

Summary
Ever since the discovery of the first microRNAs in C. elegans, increasing numbers of endogenous small RNAs have been discovered. Endogenous siRNAs (endo-siRNAs) have emerged in the last few years as a largely independent class of small RNAs that regulate endogenous gene expression, with mechanisms distinct from those of piRNAs and miRNAs. Quantification of these small RNAs and their effect on target RNAs is a powerful tool for the analysis of RNAi; however, detection of small RNAs can be difficult due to their small size and relatively low abundance. Here, we describe the novel FirePlex assay for directly detecting endo-siRNA levels in bulk, as well as an optimized qPCR method for detecting the effect of endo-siRNAs on gene targets. Intriguingly, the loss of endo-siRNAs frequently results in enhanced experimental RNAi. Thus, we also present an optimized method to assess the indirect impact of endo-siRNAs on experimental RNAi efficiency.
Keywords: siRNA quantification, endo-siRNA targets, RNAi efficacy, FirePlex assay, qPCR
1. Introduction
Since Bartel, Ambros, and colleagues first attempt to systematically clone microRNAs in C. elegans [1, 2], the definition of endogenous short-interfering RNAs has undergone many changes [3, 4]. In recent years, Mello, Fire, Ruvkun, and colleagues have defined C. elegans endo-siRNAs to refer to 22mers and 26mers that predominantly begin with a G residue. Endo-siRNAs are potentially amplified and developmentally regulated and silence endogenous gene targets with dependence on dicer-interacting genes [5–7]. Whereas the term “endo-siRNAs” is well defined in C. elegans. it is used interchangeably with or as an abbreviation for various species of endogenous small RNAs in Drosophila and mammals.
The cloning and sequencing of individual endo-siRNAs have been optimized extensively by Mello and colleagues, with robust updates in response to continuing increases in sequencing technologies [8]. However, the genetic context and the regulatory impact of many of these endo-siRNAs are still mysterious despite the many large datasets available. For instance, Ruvkun and colleagues discovered that some endo-siRNAs target duplicated genes [9], while Kennedy and colleagues found that some endo-siRNAs have a nuclear preference [10]. Hence, additional higher-order analysis is needed to unravel the extensive biological functions of endo-siRNAs.
Publicly available sequencing datasets report endo-siRNAs number from hundreds to hundreds of thousands [5, 6, 11, 12]. Because of their differential sizes and termini [13–15], confounded by an amplification processing step whereby the rarer 26Gs get amplified to become the abundant 22Gs [6], understanding the role of particular endo-siRNAs in C. elegans can be difficult. Even more challenging, the loss of endo-siRNAs in many instances induces an increase in sensitivity to experimental RNAi [16, 17]. Therefore, it is important to assay both the direct and indirect effects of endo-siRNAs in order to understand their biological significance.
Since C. elegans endo-siRNAs impact on endogenous gene expression and the efficacy of experimental RNAi, we will briefly survey the genetic resources available and outline specific methods to detect some of these effects. Here, we specifically examine the novel FirePlex method to detect endo-siRNA quantities in bulk. Additionally, we outline an optimized method for assaying validated endo-siRNA targets. Finally, we present our method for detecting subtle changes in experimental RNAi efficacy induced by the loss of endo-siRNAs.
1.1 Quantification of bulk siRNAs
Quantification of siRNA levels is useful for the analysis of endo-siRNA pathways. Due to their small size, efficient detection of siRNAs by conventional methods, such as microarrays or qPCR, is difficult. In C. elegans, the gold standard for measuring siRNA levels is small RNA sequencing. However, this approach is time-consuming, relatively expensive, and requires technical expertise and optimization.
Here, we introduce and describe the use of the FirePlex assay for the quantification of siRNAs in C. elegans. Although the FirePlex assay was originally commercialized for miRNAs, we have found that it can robustly quantify siRNA levels as well. The FirePlex platform utilizes encoded hydrogel particles to perform multiplexed detection of up to 68 targets in each well of a standard 96-well filter plate. Particles bear unique barcodes that correspond to a single target detected on each. The assay is performed in three steps – hybridization, labeling and reporting, with rinses between each step (Fig. 1). During hybridization, targets bind to siRNA-specific DNA probes embedded in the hydrogel particles. Labeling is accomplished via ligation of a biotinylated universal adaptor using the probe as a template. In the final step, a streptavidin-conjugated fluorescent reporter is added to visualize the hybridization event. The assay provides quantitative results, with the level of fluorescence on each particle corresponding to the amount of siRNA target present in the sample. The encoded particles are then scanned in a standard flow cytometer.
Figure 1.
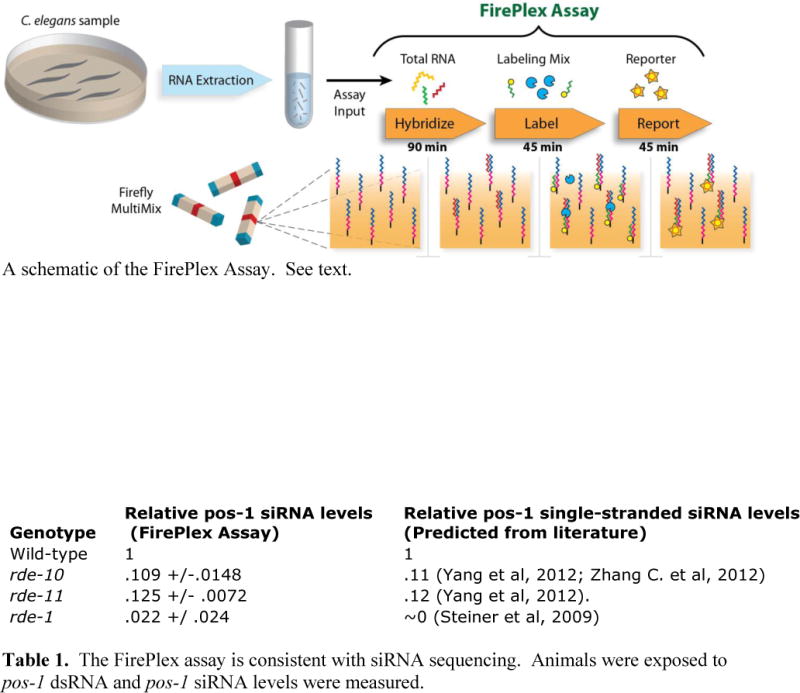
A schematic of the FirePlex Assay. See text.
We used the FirePlex assay with purified total RNA as the input, though the system may also be applied to crude cell and tissue digests. Samples can be analyzed on-site with a FirePlex kit and a conventional flow cytometer, or alternatively, sent directly to Firefly BioWorks for custom analysis with additional costs. Although the FirePlex assay is considerably simpler than conventional siRNA sequencing, it has important limitations. First, the assay is not a discovery tool – target sequences must be known and specified. Furthermore, while the FirePlex assay allows examination of up to 68 targets per well, short RNA sequencing provides a comprehensive analysis of all siRNAs in the sample. And last, the FirePlex platform has limited sensitivity; we have been able to detect sequences found at a comparable rate of 150 reads/million in a previously published siRNA sequencing dataset (Table 1) [18]. However, our previous results suggest that the FirePlex assay is likely an order of magnitude more sensitive. Despite these disadvantages, the simplicity and throughput of the FirePlex assay should make it a very attractive tool for researchers. We imagine that the FirePlex assay can be used to quickly screen through various mutants or conditions, with interesting results being followed-up with siRNA sequencing, if necessary.
Table 1.
Robust endo-siRNA targets
| Gene | Endo-siRNA loss via | Changes upon endo-siRNA loss | RT primer | F primer | R primer |
|---|---|---|---|---|---|
| F07G6.6 | rrf-3 and eri-1 mutants | Increase | ctcaaaaggtcctctctcatttg | cgcacaaaacattaaatttgctaac | ccccgacaagtcaatatttttgag |
| F14F7.5 | rrf-3 and eri-1 mutants | Increase | caagtttatccattgttcgtacttc | atggttttccgtgaatctgaag | gttgaagtgctctggattttaac |
| F39E9.7 | rrf-3 and eri-1 mutants | Increase | caagcgcctatacttagcgatgg | gattgatgtcctgaacccagtg | gagaattgcttcggcagctg |
| H16D19.4 | rrf-3 mutants | Increase | ctccgattacatcttaagtgtgtag | gattacgatcaacgcaagatac | gcaatttccttatttgaatgttgc |
| Y37E11B.2 | rrf-3 and eri-1 mutants | Increase | ggggtaaggtttcagcgaag | gtgcctgcttctcctcatc | cattgatcttggttgcaggttc |
| Y43F8B.9 | rrf-3 mutants | Increase | cagattgattgccatttcaagg | ccaccgctagctgtaaaaaatac | cactcaaagcacattggcag |
| C54G7.3 | rrf-3 mutants | Decrease | ggctcacttgctgagacac | gatcgtcggtcccggc | cattacttgcacaggttgtgatg |
| F55A4.4 | rrf-3 mutants | Decrease | gtcgggcttgcttagcgatg | aagtgcaagggagaacagaaag | gcacattggcagcaaggg |
| ZK816.5 | rrf-3 mutants | Decrease | gggattcggccgtcaatatc | gggtggaataatgtacttgttcac | attttgttttccgtcgcgc |
1.2 Direct quantification of endo-siRNA target expression
Robust amplification is a unique aspect of RNAi in C. elegans, which is absent in other organisms such as mammals [19, 20]. A possible mechanistic explanation suggests that amplification plays a significant role in C. elegans RNAi because it allows siRNAs to target more than one transcript through the process of transitive RNAi [21, 22]. However, when endo-siRNAs target more than one transcript, or one transcript is targeted by more than one endo-siRNAs, the relationship is between endo-siRNA presence and target gene expression often remains unclear. Generally, endo-siRNAs are thought to silence their complementary gene targets [5], but there are cases in which endo-siRNAs fail to silence or –in contrast- even protect their complementary gene targets [11].
Therefore, to monitor the direct effects of endo-siRNAs on endogenous gene expression, we validated nine exemplary endo-siRNA targets by qPCR. The relevant sequences derive from previously published small RNA sequencing data [22, 23]. The targets are listed below and the changes in expression upon endo-siRNA depletion are indicated. The reported targets can be used as reference genes to monitor the impact of induced changes in endo-siRNA abundance. In a related series of experiments, we describe an optimized qPCR protocol to quantitate those changes in gene expression.
1.3 Gauging endo-siRNAs’ effects on experimental RNAi
The canonical class of C. elegans endo-siRNAs was first discovered in the context of experimental RNAi. Ruvkun and colleagues performed a genetic screen for mutants with enhanced sensitivity to experimental RNAi [24]. These enhanced RNAi (Eri) mutants were later discovered to be missing a class of small RNAs that became the founding members of what are now called C. elegans endo-siRNAs [5, 6, 25]. A comprehensive list of these eri genes and their functions have been previously described [19]. Moreover, Ruvkun and colleagues recently reported that genes responsible for the transcriptional regulation of eri genes were themselves susceptible to experimental RNAi with a potential impact on endo-siRNAs [26]. In addition, there are many lesser-understood genes mediating enhanced RNAi presumably also affecting endo-siRNAs. Two recent reports identified the nuclear Argonaute nrde-3 as an essential component for nuclear exogenous and endogenous RNAi [22]. Moreover the perinuclear foci gene mut-16 was found to be required for the germline component of RNAi [27]; notably, both mutants are partially RNAi-defective.
This complex layer of gene regulation by endo-siRNAs is difficult to assess, especially when attempting to attribute direct causation. However, in most instances, perturbing the regulation of endo-siRNAs results in hyper-sensitizing or de-sensitizing experimental RNAi. Therefore, we describe here an optimized method to gauge the effect of endo-siRNAs on experimental RNAi to characterize indirect effects of endogenous gene regulation.
Note that our protocol presumes two points: First, experimental RNAi is in competition with endogenous RNAi, a well-accepted model in the field [28]; second, experimental RNAi is dosage-dependent [29]. Consequently, we assume that perturbation of endogenous RNAi affects experimental RNAi efficiency. Presented below is a protocol for detecting the differential efficiencies in experimental RNAi, described in detail from a previous study [29].
2. Materials
Always use RNase-free materials and ultrapure or DEPC-treated water.
RNA Extraction and FirePlex Assay
1.5 ml Phase-Lock Gel tubes, heavy (Eppendorf)
TRIzol Reagent (Life Technologies)
PTFE (Polytetrafluoroethylene) Tissue Grinder Douncer, 2 mL, glass vessel and serrated plunger (VWR)
Chloroform
5M NaCl: dissolve 29.22 g of NaCl in 80ml of water and fill up to 100 ml. Sterilize by autoclaving or sterile filter. Also available from commercial sources.
20 mg/ml glycogen as a carrier for RNA precipitation. Available form commercial sources.
Isopropanol
FirePlex kit (Firefly BioWorks, Inc., Cambridge, MA, USA)
A standard flow cytometer (established settings exist for Millipore Guava easyCyte 8HT, BD Accuri C6, Millipore Guava easyCyte 6HT, BD LSRFortessa and Life Technologies Attune)
Vacuum Manifold for 96-well filter plates (one optimized for FirePlex is available from Firefly BioWorks)
Direct quantification of endo-siRNA target expression
RNase-free recombinant DNase I (Roche Applied Science)
Qiagen RNeasy mini kit (Qiagen)
ThermoScript™ RT-PCR System for First-Strand cDNA Synthesis (Invitrogen)
Qiagen QuantiTect SYBR Green PCR Kit (Qiagen)
Twin.tec 96-well real-time PCR Plates, skirted, blue including Masterclear™ real-time PCR self-adhesive film, (Eppendorf)
Mastercycler® ep realplex PCR machine (Eppendorf)
Normal PCR thermocycler
PCR plate spinner
Gauging endo-siRNAs’ effects on experimental RNAi
Ahringer library of C. elegans feeding RNAi clones [30, 31] (Source BioScience, LifeSciences)
QIAprep Spin Miniprep Kit (Qiagen)
Isopropyl-β-D-thiogalactopyranoside (IPTG)
35×10 mm plates (Greiner Bio-One)
Carbenicillin
Tetracycline
NG media components; 3 g NaC1, 2.5 Bactopeptone (Difco) and 17 g Bacto-agar (Difco) are dissolved in 975 ml distilled water. After autoclaving, 1 ml cholesterol in ethanol (5 mg/ml), 1 ml M CaCl, 1 ml M MgSO, and 25 ml M potassium phosphate buffer (pH 6.0) are added [32].
LB plates
3 Methods
3.1 RNA Extraction (see Note 1)
Wash several (2–5) 10-centimeter plates of animals into a 15 ml conical centrifuge tube using water (see Note 2). Centrifuge at 11,000×g, 1 minute.
Wash 3 times with 15 ml water.
Discard the final wash. Using a Pasteur pipette, transfer 200–350 μl of worms into a 1.5 ml microfuge tube. Freeze at −80°C for one hour.
Prepare phase-lock tubes by centrifugation at 11,000×g for 1 minute.
Add 400 μl Trizol to each sample.
Vortex samples at room-temperature until the Trizol-worm mixture is a slurry.
Transfer the solution into a douncer on ice using a RNase-free glass pasteur pipette.
Dounce slurry with a twisting motion 20 times on ice. Between samples clean with RNase-Out and DEPC-treated water or use a new douncer (see Note 3).
Transfer the lysed worms to a phase-lock tube.
Add 80 μl chloroform, mix by inversion for 1 minute and incubate at room-temperature for 5 minutes.
Centrifuge at 15,000 × g for 15 minutes at 4°C.
Transfer the top aqueous layer to a siliconized 1.5 mL microfuge tube.
Add, in this order, 50 μl of 5M NaCl, 2.5 μl of 20 mg/ml glycogen and 800 μl of isoproponal to each tube.
Mix by inversion for 1 minute and place at −80°C for 1 hour.
Centrifuge at 11,000 × g for 15 minutes at 4°C.
Remove the supernatant with a pipette.
Wash with 500 μl of ice-cold 100% ethanol.
Spin at 11,000 × g at 4°C for 1 minute.
Remove the supernatant and let remaining ethanol evaporate for about 5 minutes at room temperature. Do not allow the pellet to dry out completely.
Dissolve the RNA pellet in 80 μl of water by vortexing at room temperature.
-
Quantitate the RNA concentration using a Nanodrop or an equivalent spectrophotometer.
At this point, samples can be sent directly to Firefly Bio for commercial analysis. Alternatively, samples can be processed in house using a standard flow-cytometer.
3.2 FirePlex Assay
Cut the plate seal to expose assay wells on the filter plate required for the experiment (provided in the assay kit- see Note 4).
Dilute sample to twice the final concentration, in our hands this would routinely be 200ng/μl (see Note 5).
Invert, then vortex MultiMix (provided with the assay) for 3 seconds. Add 35 μl of MultiMix to each well. Mix by pipetting up and down (see Note 6).
Vacuum filter the plate, wipe the bottom dry (see Notes 7, 8 and 9).
Add 25 μl of hybridization buffer to each well, followed by 25 μl of sample.
Hybridize the plate for 90 minutes at 37 °C shaking at 750 rpm (see Note 10).
Near the end of the hybridization step, prepare 1 × rinse buffer by mixing 0.2 ml of 10× Rinse Buffer with 1.8ml of water per assay well. Prepare the Labeling Mix by mixing 78 μl of water, 1.6 μl of 50 × labeling buffer, and 0.4 μl of labeling enzyme per assay well.
After hybridization, add 200 μl 1 × Rinse Buffer to each well, and vacuum filter the plate.
Repeat step 8. Blot dry the bottom rside of the plate.
Add 75 μl of Labeling Mix to each well and shake at room temperature at 750 rpm for 45 minutes.
Prepare the Reporter Mix by mixing 64 μl of water with 16 μl of 5 × Reporter Solution per each assay well.
After Labeling, add 200 μl 1 × Rinse Buffer to each well, and vacuum filter the plate.
Repeat step 13. Blot dry the bottom side of the plate.
Add 75 μL Reporter Mix to each well. Shake at room temperature at 750 rpm for 45 minutes. After the incubation, add 200 μl 1 × Rinse Buffer to each well and vacuum filter the plate.
Repeat step 17. Blot dry the bottom side of the plate.
Add 175 μl Run Buffer to each well.
Apply the correct scan settings for the specific flow-cytometer used (provided at http://www.fireflybio.com/productsupport).
Scan at least 100 μl of sample for each well.
Save the FCS file for analysis using the FireCode software (http://www.fireflybio.com/productsupport).
3.3 Direct quantification of endo-siRNA target expression
To remove traces of genomic DNA from the extracted RNA combine the following mixture in a PCR tube: 5 μl 10 × DNase I buffer, 4 μl DNase I, and 40 μl of the RNA sample. Incubate the sample in the PCR machine at 37°C for 20 minutes and denature the enzyme at 75 °C for 20 minutes.
Isolate the RNA using the Qiagen RNeasy kit. Transfer the reaction mix to a fresh eppendorf tube, add 50 μl of water and 350 μl of buffer RLT.
Add 250 μl of ethanol, mix, and immediately transfer to spin column. (Note 11)
Centrifuge at 11,000 × g for 15 seconds and discard the flow-through carefully.
Wash the column with 500 μl of buffer RPE centrifuge at 11,000×g for 15 seconds, and discard the flow-through carefully.
Wash the column again with 500 μL buffer RPE, centrifuge at 11,000×g for 2 minutes, and discard the flow-through carefully.
Put the column into a new collection tube, centrifuge at 11,000 × g for 1 minute, and discard flow-through.
Put the column in a 1.5 ml collection tube, add 30 μl of water, and elute the RNA by centrifugation at 11,000 × g for 1 minute.
With the column still in the same collection tube, add another 30 μl of water and centrifuge at 11,000×g for 1 minute resulting in slightly less than 60 μl of RNA containing eluate. Spec the eluted cleaned RNA on the Nanodrop.
Determine the concentration of the RNA by Nanodrop or an equivalent spectrophotometer. Dilute the samples to a concentration of about 200 ng/μl. These samples should be strored at −80 °C as a working stock, repeated freeze-thaw cycles should be avoided.
The reverse transcription (RT) reaction is assembled in a PCR tube and contains 1 μl 10 μM RT primer, 3 μl of total RNA, 2 μl of 10 mM dNTP mix, and 6 μL of water (Invitrogen Thermoscript kit). (See Note 12)
Incubate in PCR machine at 65 °C for 5 minutes, transfer the tube on ice.
Meanwhile, assemble a master mix containing per reaction, 4 μl of 5 × cDNA synthesis buffer, 1 μl of 0.1M DTT, 1 μl of RNaseOUT (RNAse inhibitor), 1 μl of water and 1 μl of Thermoscript reverse transcriptase (Invitrogen Thermoscript kit). Add 8 μL of master mix to each tube containing RNA, primer and dNTPs, mix well.
Incubate the RT reaction in a PCR machine at 50 °C for 1 hour and then denature the enzyme at 85 °C for 5 minutes. Transfer the reaction on ice.
Add 1 μl of RNase H (Invitrogen Thermoscript kit) to each tube and incubate at 37 °C for 20 minutes. Transfer the reaction on ice. The cDNA can be stored at −20°C for months.
Thaw Qiagen 2x QuantiTect SYBR Green PCR master mix, RNase-free water, cDNA, and primers and keep the vials on ice (QuantiTect SYBR Green PCR Kits).
Per Eppendorf skirted qPCR plate well, add in the following order: 20 μl of RNase-free water, 1.5 μl of forward primer (10 μM), 1.5 μl of reverse primer (10 μM), 2 μl of cDNA, and 25 μl of SYBR green master mix. Each cDNA-primer-pair combination should be done in technical triplicates. (Note 14)
Mix the contents well and seal plate with clear Eppendorf tape using a tape roller. Spin the reaction down in a PCR plate spinner.
Run the reaction on a thermocycler that allows for quantitative monitoring of the reaction. We use a Mastercycler® ep realplex PCR machine with BOTH dye and probe as SYBR green settings and the following cycling conditions: Activation at 95 °C for 15 minutes, 45 cycles of 94° C for 15 seconds, 52 °C for 30 seconds, 72 °C for 1 minute, followed by a melting curve analysis. Record all Ct values after Noiseband adjustment.
Relative transcript levels are then quantified using the −ΔΔCt method [33]. (See Note 15).
3.4 Gauging endo-siRNAs’ effects on experimental RNAi
For every liter of medium, add 17 g of bacto agar, 2.5 g of bacto peptone, and 3 g of sodium chloride. Add in stirring bar and autoclave. Stir the solution after autoclaving until you can touch the flask. Add 1 ml of 5 mg/ml cholesterol in ethanol, 1 ml of 1M CaCl2, 1 ml of 1M MgSO4, 25 ml of 1M KH2PhO4, 1 ml of 100 g/l carbenicillin, and 238 mg of IPTG while stirring. Pour a volume of 3.5 ml media per 35×10mm plate. Keep the plates at room temperature overnight; then they can be stored at 4 °C for up to two weeks.
Add and spread the antibiotics to LB plates, carbenicillin, to a final concentration 100 mg/l, and tetracycline to a final concentration of 12.5 mg/l. Leave to dry at room temperature. The plates can be stored in 4°C for up to two weeks.
From the Ahringer library, seek the RNAi vector colony against your gene of interest via the Source BioScience LifeSciences database (http://www.lifesciences.sourcebioscience.com/clone-products/mirna–rnai-resources/c-elegans-rnai-library.aspx) (see Note 16).
Take an autoclaved toothpick, and scrape a bit of bacteria carrying the vector from the desired library well, and streak in thirds onto a prewarmed LB-carb/tet plate. Grow at 37 °C overnight.
The next morning, label 5 to 10 individual colonies to be verified for correct plasmid presence.
Prepare 2 mL of LB of medium in a culture tube for each colony and add 2 μl of 100 g/l carbenicillin and 5 μl of 5 mg/ml tetracycline.
Use an autoclaved toothpick, to inoculate the culture, label both plate and culture accordingly. Grow the bacteria at 37 °C in a shaker for no longer than 15 hours. Label each tube with the colony number. Isolate the plasmids. In our hands, the QIAprep Spin Miniprep Kit and the provided protocol works well. Check the insert by sequencing using the M13 forward primer [31].
Regrow the verified colonies, either scale volume or flask size up about 5 times or grow 3–5 liquid culture tubes per RNAi gene target. Always include a colony containing the empty vector L4440 as a negative control for the knock-down experiment, grow as many liquid cultures of the negative control strain as there are RNAi-positive cultures.
After overnight incubation, take an aliquot of each culture and dilute it 1/10 × with LB media.
Measure absorbance at 600nm in the spectrometer for each culture. Based from this measurement, prepare an array of at least five different optical densities of bacteria representing different potencies of RNAi-induction (e.g. have cultures of OD600nm of 0.25, 0.5, 1.0, 1.5, 2.0) (see Note 17).
A culture with a final OD600nm lower than 1.0 should be diluted with vector L4440-containing bacteria, and a culture with a final OD600nm higher than 1.0 should be diluted with just LB media (See Note 3).
Add 50 μl of each culture of the array (RNAi gene target × dilution series of bacteria) to the middle of IPTG-carb plates (3.4.1) (See Note 18).
Leave at room temperature for 1 day allow for IPTG induction of the T7 promoters to drive dsRNA expression.
Place non-starved third-larval stage single C. elegans worms of the desired strain onto each dosage of an RNAi-bacteria containing plate, perform in triplicates (See Notes 19 and 20).
Allow worms to hatch at the desired temperature (e.g., 20 °C) until the next generation reaches young adulthood.
Score for penetrance of the RNAi knockdown phenotype on each plate, calculate the average and standard deviation for each dosage. (Note 21)
Exemplary Ahringer library targets for detecting changes in RNAi sensitivity
| Target | Phenotype | Strength of RNAi** |
|---|---|---|
| dpy-13 | Dumpy | weak |
| unc-73 | Curled dumpy | weak |
| lir-1 | Lethal or larval arrest | medium |
| dpy-11 | Dumpy | strong |
| pos-1 | Embryonic lethal | strong |
| unc-22 | Twitching | strong |
A “weak” RNAi target indicates that the bacterial colony containing that vector from the Ahringer library only induces a RNAi knockdown in an enhanced RNAi strain, whereas a “strong” RNAi target means that only a RNAi-attenuated or -defective strain would not exhibit RNAi knockdown.
As noted before, this protocol only gives a direction in which experimental RNAi is affected, which in most cases suggests that endo-siRNAs are perturbed. However, the directional relationship between an increase or decrease of endo-siRNAs and experimental RNAi is not always the inverse [27, 34]. Therefore, this protocol is appropriate only for indirectly measuring the likely presence or absence of perturbations to endo-siRNA efficacy, and not the direction of the change in efficacy.
Footnotes
RNA Extraction and FirePlex Assay
Standard procedures to avoid contamination with RNase should be used: wear gloves at all times, use filter tips, and spray work surfaces and instruments with RNase-OUT. Total RNA extraction can also be performed by any other standard method.
The typical yield from this extraction exceeds 1 mg of RNA, while we typically require 5 μg per FirePlex reaction. Therefore, the number of starting worms can be scaled down.
To avoid the cleaning of the douncer between samples, it is convenient to have multiple douncers.
The assay plate is purchased from Firefly BioWorks. It is useful to include a small RNA control that will not change between samples to normalize RNA input, for example, U18.
We use a final concentration of 100 ng/μl (5 μg total). This amount can be changed depending on the abundance of your target.
Ensure that particles are mixed well by pipetting up and down between each well.
For all filtration steps, maintain vacuum pressure below 2 PSI during filtration. Do not over-filter particles. Bubbles are OK. While performing the assay, cover the plate, but do not use a plate seal.
After all filtration steps, re-suspend particles within 30 seconds of filtering. It may be convenient to use a multichannel pipette if many samples are run in parallel.
Ensure that the bottom of the plate is always dry.
The optimal shaking speed depends on the orbit of the shaker used. The speed of 750 rpm is best suited for an orbit of 3 mm.
For steps 2–10, follow the protocol provided by Qiagen.
Perform normal PCR with cDNA and RNA-alone with F&R primers for quality control: RNA-alone should get no bands and cDNA’s amplified bands should correspond to mRNA length and not DNA lengths.
The DNase treatment of the RNA and the RT can be assessed by end-point PCR using forward and reverse primers with either cDNA or RNA as a template. Whereas amplification of cDNA should result in the expected amplified fragment the RNA should not yield any product.
Depending on the thermocycler and the reaction plate the assay volume can be reduced to 25 μl or even 10 μl.
While there are many ways to quantitate expression levels of genes after obtaining Ct values from qPCR, we favor the −ΔΔCt method because it is a relative measure. Mathematically, the method first normalizes the difference between a gene of interest against a housekeeping gene (the first Δ), then normalizes that value to the difference in a control condition/strain (the second Δ) [33]. In our hands this is an appropriate method for studying gene regulation by endo-siRNAs because endo-siRNA levels may impact other aspects of C. elegans biology [23]. Therefore, it is important that the expression of the gene of interest is normalized to a dynamic housekeeping marker which may experience some of those same hereto unknown influences of endo-siRNAs as well. Logistically speaking, it means each sample must be accompanied by an RT reaction against a housekeeping. In our experience, gpd-3, ama-1, and pmp-3 are quite robust and stable in expression throughout all worm developmental stages. For more sophisticated temporal normalizations, we found dpy-13 to be quite useful for measuring genes expressed near the first three larval molts, bli-1 useful for late L4 genes, vit-2 for early young adult genes, and mex-3 and pgl-1 for germline genes.
In the Source BioScience LifeSciences database, the library against which there are RNAi clones is listed by their gene sequence number listed on WormBase. After searching with the gene sequence number, examine the listed forward and reverse primers used to build the plasmid portion between T7 promoters driving the dsRNA, to ensure that the region covers an exonic fragment of your gene of interest.
The optical density of the bacteria carrying the RNAi-vector is used as a proxy for RNAi dosage, as previously described in two independent studies [29, 35]. A robust dilution series of RNAi dosages causes a graded RNAi phenotypic response and potentially allows to differentiate subtle sensitivities. In our experience, RNAi targets vary tremendously in their sensitivity. For example, unc-22 (RNAi target) is extremely sensitive, and bacteria at an OD600nm of 1/100,000 still produce some twitching animals. Conversely, dpy-13(RNAi target) is extremely insensitive, and OD600nm of 4.5 sometimes still does not induce Dpy phenotypic animals. Therefore, it is important to examine a wide enough range of RNAi dosages to ensure that a robust response gradient is available for comparison of sensitivity among strains. Ideally, OD600nm concentrations series ought to include concentrations which induce no and fully penetrant RNAi knockdown.
In our experience, a minimal total bacteria volume of 50 μl at a concentration of 1.0 OD600nm is necessary to ensure that the progeny of a single worm do not starve when scoring occurs.
Triplicates of triplicates provide the most robust data for dosage response curve analysis. Therefore, ensure there are enough cultures grown to accommodate the different replicates and dosages. 15 hours of growth at 37°C usually generates around an OD600nm of 2.0 in each 2 ml liquid culture.
For most phenotypes, starting the knockdown in the mother and scoring the RNAi phenotype in the offspring is the most robust and convenient method of RNAi. At the molecular level, there is both intra- and inter-generational RNAi occurring in this experimental set up [36, 37]. Furthermore, for germline RNAi targets, starting the knockdown in worms at stage L3 allows enough time for at least some level of knockdown to be observed. Under these circumstances, the reduction in brood size is scored as a read-out for RNA interference. However, gonadal RNAi targets require first-larval stage single worms to be placed to ensure the elimination of the gonad.
RNAi sensitivity is expected to result in a sigmoidal response curve which varies between strains and conditions if enough dosages are measured. The greatest variability should occur in the intermediate dosages. If the RNAi knockdown phenotype does not vary significantly across the dilution series, a different range of RNAi-bacteria concentration dosages should be assessed.
References
- 1.Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294(5543):858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- 2.Ambros V, Lee RC, Lavanway A, Williams PT, Jewell D. MicroRNAs and other tiny endogenous RNAs in C-elegans. Current Biology. 2003;13(10):807–818. doi: 10.1016/s0960-9822(03)00287-2. [DOI] [PubMed] [Google Scholar]
- 3.Ruby JG, Jan C, Player C, Axtell MJ, Lee W, Nusbaum C, Ge H, Bartel DP. Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans. Cell. 2006;127(6):1193–1207. doi: 10.1016/j.cell.2006.10.040. [DOI] [PubMed] [Google Scholar]
- 4.Lee RC, Hammell CM, Ambros V. Interacting endogenous and exogenous RNAi pathways in Caenorhabditis elegans. Rna. 2006;12(4):589–597. doi: 10.1261/rna.2231506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vasale JJ, Gu W, Thivierge C, Batista PJ, Claycomb JM, Youngman EM, Duchaine TF, Mello CC, Conte D., Jr Sequential rounds of RNA-dependent RNA transcription drive endogenous small-RNA biogenesis in the ERGO-1/Argonaute pathway. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(8):3582–3587. doi: 10.1073/pnas.0911908107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gent JI, Lamm AT, Pavelec DM, Maniar JM, Parameswaran P, Tao L, Kennedy S, Fire AZ. Distinct phases of siRNA synthesis in an endogenous RNAi pathway in C. elegans soma. Molecular cell. 2010;37(5):679–689. doi: 10.1016/j.molcel.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang C, Montgomery TA, Gabel HW, Fischer SEJ, Phillips CM, Fahlgren N, Sullivan CM, Carrington JC, Ruvkun G. mut-16 and other mutator class genes modulate 22G and 26G siRNA pathways in Caenorhabditis elegans. P Natl Acad Sci USA. 2011;108(4):1201–1208. doi: 10.1073/pnas.1018695108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu WF, Lee HC, Chaves D, Youngman EM, Pazour GJ, Conte D, Mello CC. CapSeq and CIP-TAP Identify Pol II Start Sites and Reveal Capped Small RNAs as C. elegans piRNA Precursors. Cell. 2012;151(7):1488–1500. doi: 10.1016/j.cell.2012.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fischer SE. Small RNA-mediated gene silencing pathways in C. elegans. The international journal of biochemistry & cell biology. 2010;42(8):1306–1315. doi: 10.1016/j.biocel.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 10.Guang S, Bochner AF, Burkhart KB, Burton N, Pavelec DM, Kennedy S. Small regulatory RNAs inhibit RNA polymerase II during the elongation phase of transcription. Nature. 2010;465(7301):1097–1101. doi: 10.1038/nature09095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Claycomb JM, Batista PJ, Pang KM, Gu W, Vasale JJ, van Wolfswinkel JC, Chaves DA, Shirayama M, Mitani S, Ketting RF, et al. The Argonaute CSR-1 and its 22G-RNA cofactors are required for holocentric chromosome segregation. Cell. 2009;139(1):123–134. doi: 10.1016/j.cell.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conine CC, Batista PJ, Gu W, Claycomb JM, Chaves DA, Shirayama M, Mello CC. Argonautes ALG-3 and ALG-4 are required for spermatogenesis-specific 26G-RNAs and thermotolerant sperm in Caenorhabditis elegans. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(8):3588–3593. doi: 10.1073/pnas.0911685107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamminga LM, van Wolfswinkel JC, Luteijn MJ, Kaaij LJ, Bagijn MP, Sapetschnig A, Miska EA, Berezikov E, Ketting RF. Differential impact of the HEN1 homolog HENN-1 on 21U and 26G RNAs in the germline of Caenorhabditis elegans. PLoS genetics. 2012;8(7):e1002702. doi: 10.1371/journal.pgen.1002702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Montgomery TA, Rim YS, Zhang C, Dowen RH, Phillips CM, Fischer SE, Ruvkun G. PIWI associated siRNAs and piRNAs specifically require the Caenorhabditis elegans HEN1 ortholog henn-1. PLoS genetics. 2012;8(4):e1002616. doi: 10.1371/journal.pgen.1002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Billi AC, Alessi AF, Khivansara V, Han T, Freeberg M, Mitani S, Kim JK. The Caenorhabditis elegans HEN1 ortholog, HENN-1, methylates and stabilizes select subclasses of germline small RNAs. PLoS genetics. 2012;8(4):e1002617. doi: 10.1371/journal.pgen.1002617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gent JI, Schvarzstein M, Villeneuve AM, Gu SG, Jantsch V, Fire AZ, Baudrimont A. A Caenorhabditis elegans RNA-directed RNA polymerase in sperm development and endogenous RNA interference. Genetics. 2009;183(4):1297–1314. doi: 10.1534/genetics.109.109686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pavelec DM, Lachowiec J, Duchaine TF, Smith HE, Kennedy S. Requirement for the ERI/DICER complex in endogenous RNA interference and sperm development in Caenorhabditis elegans. Genetics. 2009;183(4):1283–1295. doi: 10.1534/genetics.109.108134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang C, Montgomery TA, Fischer SEJ, Garcia SMDA, Riedel CG, Fahlgren N, Sullivan CM, Carrington JC, Ruvkun G. The Caenorhabditis elegans RDE-10/RDE-11 Complex Regulates RNAi by Promoting Secondary siRNA Amplification. Current Biology. 2012;22(10):881–890. doi: 10.1016/j.cub.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhuang JJ, Hunter CP. RNA interference in Caenorhabditis elegans: uptake, mechanism, and regulation. Parasitology. 2012;139(5):560–573. doi: 10.1017/S0031182011001788. [DOI] [PubMed] [Google Scholar]
- 20.Pak J, Fire A. Distinct populations of primary and secondary effectors during RNAi in C. elegans. Science. 2007;315(5809):241–244. doi: 10.1126/science.1132839. [DOI] [PubMed] [Google Scholar]
- 21.Alder MN, Dames S, Gaudet J, Mango SE. Gene silencing in Caenorhabditis elegans by transitive RNA interference. Rna. 2003;9(1):25–32. doi: 10.1261/rna.2650903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhuang JJ, Banse SA, Hunter CP. The nuclear argonaute NRDE-3 contributes to transitive RNAi in Caenorhabditis elegans. Genetics. 2013;194(1):117–131. doi: 10.1534/genetics.113.149765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhuang JJ, Hunter CP. The Influence of Competition Among Small RNA Pathways on Development. Genes. 2012;3(4) doi: 10.3390/genes3040671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kennedy S, Wang D, Ruvkun G. A conserved siRNA-degrading RNase negatively regulates RNA interference in C. elegans. Nature. 2004;427(6975):645–649. doi: 10.1038/nature02302. [DOI] [PubMed] [Google Scholar]
- 25.Fischer SE, Montgomery TA, Zhang C, Fahlgren N, Breen PC, Hwang A, Sullivan CM, Carrington JC, Ruvkun G. The ERI-6/7 helicase acts at the first stage of an siRNA amplification pathway that targets recent gene duplications. PLoS genetics. 2011;7(11):e1002369. doi: 10.1371/journal.pgen.1002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu X, Shi Z, Cui M, Han M, Ruvkun G. Repression of germline RNAi pathways in somatic cells by retinoblastoma pathway chromatin complexes. PLoS genetics. 2012;8(3):e1002542. doi: 10.1371/journal.pgen.1002542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Phillips CM, Montgomery TA, Breen PC, Ruvkun G. MUT-16 promotes formation of perinuclear mutator foci required for RNA silencing in the C. elegans germline. Genes & development. 2012;26(13):1433–1444. doi: 10.1101/gad.193904.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duchaine TF, Wohlschlegel JA, Kennedy S, Bei Y, Conte D, Jr, Pang K, Brownell DR, Harding S, Mitani S, Ruvkun G, et al. Functional proteomics reveals the biochemical niche of C. elegans DCR-1 in multiple small-RNA-mediated pathways. Cell. 2006;124(2):343–354. doi: 10.1016/j.cell.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 29.Zhuang JJ, Hunter CP. Tissue-specificity of Caenorhabditis elegans Enhanced RNAi Mutants. Genetics. 2011 doi: 10.1534/genetics.111.127209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Timmons L, Fire A. Specific interference by ingested dsRNA. Nature. 1998;395(6705):854. doi: 10.1038/27579. [DOI] [PubMed] [Google Scholar]
- 31.Kamath RS, Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30(4):313–321. doi: 10.1016/s1046-2023(03)00050-1. [DOI] [PubMed] [Google Scholar]
- 32.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77(1):71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 34.Tabach Y, Billi AC, Hayes GD, Newman MA, Zuk O, Gabel H, Kamath R, Yacoby K, Chapman B, Garcia SM, et al. Identification of small RNA pathway genes using patterns of phylogenetic conservation and divergence. Nature. 2013;493(7434):694–698. doi: 10.1038/nature11779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rea SL, Ventura N, Johnson TE. Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biology. 2007;5:2312–2329. doi: 10.1371/journal.pbio.0050259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burton NO, Burkhart KB, Kennedy S. Nuclear RNAi maintains heritable gene silencing in Caenorhabditis elegans. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(49):19683–19688. doi: 10.1073/pnas.1113310108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buckley BA, Burkhart KB, Gu SG, Spracklin G, Kershner A, Fritz H, Kimble J, Fire A, Kennedy S. A nuclear Argonaute promotes multigenerational epigenetic inheritance and germline immortality. Nature. 2012;489(7416):447–451. doi: 10.1038/nature11352. [DOI] [PMC free article] [PubMed] [Google Scholar]