and 15 mouse strains. Exp Clin Psychopharmacol. 2011;19:321–41. doi: 10.1037/a0024798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang HY, Burns LH. Naloxone’s pentapeptide binding site on filamin A blocks Mu opioid receptor-Gs coupling and CREB activation of acute morphine. PLoS One. 2009;4:e4282. doi: 10.1371/journal.pone.0004282. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]