

Abstract
In this study we used a next generation sequencing-based approach to profile gene mutations in therapy-related myelodysplastic syndromes (t-MDS) and acute myeloid leukemia (t-AML); and compared these findings with de novo MDS/AML. Consecutive bone marrow samples of 498 patients, including 70 therapy-related (28 MDS and 42 AML) and 428 de novo (147 MDS and 281 AML) were analyzed using a modified-TruSeq Amplicon Cancer Panel (Illumina) covering mutation hotspots of 53 genes. Overall, mutation(s) were detected in 58.6% of t-MDS/AML and 56.8% of de novo MDS/AML. Of therapy-related cases, mutations were detected in 71.4% of t-AML versus 39.3% t-MDS (p=0.0127). TP53 was the most common mutated gene in t-MDS (35.7%) as well as t-AML (33.3%), significantly higher than de novo MDS (17.7%) (p=0.0410) and de novo AML (12.8%) (p=0.0020). t-AML showed more frequent PTPN11 but less NPM1 and FLT3 mutations than de novo AML. In summary, t-MDS/AML shows a mutation profile different from their de novo counterparts. TP53 mutations are highly and similarly prevalent in t-MDS and t-AML but mutations in genes other than TP53 were more frequent in t-AML than t-MDS. The molecular genetic profiling further expands our understanding in this group of clinically aggressive yet heterogeneous myeloid neoplasms.
Keywords: Next generation sequencing, therapy-related, MDS, AML, karyotype and TP53
1. Introduction
Therapy-related myelodysplastic syndromes (t-MDS) and acute myeloid leukemia (t-AML) are a group of clinically aggressive diseases that occur after various cytotoxic chemotherapy regimens and/or radiation therapy administered previously for prior neoplastic, or rarely, non-neoplastic diseases [1]. Patients with t-MDS/AML commonly respond poorly to conventional therapies and show rapid disease progression [2, 3]. Chromosomal abnormalities, present in 40%–60% of patients with de novo MDS/AML [4], are observed in up to 80–90% t-MDS/AML patients [5, 6] with frequent high-risk cytogenetic abnormalities. The presence of frequent and high-risk cytogenetic abnormalities is thought to be one main reason for clinical aggressiveness of t-MDS/AML. However, in our recent study of 411 t-MDS/oligoblastic AML patients stratified by the Revised International Scoring System (IPSS-R), we showed that for a same IPSS-R score (a combination of blasts, severity of cytopenias and cytogenetic risk), patients with t-MDS/oligoblastic AML did significantly worse than their de novo counterparts. These differences were more pronounced in patients in the “low” and “very low” IPSS-R risk categories [7]. This observation indicates that genetic events in addition to chromosomal alterations may be involved in the pathogenesis and contribute to clinical aggressiveness of patients with t-MDS/AML.
In the past decade, massive parallel next generation sequencing (NGS) technology has been developed and more recently has been integrated into the routine laboratory work-up of MDS and AML cases at our institution. NGS assays allow the analysis of numerous genes in a quicker, more scalable fashion and the cost of NGS assays has dropped substantially in recent years. Application of NGS to samples from patients with de novo MDS [8] and AML [9] has expanded our understanding in these diseases. Using these data, several risk stratification models for de novo MDS and AML have been proposed [10–12]. In contrast, systematic mutational analysis of therapy-related (t) MDS and AML has only been performed in a small number of cases [13, 14].
In this study, our goal was to improve our understanding in molecular genetic events in t-MDS/AML and to contrast these findings with de novo MDS/AML. To achieve this goal we assessed the mutational profiles of a large number of t-MDS/AML cases using next generation sequencing methods. We also correlated the mutation results with cytogenetic data.
2. Materials and Methods
2.1 Patients
We studied a cohort of 498 patients with MDS/AML from October, 2012 through August, 2013 at The University of Texas MD Anderson Cancer Center. Clinical, hematological and cytogenetic data were collected for all patients. For patients with t-MDS/AML, primary malignant diseases were collected from the medical record. Brachytherapy, radioisotopes, and radiation therapy in patients in whom the field did not include active hematopoietic bone marrow were not considered as radiation therapy. All cases were collected consecutively and classified according to the World Health Organization (WHO) classification system. An informed consent was obtained from the patients or their guardians. This study was conducted in accord with the Declaration of Helsinki and was approved by the IRB at The University of Texas MD Anderson Cancer Center in Houston, Texas, USA.
2.2 Next generation sequencing
Genomic DNA (gDNA) was extracted from bone marrow aspirate or peripheral blood of each case using an Autopure extractor (Qiagen, Valencia, CA) and was quantified using a Qubit DNA BR assay kit (Life Technologies, Carlsbad, CA). The genomic library was prepared using 250 ng of DNA template and a commercially available 48-gene TruSeq Amplicon Cancer Panel (Illumina Inc., San Diego, CA) to which custom-designed probe pairs for 5 genes were added (Table 1).
Table 1.
Complete list of 53 genes in our panel with exons/codons tested in each gene.
| Gene | Exons (codons) tested | Amplicon (number) | |
|---|---|---|---|
| 48 gene TruSeq Amplicon Cancer Panel (Illumina) | ABL1 | 4 (243–274), 5 (275–303), 6 (303–321), 6 (321–362), 7 (395–424) | 5 |
| AKT1 | 3 (16–49) | 1 | |
| ALK | 23 (1172–1175), 25 (1248–1275) | 2 | |
| APC | 16 (875–918), 16 (1113–1153), 16 (1257–1297), 16 (1288–1328), 16 (1318–1357), 16 (1349–1386), 16 (1377–1416), 16 (1416–1456), 16 (1456–1494), 16 (1493–1530), 16 (1530–1575) | 11 | |
| ATM | 8 (353–355), 9 (409–412), 12 (601–633), 17 (846–880), 26 (1308–1331), 34 (1678–1719), 35 (1741–1773), 36 (1792–1832), 39 (1940–1973), 50 (2441–2479), 54 (2665–2670), 55 (2694–2717), 56 (2725–2756), 59 (2889–2891), 61 (2946–2950), 63 (3007–3051) | 16 | |
| BRAF | 11 (439–471), 15 (581–606) | 2 | |
| CDH1 | 3 (77–117), 8 (369–379), 9 (399–439) | 3 | |
| CDKN2A | 2 (51–70)* | 1 | |
| CSF1R | 7 (297–301), 22 (926–970) | 2 | |
| CTNNB1 | 3 (12–50) | 1 | |
| EGFR | 3 (108–142), 7 (288–297), 15 (598–627), 18 (708–728), 19 (729–761), 20 (762–775), 20 (775–817), 21 (857–875) | 8 | |
| ERBB2 | 19 (754–769), 20 (772–818), 21 (839–883) | 3 | |
| ERBB4 | 3 (98–140), 4 (153–186), 6 (208–244), 7 (248–287), 8 (295–306), 9 (333–350), 15 (579–619), 23 (907–936) | 8 | |
| FBXW7 | 5 (243–278), 8 (375–394), 9 (429–471), 10 (473–508), 11 (549–583) | 5 | |
| FGFR1 | 4 (120–126), 7 (247–250) | 2 | |
| FGFR2 | 7 (250–273), 7 (273–311), 7 (302–313), 9 (362–382), 12 (521–550) | 5 | |
| FGFR3 | 7 (247–288), 9 (379–422), 14 (639–653), 15 (654–659), 18 (792–807) and 16 (692–723)* | 6 | |
| FLT3 | 11 (437–456), 14 (569–605), 16 (648–683), 20 (807–843) | 4 | |
| GNA11 | 4 (172–202), 5 (202–216), 6 (255–297), 7 (297–304), 7 (304–349), 7 (349–360) and 4 (159–172)* | 7 | |
| GNAQ | 4 (159–202), 5 (202–210), 5 (210–245), 5 (241–245), 6 (246–263), 6 (263–297), 6 (291–297), 7 (297–324), 7 (324–360), 7 (355–360) | 10 | |
| GNAS | 8 (200–220) | 1 | |
| HNF1A | 3 (205–238), 4 (271–314) | 2 | |
| HRAS | 2 (1–15), 3 (38–63) | 2 | |
| IDH1 | 4 (90–132) | 1 | |
| JAK2 | 14 (615–622) | 1 | |
| JAK3 | 13 (568–573), 16 (683–723) | 2 | |
| KDR | 6 (220–248), 7 (267–276), 11 (471–476), 19 (872–874), 21 (946–985), 26 (1135–1146), 27 (1171–1211), 30 (1308–1352), 30 (1352–1357) | 9 | |
| KIT | 2 (51–93), 9 (502–514), 10 (514–547), 10 (540–549), 11 (550–550), 11 (550–592), 13 (641–664), 14 (670–712), 15 (714–745), 17 (815–828), 18 (838–866) | 11 | |
| KRAS | 2 (1–22), 3 (38–63), 4 (103–147) | 3 | |
| MET | 2 (168–209), 2 (375–400), 14 (1008–1028), 16 (1110–1132), 19 (1247–1284) | 5 | |
| MLH1 | 12 (383–426) | 1 | |
| MPL | 10 (514–522) | 1 | |
| NOTCH1 | 26 (1562–1601), 27 (1673–1679) and 34 (2467–2526)* | 3 | |
| NPM1 | 11 (283–295) | 1 | |
| NRAS | 2 (1–18), 3 (38–62) | 2 | |
| PDGFRA | 12 (552–592), 14 (659–668), 15 (673–717), 18 (823–854) | 4 | |
| PIK3CA | 2 (83–118), 5 (345–353), 8 (418–445), 10 (538–555), 14 (701–729), 21 (988–1027), 21 (1027–1069) | 7 | |
| PTEN | 1 (5–27), 3 (67–70), 6 (170–210), 7 (212–221), 7 (221–266), 8 (287–332), 8 (332–342) | 7 | |
| PTPN11 | 3 (59–104), 13 (501–533) | 2 | |
| RB1 | 4 (127–158), 6 (199–203), 11 (357–376), 17 (550–565), 18 (570–605), 20 (659–700), 21 (703–733), 22 (746–775) | 8 | |
| RET | 10 (610–627), 11 (628–667), 13 (766–798), 15 (880–910), 16 (918–934) | 5 | |
| SMAD4 | 3 (119–142), 5 (167–208), 6 (243–263), 8 (310–319), 9 (329–373), 10 (385–424), 11 (443–480), 12 (496–535) | 8 | |
| SMARCB1 | 2 (39–78), 4 (156–167), 5 (199–210), 9 (381–386) | 4 | |
| SMO | 3 (197–242), 5 (323–366), 6 (403–422), 11 (639–646) and 9 (533–551)* | 5 | |
| SRC | 14 (530–537)* | 1 | |
| STK11 | 1 (36–77), 6 (261–288), 8 (332–370) and 4 (193–199), 5 (200–211)* | 5 | |
| TP53 | 2 (1–12), 4 (69–112), 5 (126–147), 5 (147–186), 5 (181–187), 6 (187–192), 6 (187–223), 6 (214–224), 7 (225–253), 8 (267–306), 10 (332–342) | 11 | |
| VHL | 1 (88–114), 2 (129–155), 3 (157–200) | 3 | |
| 5 custom-designed gene panel | DNMT3A | 23 (866–913) | 1 |
| EZH2 | 16 (613–644) | 1 | |
| IDH2 | 4 (125–178) | 1 | |
| KLHL6 | 1 (1–13), 1 (13–73), 1 (73–98) | 3 | |
| XPO1 | 14 (501–522), 15 (523–539), 15 (539–575) | 3 |
Indeterminate amplicons, defined as those having total coverage depth below 250 reads. The remaining are adequately covered amplicons, defined as those having total coverage depth of greater than or equal to 250 reads, or for which orthogonal mutation analysis testing has been performed.
The generated library was purified using AMPure magnetic beads (Agencourt, Brea, CA) and then subjected to next generation sequencing (NGS) using a MiSeq sequencer (Illumina Inc., San Diego, CA) [15]. In our clinical practice, a minimum quality score of AQ30 is required for a minimum of 75% of bases sequenced ensuring high quality sequencing results. In practice, >85% of the bases consistently show quality scores of >AQ30 and 90–95% bases show quality score of >AQ20. Using human genome build 19 (hg 19) as a reference, variant calling was performed with Illumina MiSeq Reporter Software 1.3.17. To visualize read alignment and confirm the variant calls, Integrative Genomics Viewer (IGV, Broad Institute, MA) was used [16]. For clinical reporting, a sequencing coverage of 250X (bi-directional true paired-end sequencing) and a variant frequency of 5% in the background of wild-type were used as cutoffs. To annotate sequence variant, custom-developed, in-house software (OncoSeek) was used to interface the data with IGV. OncoSeek has several functionalities including mapping of variants directly to COSMIC and dbSNP, automatic translation of variants and their genomic position to Human Genome Variation Society compliant nomenclature, identification of amplicons with suboptimal coverage for clinical reporting, and self-updating population analysis, which identified reference genome polymorphisms or sequencing artifacts [17]. Among the 53 genes in the panel, NPM1 mutations and FLT3 internal tandem duplication (ITD) were confirmed with fragment analysis using capillary electrophoresis. Pyrosequencing was performed to confirm mutations in BRAF codons 599 and 600, JAK2 codon 617, KRAS codons 12, 13 and 61, NRAS codons 12, 13 and 61. Sanger sequencing was performed to confirm mutations in the remaining genes in the panel. Confirmation tests were generally triggered when the sequencing coverage was less than 250X or mutational frequency in major genes such as DNMT3A, IDH1, IDH2, KRAS, NPM1 and NRAS was less than 5%. Fragment analysis for FLT3-ITD was performed in all cases [18].
2.3 Cytogenetic Analysis
Conventional cytogenetic analysis was performed using standard methods as previously described [19]. Twenty metaphases were analyzed and the results were reported using the current International System for Human Cytogenetic Nomenclature [20]. Only karyotypes with adequate metaphases for analysis were included, except in some cases where a lesser number of metaphases were available in which fluorescence in situ hybridization (FISH) was performed to confirm clonal cytogenetic abnormalities. For MDS patients, the cytogenetic risk was stratified according to the International Prognostic Scoring System (IPSS) [21] and for AML patients the risk was categorized by the revised cytogenetic classification proposed by the United Kingdom Medical Research Council (UKMRC) [22].
2.4 Statistical analysis
For continuous variables, data were reported as a median and range. For nominal variables, data were reported as the number of patients if not otherwise specified. Fisher’s exact test was used for categorical variables and the Mann-Whitney U test for continuous variables. All differences with p<0.05 were considered to be statistically significant (two-tailed). GraphPad Prism 6.0 (La Jolla, CA, USA) was used for statistical analyses.
3. Results
3.1 Patient Characteristics
The study cohort consisted of 28 patients with t-MDS, 42 with t-AML, 147 with de novo MDS, and 281 with de novo AML. In the therapy-related patient group, the prior diseases were hematological malignancies (n=33); carcinoma (n=32), sarcoma (n=4), and medulloblastoma (n=1). The cytotoxic therapy used to treat these tumors included radiation therapy only (n=5), chemotherapy only (n=41), and combined chemoradiation therapy (n=24). The WHO categories of de novo MDS as well t-MDS are shown in Table 2.
Table 2.
Patient characteristics
| t-MDS (n=28) | de novo MDS (n=147) | p value | |
|---|---|---|---|
| Age (years) | 69 (37–81) | 70 (14–91) | 0.2061 |
| Men:Women | 17:11 | 106:41 | 0.2610 |
| Hemoglobin (g/L) | 96 (72–128) | 97 (60–143) | 0.5798 |
| Mean corpuscular volume (femtoliter) | 92 (79–109) | 94 (70–120) | 0.6973 |
| White blood cell (× 109/L) | 3.1 (0.5–24.9) | 2.9 (0.1–14.3) | 0.9497 |
| Absolute neutrophil count (× 109/L) | 1.2 (0.3–15.4) | 1.4 (0–10.0) | 0.9641 |
| Platelet (× 109/L) | 46.5 (17.0–257.0) | 65.0 (3.0–614.0) | 0.1423 |
| BM blasts (%) | 4.5 (0–18.0) | 5.0 (0–19.0) | 0.4926 |
| BM cellularity (%) | 40 (20–100) | 60 (5–100) | 0.2364 |
| Cytogenetic risk, n (%) | 0.0005 | ||
| Good | 4 (14.3%) | 74 (50.3%) | |
| Intermediate | 4 (14.3%) | 28 (19.0%) | |
| Poor | 18 (64.3%) | 45 (30.6%) | |
| n/a | 2 (7.1%) | 0 (0.0%) | |
| WHO classification, n (%) | |||
| RCUD | 0 | 1 (0.7%) | |
| RARS | 0 | 2 (1.4%) | |
| RCMD | 12 (42.8%) | 58 (39.5%) | |
| RAEB-1 | 8 (28.6%) | 37 (25.2%) | |
| RAEB-2 | 8 (28.6%) | 47 (32.0%) | |
| MDS with isolated del 5q | 0 | 2 (1.4%) |
| t-AML (n=42) | de novo AML (n=281) | p value | |
|---|---|---|---|
| Age (years) | 66.5 (18.0–87.0) | 65.0 (1.0–92.0) | 0.3842 |
| Men:Women | 24:18 | 165:116 | 0.8678 |
| Hemoglobin (g/L) | 91 (60–118) | 9.3 (56–154) | 0.5081 |
| Mean corpuscular volume (femtoliter) | 91.5 (78.0–112.0) | 91 (68–112) | 0.6599 |
| White blood cell (× 109/L) | 2.3 (0.3–93.9) | 3.5 (0.3–255.0) | 0.0819 |
| Platelet (× 109/L) | 33.0 (7.0–394.0) | 47.0 (3.0–1069.0) | 0.1446 |
| BM blasts (%) | 31 (2–91) | 39 (0–99) | 0.1883 |
| BM cellularity (%) | 60 (5–100) | 80 (5–100) | 0.1693 |
| UKMRC cytogenetic risk, n (%) | 0.0006 | ||
| Favorable | 2 (4.8%) | 26 (9.3%) | |
| Intermediate | 11 (26.2%) | 154 (54.8%) | |
| Unfavorable | 28 (66.7%) | 94 (33.5%) | |
| n/a | 1 (2.4%) | 7 (2.5%) |
BM, bone marrow; n/a, non-available; WHO, World Health Organization; UKMRC, United Kingdom Medical Research Council; RCUD; refractory cytopenia with unilineage dysplasia, RARS; refractory anemia with ring sideroblasts, RCMD; refractory cytopenia with multilineage dysplasia, RAEB; refractory anemia with excess blasts
De novo AML included 44 (16.7%) cases of AML with recurrent cytogenetic abnormalities, 127 (45.2%) cases of AML with myelodysplasia related changes (AML-MRC), and 110 (39.1%) cases of AML, not otherwise specified (AML, NOS). There were no differences in demographic and hematological findings between patients with t-MDS and de novo MDS or between patients with t-AML and de novo AML (Table 2). However, the distribution of cytogenetic risk was significantly different between patients with therapy-related versus de novo diseases. Patients with good, intermediate and poor risk cytogenetics represented 14.3%, 14.3% and 64.3% of cases in t-MDS. In contrast, 50.3% of patients had good risk, 19.0% intermediate risk, and 30.6% poor risk cytogenetics in de novo MDS (p=0.0005). Similarly, t-AML patients with favorable, intermediate and unfavorable risk cytogenetics were 4.8%, 26.2% and 66.7%, respectively in t-AML; in contrast with 9.3%, 54.8% and 33.5%, respectively, in de novo AML (p=0.0006).
3.2 Mutational profiles
In all patients, mutations were found involving 19 different genes in 284 (57.0%) patients. Of these 284 patients, 193 patients (68.0%) had mutations involving one single gene, 59 (20.8%) had mutations in two genes, 24 (8.5%) had mutations in 3 genes and 8 (2.8%) had mutations in 4 genes.
As a group, t-MDS/t-AML, mutations were detected in 41 (58.6%) patients, involving 13 genes. These rates were not significantly different from de novo MDS/AML that mutations were detected in 243 patients (56.8%) (p=0.7962) and involved 17 genes.
3.2.1 Comparing t-MDS with t-AML
In t-MDS, mutations were found in 11 of 28 (39.3%) patients and these mutations involved only 2 genes, TP53 and IDH1 (Figure 1A).
Figure 1.
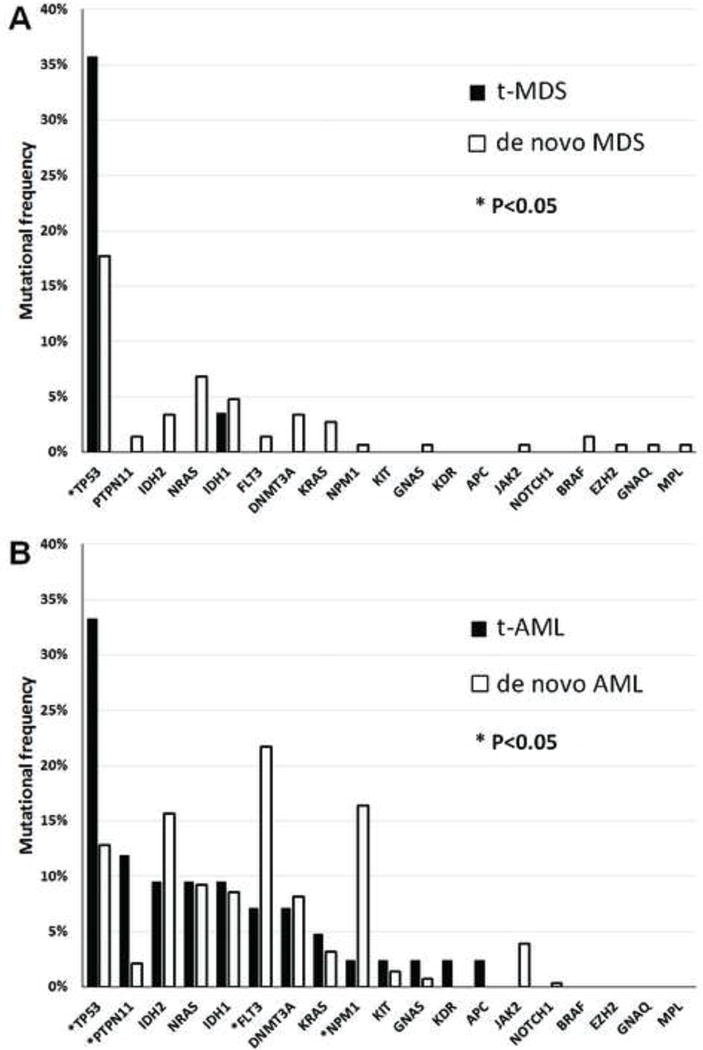
Mutational profiles in myeloid neoplasms. 1A. mutational profile of therapy-related myelodysplastic syndromes (t-MDS) versus de novo MDS. 1B. mutational profile of therapy-related acute myeloid leukemia (t-AML) versus de novo AML. Asterisk denotes genes with significant difference between therapy-related versus de novo MDS/AML.
In contrast, mutations were detected in 30 of 42 (71.4%) t-AML patients and involved 13 different genes, significantly more frequent as well as more diverse than t-MDS (p=0.0127 and p=0.0040 respectively). The most frequently mutated gene in t-AML was TP53 (35.7%). Other genes in t-AML in a decreasing frequency were: PTPN11, 11.9%; IDH1, 9.5%; IDH2, 9.5%; NRAS, 9.5%; FLT3, 7.1%; DNMT3A, 7.1%; and KRAS, 4.8% (Figure 1B). Mutations in GNAS, KDR, KIT, NPM1 and APC were rare (<3.0%).
3.2.2 Comparing t-MDS/AML with de novo MDS/AML
In de novo MDS (n=147), mutations were found in 58 cases (39.5%) and mutation frequency was commensurate with the risk grade of MDS. For example, mutations were found in 24% low-grade MDS (RCUD, RARS, RCMD, and MDS with isolated deletion 5q) and 51% in high-grade MDS (RAEBs), respectively (p=0.0011). The overall mutation frequency was very similar to t-MDS (39.3%, not significant); however, mutations in de novo MDS involving 15 different genes, significantly more diverse than t-MDS (p=0.001) that only two genes were found mutated (p=0.0010). For individual mutations, the frequency of TP53 mutations (17.7%) was significantly lower than t-MDS (p=0.0410). Other mutated genes in de novo MDS were: NRAS, 6.8%; IDH1, 4.8%; DNMT3A, 3.4%; IDH2, 3.4%; and KRAS, 2.7%. Mutations in PTPN11, FLT3, BRAF, JAK2, EZH2, GNAQ, GNAS, MPL, and NPM1 were rarely (< 2.0%) detected (Figure 1A).
Mutation frequency was similar between de novo AML and t-AML (65.8% versus 71.4%, not significant). However, compared with de novo AML, t-AML showed a higher frequency of mutations in TP53 (35.7% vs 12.8%, p=0.0020) and PTPN11 (11.9% vs 2.1%, p=0.0075), whereas a lower frequency in mutations in FLT3 (7.1% vs 21.7%, p=0.0357) and NPM1 (2.5% vs 16.4%, p=0.0165). Compared to de novo MDS, mutations in de novo AML (n=281) were more frequently detected (65.8% versus 39.5%, p<0.0001), and involved 14 different genes, with FLT3 (21.7%) being the most frequently mutated gene. Other mutated genes were: NPM1, 16.4%; IDH2, 15.7%; TP53, 12.8%; NRAS, 9.3%; IDH1, 8.5%; DNMT3A, 8.2%; JAK2, 3.9%; KRAS, 3.2%; and PTPN11, 2.1% (Figure 1B). Mutations in KIT, GNAS, NOTCH1 and EGFR were rarely detected (< 2.0%). By the WHO group, mutations were less frequent in AML with recurrent cytogenetic abnormalities compared to AML-MRC and AML, NOS (44% vs. 70%, respectively, p=0.0009).
Co-mutations are shown by Circos plots using the common mutated genes in de novo AML, de novo MDS, t-MDS and t-AML, including DNMT3A, FLT3, IDH1, IDH2, KRAS, NPM1, NRAS and TP53 (Figure 2). Differences in co-mutational patterns were observed between t-MDS/AML (Figure 2A) and de novo MDS/AML (Figure 2B). The common co-mutations in t-MDS/AML were TP53 with IDH1, NRAS and FLT3; whereas, in de novo MDS/AML, the common co-mutations were FLT3 with NPM1, IDH2 and DNMT3A.
Figure 2.
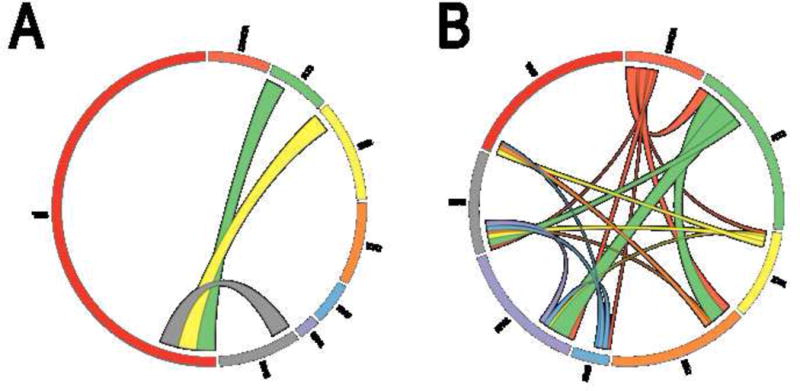
Difference of mutational pattern between t-MDS/AML and de novo MDS/AML. 2A. Mutational pattern of t-MDS/AML in Circos plot. 2B. Mutational pattern of de novo MDS/AML in Circos plot. The thickness of connecting lines between two genes is proportional of the number of such cases. Areas without connecting lines denote cases with single gene mutation.
3.3 Mutation profile and Karyotypical Abnormalities
Karyotype information was available in 26 t-MDS, 147 de novo MDS, 41 t-AML and 274 de novo AML. Cases with a normal, complex karyotype and all other abnormalities were shown in Figure 3.
Figure 3.
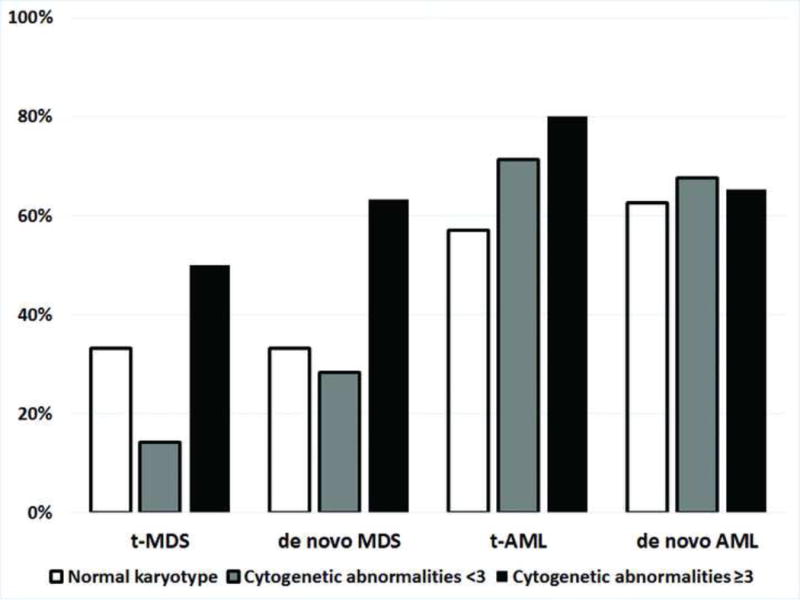
Percentages of mutations in cases with a normal karyotype, an abnormal non-complex karyotype and a complex karyotype, in different disease subtypes. TP53 denotes mutations in TP53 gene, and non-TP53 denotes mutations other than TP53 gene.
t-MDS showed a higher risk karyotypic distribution than de novo MDS (p=0.0006); similarly, t-AML showed a high risk distribution of karyotypic abnormalities than de novo AML (p=0.0085). The mutation frequencies for cases with a normal karyotype, a non-complex abnormal karyotype and a complex karyotype in each subgroup are shown in Figure 2. Of 3 t-MDS patients with a normal karyotype (13.5%), one patient had an IDH1 mutation. Of de novo MDS patients, 21 out of 63 cases with a normal karyotype were found to have mutations (33.3%). The most frequently mutated genes in a decreased frequency were NRAS (n=7), DNMT3A (n=4), IDH1 (n=4), IDH2 (n=2), FLT3 (n=1), BRAF (n=1), EZH2 (n=1), GNAQ (n=1), GNAS (n=1), JAK2 (n=1) and KRAS (n=1). Of t-AML, 4 of the 7 patients with a normal karyotype harbored mutations, involving IDH2 (n=3), TP53 (n=1), FLT3 (n=1), DNMT3A (n=1), NPM1 (n=1) and GNAS (n=1); whereas, in de novo AML with a normal karyotype (n=91), mutations were detected in 57 cases (62.6%), involving FLT3 (n=30), NPM1 (n=26), IDH2 (n=19), DNMT3A (n=12), IDH1 (n=10), NRAS (n=5), TP53 (n=4), KRAS (n=2), PTPN11 (n=2), GNAS (n=1), JAK2 (n=1) and KIT (n=1) in a decreased frequencies. TP53 was the most frequently mutated gene in all subsets of cases with a complex karyotype, 50% in t-MDS, 60% in t-AML, 63.2% in de novo MDS, and 41.7% of in de novo AML. Of cases with a non-complex abnormal karyotype, one patient with t-MDS had TP53 mutation; 13 patients (28.2%) with de novo MDS had mutations involving 10 genes. In t-AML, 13 patients (71.4%) had mutations, involving 8 genes; whereas, 75 patients with de novo AML (67.8%) had mutations involving 13 genes.
4. DISCUSSION
We assessed the bone marrow samples of 70 t-MDS/AML using a NGS of 53 selected gene panel and compared the data with 428 de novo MDS/AML patients. With this large dataset and the genes we tested, we showed that the mutation profiles of t-MDS/AML are conspicuously different from de novo MDS/AML (Figures 1 and 2). TP53 was almost the only mutated gene in t-MDS (except for one case with IDH1 mutation), and found in about 1/3 of our t-MDS cases. Similarly, IDH1 mutations were rarely reported in t-MDS [23]. In contrast, we identified mutations involving 15 different genes in de novo MDS; and TP53 gene mutation frequency in de novo MDS was significantly lower than t-MDS. Of note, the overall mutation frequency in our de novo MDS was ~40%, slightly lower than what reported by others [8, 24]. This lower frequency is likely attributable to the methods we used as our gene panel focused on selected exons in 53 genes and did not include some genes known to be mutated in MDS. For example, DNMT3A and EZH2 mutations were identified in <5% in our de novo MDS patients, slightly lower than 6–8% reported by others [8, 25–27], and this could be due to some regions not covered by our assay (Table 1). TET2, ASXL1 and spliceosome genes (U2AF35, SF3B1 and SRSF2) were not included in our panel. These genes are reported frequently mutated in de novo MDS [24, 27, 28], but, infrequently in t-MDS [13].
In t-AML, like t-MDS, TP53 was frequently mutated with a frequency significantly higher than de novo AML (p=0.0020). PTPN11 mutations were seen in 11.9% of t-AML patients in this series, significantly higher than de novo AML (2.1%) patients. The PTPN11 gene encodes a protein tyrosine phosphatase (SHP-2) in the RAS/MEK/ERK kinase pathway [29], and has been reported mutated in approximately 4–5% t-MDS/t-AML [14, 30]. NPM1 mutations were detected in only 5% of t-AML cases in this study. In the literature, NPM1 mutations have been reported in about 30% of all AML cases and 50–60% of cytogenetically normal AML [31–34]. In our de novo AML group, NPM1 mutations were shown in 16.4% of all patients and 29.2% of AML with a normal karyotype. This slightly lower NPM1 mutation frequency in our de novo AML group was likely due to higher risk patients in our cohort as a result of a tertiary referral cancer center. FLT3 mutations were also significantly lower in t-AML, compared with de novo AML. These data are similar to that reported by Pedersen-Bjergaard and colleagues in their study of t-MDS/AML [14]. In addition, we identified 1 case of t-AML with mutations in DNMT3A, NPM1, and FLT3. This clustering of mutations has been shown in a small subset of de novo AML patients [34].
Due to a general aggressive clinical course related to t-MDS and AML, in the 2008 WHO classification, t-MDS and t-AML are combined under the umbrella term “therapy-related myeloid neoplasms (t-MN)” [1]. Recent studies have shown that t-MDS with a low blast count is less aggressive than t-AML [7, 35]. In this study, we showed that the frequency of TP53 mutations were similarly high in both t-MDS and t-AML; however, mutations in genes other than TP53 were significantly higher in t-AML than t-MDS. Our data also suggests that TP53 mutation may be heavily involved in the early pathogenesis of myeloid neoplasms post cytotoxic exposure, but mutations in other genes likely provide proliferative advantage in cases of t-AML. These findings also illustrate that disease progression, such as from t-MDS to t-AML, is coupled with a step-wise molecular genetic evolution.
In summary, we showed that the mutational profiles of therapy-related MDS and AML differed substantially from their de novo MDS and AML counterparts. t-AML and t-MDS both harbored a high frequency of TP53 mutations; and PTPN11 mutations were more frequent, but FLT3 and NPM1 mutations were less frequent in t-AML than de novo AML. Within the therapy-related myeloid neoplasms, t-AML harbored more frequent mutations in genes other than TP53. Overall, TP53 mutations were strongly associated with a complex karyotype and the complexity degree of karyotypic abnormalities in t-MDS/AML as well as de novo MDS/AML. The information we provide here contribute to our further understanding of the molecular-genetic basis of therapy-related myeloid neoplasms, the similarity and difference between t-MDS and t-AML and how they differ from de novo counterparts.
*Highlights (for review).
Mutation profile of t-MDS/AML is different from their de novo counterparts.
Mutation profile is different between t-MDS and t-AML.
TP53 mutations are associated with a complex karyotype in both groups of MDS/AML.
Different molecular profiling may contribute to clinical heterogeneity of MDS/AML.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure/Conflict of Interest
Authors do not report conflict of interest.
Authors’ contributions. C.Y.O. and S.A.W. designed the study; G.G-M., S.A.P. and H.M.K. provided patient information; C.Y.O., K.P.P., M.J.R., B.F., G.T., M.G., R.S., R.K-S., R.L. and S.A.W. collected and analyzed data; C.Y.O. and S.A.W. performed the statistical analysis; C.Y.O., K.P.P., K.H.Y., L.J.M., R.L. and S.A.W. wrote the manuscript and created the figures and tables; and all authors critically reviewed the manuscript and read and approved the final version of the manuscript.
References
- 1.Vardiman JW, Arber DA, Brunning RD, Larson RA, Matutes E, Baumann I, et al. Therapy-related myeloid neoplasms. In: Swerdlow SH, Campo E, Harris L, Jaffe ES, Pileri SA, Stein H, et al., editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th. Lyon: International Agency for Research on Cancer (IARC); 2008. pp. 127–9. [Google Scholar]
- 2.Greco M, D’Alo F, Scardocci A, Criscuolo M, Fabiani E, Guidi F, et al. Promoter methylation of DAPK1, E-cadherin and thrombospondin-1 in de novo and therapy-related myeloid neoplasms. Blood cells, molecules & diseases. 2010;45:181–5. doi: 10.1016/j.bcmd.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 3.Voso MT, D’Alo F, Greco M, Fabiani E, Criscuolo M, Migliara G, et al. Epigenetic changes in therapy-related MDS/AML. Chemico-biological interactions. 2010;184:46–9. doi: 10.1016/j.cbi.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 4.Olney HJ, Le Beau MM. The cytogenetics of myelodysplastic syndromes. Best Pract Res Clin Haematol. 2001;14:479–95. doi: 10.1053/beha.2001.0151. [DOI] [PubMed] [Google Scholar]
- 5.Smith SM, Le Beau MM, Huo D, Karrison T, Sobecks RM, Anastasi J, et al. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood. 2003;102:43–52. doi: 10.1182/blood-2002-11-3343. [DOI] [PubMed] [Google Scholar]
- 6.Zhou Y, Tang G, Medeiros LJ, McDonnell TJ, Keating MJ, Wierda WG, et al. Therapy-related myeloid neoplasms following fludarabine, cyclophosphamide, and rituximab (FCR) treatment in patients with chronic lymphocytic leukemia/small lymphocytic lymphoma. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2012;25:237–45. doi: 10.1038/modpathol.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ok CY, Hasserjian RP, Fox PS, Stingo F, Zuo Z, Young KH, et al. Application of the International Prognostic Scoring System-Revised in therapy-related myelodysplastic syndromes and oligoblastic acute myeloid leukemia. Leukemia. 2014;28:185–9. doi: 10.1038/leu.2013.191. [DOI] [PubMed] [Google Scholar]
- 8.Walter MJ, Shen D, Shao J, Ding L, White BS, Kandoth C, et al. Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia. 2013;27:1275–82. doi: 10.1038/leu.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grossmann V, Schnittger S, Kohlmann A, Eder C, Roller A, Dicker F, et al. A novel hierarchical prognostic model of AML solely based on molecular mutations. Blood. 2012;120:2963–72. doi: 10.1182/blood-2012-03-419622. [DOI] [PubMed] [Google Scholar]
- 11.Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–89. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bejar R, Stevenson KE, Caughey BA, Abdel-Wahab O, Steensma DP, Galili N, et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J Clin Oncol. 2012;30:3376–82. doi: 10.1200/JCO.2011.40.7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shih AH, Chung SS, Dolezal EK, Zhang SJ, Abdel-Wahab OI, Park CY, et al. Mutational analysis of therapy-related myelodysplastic syndromes and acute myelogenous leukemia. Haematologica. 2013;98:908–12. doi: 10.3324/haematol.2012.076729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pedersen-Bjergaard J, Andersen MK, Andersen MT, Christiansen DH. Genetics of therapy-related myelodysplasia and acute myeloid leukemia. Leukemia. 2008;22:240–8. doi: 10.1038/sj.leu.2405078. [DOI] [PubMed] [Google Scholar]
- 15.Junemann S, Sedlazeck FJ, Prior K, Albersmeier A, John U, Kalinowski J, et al. Updating benchtop sequencing performance comparison. Nat Biotechnol. 2013;31:294–6. doi: 10.1038/nbt.2522. [DOI] [PubMed] [Google Scholar]
- 16.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Routbort M, Handal B, Patel KP, Singh R, Aldape K, Reddy N, et al. OncoSeek: a versatile annotation and reporting system for next generation sequencing-based clinical mutation analysis of cancer specimens. AMP 2012 Meeting Abstact. J Mol Diagn. 2012;14:637–748. Abstract TT79. [Google Scholar]
- 18.Luthra R, Patel KP, Reddy NG, Haghshenas V, Routbort MJ, Harmon MA, et al. Next-generation sequencing-based multigene mutational screening for acute myeloid leukemia using MiSeq: applicability for diagnostics and disease monitoring. Haematologica. 2014;99:465–73. doi: 10.3324/haematol.2013.093765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khoury JD, Sen F, Abruzzo LV, Hayes K, Glassman A, Medeiros LJ. Cytogenetic findings in blastoid mantle cell lymphoma. Hum Pathol. 2003;34:1022–9. doi: 10.1053/s0046-8177(03)00412-x. [DOI] [PubMed] [Google Scholar]
- 20.Shaffer LG, McGowan-Jordan J,MS. An International System for Human Cytogenetic Nomenclature. Basel: S. Karger; 2013. [Google Scholar]
- 21.Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89:2079–88. [PubMed] [Google Scholar]
- 22.Grimwade D, Hills RK, Moorman AV, Walker H, Chatters S, Goldstone AH, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010;116:354–65. doi: 10.1182/blood-2009-11-254441. [DOI] [PubMed] [Google Scholar]
- 23.Westman MK, Pedersen-Bjergaard J, Andersen MT, Andersen MK. IDH1 and IDH2 mutations in therapy-related myelodysplastic syndrome and acute myeloid leukemia are associated with a normal karyotype and with der(1;7)(q10;p10) Leukemia. 2013;27:957–9. doi: 10.1038/leu.2012.347. [DOI] [PubMed] [Google Scholar]
- 24.Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28:241–7. doi: 10.1038/leu.2013.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122:3616–27. doi: 10.1182/blood-2013-08-518886. quiz 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walter MJ, Ding L, Shen D, Shao J, Grillot M, McLellan M, et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia. 2011;25:1153–8. doi: 10.1038/leu.2011.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364:2496–506. doi: 10.1056/NEJMoa1013343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thol F, Kade S, Schlarmann C, Loffeld P, Morgan M, Krauter J, et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood. 2012;119:3578–84. doi: 10.1182/blood-2011-12-399337. [DOI] [PubMed] [Google Scholar]
- 29.Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29:465–8. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 30.Christiansen DH, Desta F, Andersen MK, Pedersen-Bjergaard J. Mutations of the PTPN11 gene in therapy-related MDS and AML with rare balanced chromosome translocations. Genes Chromosomes Cancer. 2007;46:517–21. doi: 10.1002/gcc.20426. [DOI] [PubMed] [Google Scholar]
- 31.Cancer Genome Atlas Research N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marcucci G, Haferlach T, Dohner H. Molecular genetics of adult acute myeloid leukemia: prognostic and therapeutic implications. J Clin Oncol. 2011;29:475–86. doi: 10.1200/JCO.2010.30.2554. [DOI] [PubMed] [Google Scholar]
- 33.Martelli MP, Sportoletti P, Tiacci E, Martelli MF, Falini B. Mutational landscape of AML with normal cytogenetics: biological and clinical implications. Blood Rev. 2013;27:13–22. doi: 10.1016/j.blre.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 34.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–33. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bacher U, Haferlach C, Alpermann T, Schnittger S, Kern W, Haferlach T. Patients with therapy-related myelodysplastic syndromes and acute myeloid leukemia share genetic features but can be separated by blast counts and cytogenetic risk profiles into prognostically relevant subgroups. Leuk Lymphoma. 2013;54:639–42. doi: 10.3109/10428194.2012.717275. [DOI] [PubMed] [Google Scholar]