

Abstract
Study Objective
As we previously observed a significant 41% reduction in gemfibrozil exposure after 2 weeks of lopinavir-ritonavir administration, we sought to determine the influence of lopinavir-ritonavir and ritonavir alone on the pharmacokinetics of fenofibric acid, an alternative to gemfibrozil for the treatment of elevated triglyceride levels.
Design
Open-label, single-sequence pharmacokinetic study.
Setting
Clinical Research Center at the National Institutes of Health.
Subjects
Thirteen healthy adult volunteers.
Intervention
Subjects received a single oral dose of fenofibrate 145 mg during three study phases: before ritonavir administration, after 2 weeks of administration of ritonavir 100 mg twice daily, and after 2 weeks of administration of lopinavir 400 mg–ritonavir 100 mg twice daily.
Measurements and Main Results
Serial blood samples were collected over 120 hours for determination of fenofibric acid concentrations. Fenofibric acid pharmacokinetic parameter values were compared before and after concomitant ritonavir or lopinavir-ritonavir administration. The geometric mean ratios (90% confidence intervals) for fenofibric acid area under the plasma concentration–time curve were 0.89 (0.77–1.01) after 14 days of ritonavir alone compared with baseline (p>0.05) and 0.87 (0.69–1.05) after 14 days of lopinavir-ritonavir compared with baseline (p>0.05). Study drugs were generally well tolerated; all adverse events were mild or moderate, transient, and resolved without intervention.
Conclusion
In contrast to a significant interaction between gemfibrozil and lopinavir-ritonavir, neither lopinavir-ritonavir nor ritonavir alone altered the pharmacokinetics of fenofibric acid in healthy volunteers. These data suggest that fenofibrate remains an important option in human immunodeficiency virus–infected patients receiving common ritonavir-boosted therapy.
Keywords: hypertriglyceridemia, HIV, protease inhibitor, fenofibrate, lopinavir-ritonavir, pharmacokinetics
Potent combination antiretroviral therapy (cART) has transformed human immunodeficiency virus (HIV) infection from a terminal illness to a chronic, treatable condition. Despite the recent update to the United States Department of Health and Human Services treatment guidelines,1 HIV protease inhibitors remain a key component of regimens for both treatment-naïve and treatment-experienced patients globally. Despite reductions in HIV-related morbidity and mortality secondary to cART, certain antiretroviral drugs, including most protease inhibitors, have been associated with lipid perturbations such as increases in triglyceride, low-density lipoprotein cholesterol, and total cholesterol levels.2–9 Furthermore, HIV infection itself is characterized by elevated triglyceride levels and reduced high-density lipoprotein cholesterol levels.7,8,10 Hyperlipidemia and severe isolated hypertriglyceridemia (triglyceride level > 1000 mg/dL) require intervention to reduce the risk of cardiovascular events and pancreatitis, respectively, in persons living with HIV.8,11–13
The fibric acid derivatives, gemfibrozil and fenofibrate, are commonly used for the management of severe or isolated hypertriglyceridemia.14 In a retrospective cohort study, over 100 hypertriglyceridemic, HIV-infected patients who were treated with gemfibrozil and also receiving protease inhibitor–based cART, had a smaller mean change in triglyceride levels (−44.0%) compared with HIV-infected patients who received nonnucleoside reverse transcriptase inhibitor (NNRTI)-based cART (–60.3%) or non–HIV-infected controls (–59.3%).15 Gemfibrozil is a well-recognized substrate for uridine diphosphate–glucuronosyltransferase (UGT) enzymes; several isoforms of UGT are known to undergo induction in the presence of the HIV protease inhibitor, ritonavir, which is commonly used in numerous protease inhibitor–containing regimens as a pharmacokinetic enhancer.16,17 To determine whether a drug-drug interaction between protease inhibitors and gemfibrozil was partially responsible for gemfibrozil’s suboptimal activity, we previously conducted a pharmacokinetic study in healthy volunteers in which we observed a 41% reduction (P = 0.001) in gemfibrozil exposure after 2 weeks of administration of lopinavir 400 mg–ritonavir 100 mg twice daily.17 Given the dose-response relationship previously identified between gemfibrozil and its ability to lower serum triglyceride levels, the 41% reduction we observed in gemfibrozil exposure is likely clinically relevant. Contrary to our initial hypothesis of ritonavir-mediated UGT2B7 induction of gemfibrozil metabolism, the mechanism of this interaction appeared to be a reduction in gemfibrozil absorption with concurrent lopinavir-ritonavir.
In light of the observed drug-drug interaction between gemfibrozil and lopinavir-ritonavir, clinicians are likely to choose fenofibrate instead of gemfibrozil in HIV-infected patients receiving ritonavir-boosted protease inhibitor–based cART who require triglyceride-lowering therapy. Like gemfibrozil, fenofibrate is chemically similar in that it is also a fibric acid derivative. Fenofibric acid, the active moiety of fenofibrate, is formed by rapid hydrolysis of the prodrug; whereas fenofibrate does not undergo metabolism by cytochrome P450 or UGT pathways, fenofibric acid is metabolized through UGT conjugation pathways, mainly UGT2B7, with UGT1A3, UGT1A6, and UGT1A9 playing minor roles.18,19 Whether the systemic exposure of fenofibric acid is also reduced in the presence of ritonavir or lopinavir-ritonavir, through ritonavir-mediated UGT induction or reduced absorption, is unknown. Therefore, the purpose of the current investigation was to determine the influence of lopinavir-ritonavir and ritonavir alone on the pharmacokinetics of fenofibric acid in healthy volunteers.
Methods
Study Subjects
The study enrolled HIV-negative (determined by enzyme-linked immunosorbent assay) individuals aged 18–60 years who were receiving no concomitant medications, including prescription, over-the-counter, or herbal preparations, and were free of concurrent illnesses according to their medical history, physical examination, and screening laboratory values, including liver function tests, serum creatinine concentration, total and direct bilirubin levels, and hemoglobin level. Screening laboratory values were required to be within institutional normal ranges, except for fasting total cholesterol and triglyceride levels, which were required to be below 270 mg/dL and 400 mg/dL, respectively. Women of childbearing potential were required to have a negative urine or serum pregnancy test before beginning each study phase and to practice abstinence or use effective nonhormonal methods of birth control throughout the investigation. The use of tobacco products was not permitted.
All participants gave written informed consent, and clinical research was conducted according to guidelines for human experimentation as specified by the U.S. Department of Health and Human Services. This study was approved by the National Institute of Allergy and Infectious Diseases Institutional Review Board.
Study Design and Setting
This study was a single-sequence, open-label investigation to evaluate the individual effects of ritonavir alone and lopinavir-ritonavir, each administered for 19.5 days, on the pharmacokinetics of fenofibric acid in healthy volunteers (Figure 1). This study was conducted at the Clinical Research Center at the National Institutes of Health (Bethesda, MD, USA).
Figure 1.
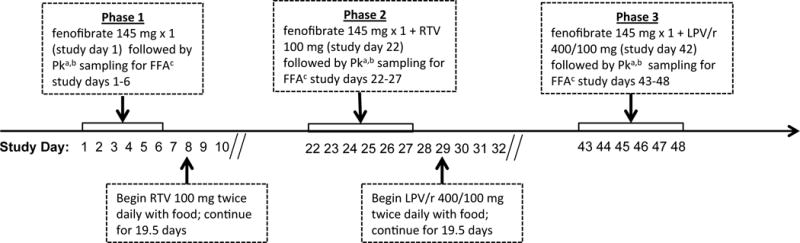
Study schematic for fenofibrate administered alone (phase 1), fenofibrate in combination with ritonavir (phase 2), and fenofibrate in combination with lopinavir-ritonavir (phase 3) in 13 healthy human subjects. PK = pharmacokinetic; FFA = fenofibric acid; RTV = ritonavir; LPV/r = lopinavir-ritonavir. aPharmacokinetic sampling times for fenofibric acid were at time 0 (predose) and at 1, 2, 3, 4, 5, 6, 8, 10, 12, 24, 48, 72, 96, and 120 hours following fenofibrate administration.
Treatment and Blood Sampling
After an overnight fast, subjects received a single oral dose of fenofibrate 145 mg (Tricor; AbbVie Inc., North Chicago, IL) following a standardized breakfast. Blood samples for determination of fenofibric acid plasma concentrations were collected into heparinized tubes at time 0 (predose) and at 1, 2, 3, 4, 5, 6, 8, 10, 12, 24, 48, 72, 96, and 120 hours after dosing (phase 1). Blood was centrifuged after collection, and plasma was harvested and frozen at –80°C until the time of analysis.
On study day 8, subjects took ritonavir (Norvir; AbbVie Inc.) 100 mg twice daily with food for 19.5 days. On the morning of day 15 of ritonavir dosing (study day 22), subjects arrived at the clinic and took their morning dose of ritonavir with a single dose of fenofibrate 145 mg following the same standardized breakfast (phase 2). Subjects then underwent plasma concentration sampling for fenofibric acid as described for phase 1. Subjects continued to take ritonavir for an additional 5.5 days (for a total of 19.5 days) throughout the entire pharmacokinetic sampling period.
After phase 2 completion, subjects took lopinavir 400 mg–ritonavir 100 mg (Kaletra, AbbVie Inc, North Chicago, IL), given as two lopinavir 200–ritonavir 50 mg tablets, twice daily with food for 19.5 days. On day 15 of lopinavir-ritonavir dosing, subjects took their morning lopinavir-ritonavir dose with a single dose of fenofibrate 145 mg following the same standardized breakfast (phase 3). Subjects then underwent plasma concentration sampling for fenofibric acid as described for phases 1 and 2. Subjects continued to take lopinavir-ritonavir for an additional 5.5 days (for a total of 19.5 days) throughout the entire pharmacokinetic sampling period.
Blood was collected throughout the study and on the last day of pharmacokinetic sampling for laboratory safety monitoring. Additionally, subjects were assessed for adverse events throughout the course of the investigation by routine questioning. Adherence was assessed by self-report and examination of diary cards and pill counts at scheduled study visits.
Fenofibric Acid Analysis
Fenofibric acid and clofibric acid internal standards were separated by using a newly developed high-performance liquid chromatography (HPLC) method and detected by HPLC with tandem mass spectrometry (HPLC–MS-MS) using multiple reaction monitoring. The HPLC–MS-MS analysis was performed by using an Acquity Ultra Performance Liquid Chromatography liquid handling system and a Quattro Premier XE triple quadrupole mass spectrometer (Waters Corp., Milford, MA, USA). The separation was performed on an Acquity Shield RP18, 2.1 × 50-mm, 1.7-μm analytical column preceded by an Vanguard Shield RP18, 2.1 × 5-mm, 1.7-μm pre-column (Waters Corp.) by using an isocratic method with a mobile phase ratio of 47:53 (v/v) acetonitrile and 2.0-mM ammonium formate (buffer) adjusted to pH 3.4 with formic acid at a flow rate of 0.500 ml/minute. Extraction of fenofibric acid and clofibric acid were performed by using an off-line solid-phase extraction method employing Evolute AX 1cc/25mg cartridges (Biotage, Uppsala, Sweden). Calibration curves for fenofibric acid were linear from 0.010–10.0 μg/ml with R2 > 0.99. Percent errors, as a measure of accuracy, were <15% and the inter- and intraassay coefficients of variation for fenofibric acid were 4.2–9.8% and 6.4–7.4%, respectively, at three different drug concentrations. The limit of quantitation for fenofibric acid was 0.010 μg/ml and the limit of detection was 0.005 μg/ml. The overall recovery of fenofibric acid and clofibric acid was >90%.
Pharmacokinetic Analysis
Fenofibric acid pharmacokinetic parameter values were determined by noncompartmental analysis using Phoenix WinNonlin software, version 6.3 (Pharsight Corp., Mountain View, CA, USA). Maximum plasma concentration (Cmax) and time to reach Cmax were determined by visual inspection of the concentration-time profiles. The elimination rate constant (λz) was estimated as the absolute value of the slope of a linear regression of a natural logarithm of concentration versus time using at least three points on the line. Half-life (t½) was calculated as ln2/λz. Area under the plasma concentration–time curve from time zero to the last quantifiable concentration (AUC0-last) was determined by using the linear trapezoidal rule. The AUC from time zero extrapolated to infinity (AUC0-∞) was determined by dividing the last measured concentration by λz and adding this value to AUC0-last. Apparent oral clearance (CL/F) was estimated as the dose divided by AUC0-∞.
Statistical Analysis
Sample size was calculated with regard to reported variability in fenofibric acid AUC in healthy volunteers (mean ± SD 92 ± 26 μg•hr/mL).20 Based on these data and α = 0.05, a sample size of 13 yielded > 90% power to detect a change of 25% in fenofibric acid AUC0-∞ with concomitant ritonavir or lopinavir-ritonavir. Fenofibric acid pharmacokinetic parameter values derived following fenofibrate administration alone were compared with values derived following ritonavir or lopinavir-ritonavir administration by using a paired, two-tailed Student t test. Statistical significance was defined a priori as p < 0.05; adjustments were not implemented for multiple comparisons. Geometric mean ratios (GMRs) and 90% confidence intervals (CI) for pharmacokinetic parameter values were generated for concomitant ritonavir and fenofibric acid (phase 2:phase 1) and concomitant lopinavir-ritonavir and fenofibric acid (phase 3:phase 1). SYSTAT software, version 11 (Systat Software Inc., Richmond, CA) was used for sample size calculation and inferential statistics, and Microsoft Excel (Microsoft Corp. Redmond, WA) was used to generate descriptive statistical data.
Results
Pharmacokinetic Analysis
Between July 2010 and October 2011 14 subjects were screened (3 women, 11 men), and 13 completed all three phases of the protocol. Demographic information for the study participants is presented in Table 1. One subject was removed from the study after completing phase 1 due to complications from a motor vehicle accident (described in further detail below). Pharmacokinetic data from this individual were not included in the analysis.
Table 1.
Baseline Demographic Characteristics of the 13 Healthy Adult Volunteers
| Subject | Age (yrs) | Sex | Race/Ethnicity | Weight (kg) | Body Mass Index (kg/m2) |
|---|---|---|---|---|---|
| 1 | 36 | Male | Black/non-Hispanic or Latino | 77 | 25.0 |
| 2 | 32 | Male | White/non-Hispanic or Latino | 86 | 27.1 |
| 3 | 36 | Male | Black/non-Hispanic or Latino | 114 | 24.6 |
| 4 | 57 | Male | White/non-Hispanic or Latino | 90 | 29.0 |
| 5 | 52 | Male | White/non-Hispanic or Latino | 72 | 23.4 |
| 6 | 22 | Female | Asian/non-Hispanic or Latino | 61 | 22.7 |
| 7 | 33 | Female | Black/non-Hispanic or Latino | 128 | 41.0 |
| 8 | 58 | Male | Black/non-Hispanic or Latino | 98 | 31.8 |
| 9 | 55 | Male | White/non-Hispanic or Latino | 85 | 27.2 |
| 10 | 40 | Male | White/non-Hispanic or Latino | 76 | 24.3 |
| 11 | 53 | Male | Asian/non-Hispanic or Latino | 67 | 22.4 |
| 12 | 44 | Male | White/Hispanic or Latino | 73 | 25.5 |
| 13 | 24 | Female | Multiple race/Hispanic or Latino | 58 | 23.5 |
| Median | 40 | 77 | 25.0 |
Fenofibric acid geometric mean pharmacokinetic parameter values and GMRs are displayed in Table 2, and the plasma concentration–time profiles for fenofibric acid are shown in Figure 2. No statistically significant changes were noted in any of the pharmacokinetic parameter values for fenofibric acid when it was coadministered with ritonavir (phase 2) or lopinavir-ritonavir (phase 3) compared with when fenofibrate was administered by itself (baseline) in phase 1. Geometric mean ratios (90% CIs) for fenofibric acid AUC0-∞ and Cmax were 0.89 (0.77–1.01) and 1.0 (0.88–1.10) after ritonavir administration versus baseline, respectively (p > 0.05 for both comparisons). Similarly, GMRs (90% CIs) for fenofibric acid AUC0-∞ and Cmax were 0.87 (0.69–1.05) and 1.0 (0.79–1.15) after lopinavir-ritonavir administration versus baseline, respectively (p > 0.05 for both comparisons).
Table 2.
Geometric Mean Ratios and Geometric Mean Fenofibrate Pharmacokinetic Parameter Values for Fenofibrate Administration Alone, After 2 Weeks of Ritonavir, and After 2 Weeks of Lopinavir-Ritonavir in the 13 Healthy Adult Volunteersa
| Pharmacokinetic Parameter | AUC0–∞ (μg·hr/mL) | Cmax (μg/mL) | Tmax (hrs) | CL/F (L/hr) | t½ (hrs) |
|---|---|---|---|---|---|
| Geometric Mean Value (90% CI) | |||||
| Phase 1 (fenofibrate alone) | 165 (114 – 216) | 7.6 (6.56 – 8.66) | 2.7 (1.95 – 3.44) | 0.88 (0.69 – 1.07) | 19.7 (15.6 – 23.8) |
| Phase 2 (fenofibrate + ritonavir) | 146 (125 – 167) | 7.5 (6.78 – 8.27) | 2.5 (2.16 – 2.93) | 0.99 (0.87 – 1.12) | 19.7 (15.9 – 23.4) |
| Phase 1 (fenofibrate + lopinavir-ritonavir) | 143 (131 – 156) | 7.4 (6.50 – 8.28) | 2.2 (1.68 – 2.69) | 1.01 (0.89 – 1.13) | 18.5 (16.3 – 20.7) |
| Geometric Mean Ratio (90% CI) | |||||
| Phase 2:phase 1 | 0.89 (0.77 – 1.01) | 1.0 (0.88 – 1.10) | 1.0 (0.60 – 1.29) | 1.13 (0.96 – 1.29) | 1.00 (0.87 – 1.12) |
| p valueb | 0.11 | 0.75 | 0.24 | 0.49 | 0.83 |
| Phase 3:phase 1 | 0.87 (0.69 – 1.05) | 1.0 (0.79 – 1.15) | 0.8 (0.48 – 1.14) | 1.15 (0.91 – 1.39) | 0.94 (0.84 – 1.04) |
| p valueb | 0.14 | 0.77 | 0.18 | 0.49 | 0.16 |
AUC0-∞ = area under the plasma concentration–time curve from time zero extrapolated to infinity; Cmax = maximum plasma concentration; Tmax = time to reach Cmax; CL/F = apparent oral clearance; t½ = half-life; CI = confidence interval.
Fenofibrate 145 mg was administered to the 13 healthy human volunteers as a single dose by itself (phase I), after 2 weeks of lopinavir 400 mg–ritonavir 100 mg twice daily (phase 2), and after 2 weeks of ritonavir 100 mg twice daily for 2 weeks (phase 3).
Determined by using a two-tailed, paired Student t test.
Figure 2.
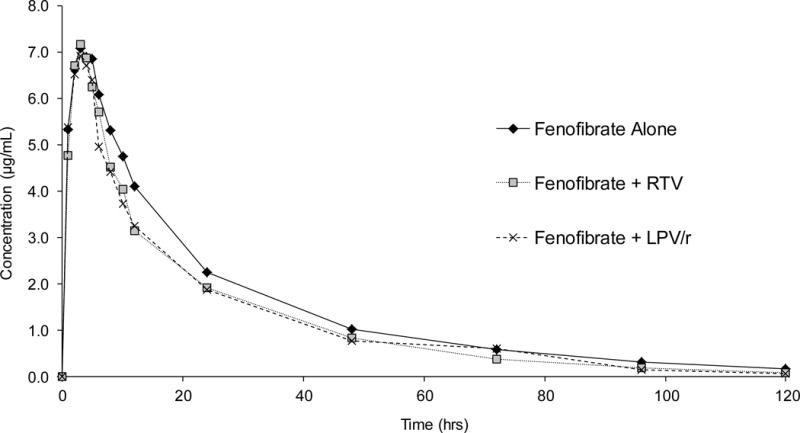
Fenofibrate mean plasma concentration–time curves following fenofibrate administration alone, fenofibrate in combination with ritonavir, and fenofibrate in combination with lopinavir-ritonavir in 13 healthy human subjects. RTV = ritonavir; LPV/r = lopinavir-ritonavir.
Safety and Tolerability
One subject was removed from the study prior to completion due to involvement in a motor vehicle accident and the emergent need for corticosteroid use. This subject experienced grade 3 gastric ulcers and occult blood in his stool while taking concomitant methylprednisolone and ritonavir during phase 2 of the study. Neither the study team nor the subject’s personal physician were aware he was taking these medications in combination, as he did not alert either party. When investigators became aware of the situation, the subject was removed from the study and treated for his medical conditions.
All study medications were generally well tolerated among the remaining 13 subjects who completed all phases of the investigation. Adverse events were mild or moderate (grade 1 or 2), and included diarrhea (3 patients), rash (2 patients), indigestion (1 patient), nausea (1 patient), anorexia (1 patient), and headache (1 patient). Laboratory abnormalities included elevations in serum cholesterol (2 patients), total bilirubin (1 patient, prior to receiving any study medications), and alanine aminotransferase levels, and decreases in hemoglobin level (2 patients). All adverse events were transient and resolved without intervention. No cases of myopathy or signs and symptoms consistent with skeletal muscle toxicity were reported.
Discussion
The current study demonstrated lack of a drug-drug interaction with the concomitant administration of oral fenofibrate with lopinavir-ritonavir or ritonavir alone. Fenofibric acid pharmacokinetic parameter values in all three phases were consistent with those previously reported for fenofibrate when administered as a single agent.21 We previously showed that lopinavir-ritonavir significantly reduced the systemic exposure of the other commercially available fibric acid derivative, gemfibrozil, in healthy human subjects; when administered twice daily for 2 weeks, lopinavir 400 mg–ritonavir 100 mg reduced the AUC0-∞ of single-dose gemfibrozil 600 mg by 41% (P < 0.05).17 Moreover, the magnitude and consistency of the interaction (all 15 subjects experienced a decrease in gemfibrozil AUC0-∞) suggest the need for an alternative to gemfibrozil in HIV-infected patients who are receiving ritonavir and also require triglyceride-lowering therapy.17
The lack of an interaction between fenofibrate and lopinavir-ritonavir, and fenofibrate and ritonavir, is likely explained by fenofibric acid being largely glucuronidated by UGT2B7, which does not appear to undergo induction by ritonavir, despite ritonavir’s ability to modulate the activity of other UGT enzymes such as UGT1A1 and UGT1A4.18,22 Additionally, in contrast to the reduction in gemfibrozil absorption that was previously observed with concurrent administration of lopinavir-ritonavir, neither lopinavir-ritonavir nor ritonavir alone significantly affected fenofibric acid absorption in the current investigation.
Data are limited regarding the role of membrane transporters in fenofibric acid absorption and disposition. However, studies have shown that fenofibric acid is not a substrate for the efflux transporter P-glycoprotein (P-gp). Since ritonavir has been shown to modulate P-gp, this is consistent with the lack of an effect of ritonavir (and lopinavir-ritonavir) on fenofibric acid absorption in this investigation.23,24 Whether commonly recognized intestinal transport proteins, such as multidrug resistance proteins (MRP2, MRP3), Breast Cancer-Related Protein (BCRP), and organic cation transporter (OCT1), are involved in fenofibric acid absorption is unclear. Nonetheless, it appears that whichever transport processes are involved in fenofibric acid absorption, they are not significantly altered by concomitant lopinavir-ritonavir or ritonavir administration.
Lopinavir-ritonavir and ritonavir alone were chosen for coadministration with fenofibrate in this study based on the previously described interaction between lopinavir-ritonavir and gemfibrozil (a fibric acid derivative that is chemically similar to fenofibrate) and because fenofibric acid is largely biotransformed through glucuronidation, and several glucuronidation pathways are known to undergo induction in the presence of ritonavir and/or lopinavir-ritonavir.22, 25, 26 In addition, although lopinavir-ritonavir is no longer considered a recommended protease inhibitor for inclusion in cART regimens, it continues to be a widely used protease inhibitor throughout the world and therefore remains clinically relevant.27 Indeed, due to the advent of generic lopinavir-ritonavir formulations, lopinavir-ritonavir is often a first-line protease inhibitor in a number of international settings.28 Moreover, this study design allowed for direct comparisons between the fenofibrate and gemfibrozil investigations. It also allowed us to assess the impact of ritonavir by itself on fenofibric disposition, since it is currently included in most protease inhibitor–based regimens.
In contrast to the significant interaction between gemfibrozil and lopinavir-ritonavir, neither lopinavir-ritonavir nor ritonavir alone altered the pharmacokinetics of fenofibric acid in healthy volunteers. Despite the lack of a pharmacokinetic interaction between ritonavir alone and fenofibrate, it remains unclear whether these results can likely be extrapolated to ritonavir-boosted atazanavir and darunavir-based cART. Ritonavir-boosted atazanavir has been largely been described as an inhibitor of UGT1A1 and has not been noted to induce UGT2B7.29,30 However, a recent investigation of concomitant buprenorphine and ritonavir-boosted darunavir demonstrated darunavir-mediated induction of buprenorphine glucuronidation (by UGT1A1 and/or UGT2B7).31, 32 This investigation did not include ritonavir-boosted atazanavir, which may have provided further insight regarding the specific mechanism (UGT2B7 compared with UGT1A1) of glucuronidation induction by darunavir.31 Given these data, further pharmacokinetic investigations, inclusive of ritonavir-boosted darunavir in particular, are likely warranted to ensure appropriate management of HIV-infected patients who require triglyceride-lowering therapy.
Conclusion
Neither lopinavir-ritonavir nor ritonavir alone altered the pharmacokinetics of fenofibric acid in healthy volunteers. The lack of an interaction observed with lopinavir-ritonavir and ritonavir alone suggests that fenofibrate remains an important option in HIV-infected patients who are receiving ritonavir-boosted cART.
Acknowledgments
Source of Funding: Funding for this study was provided by the Intramural Research Programs of the National Institutes of Health Clinical Center and the National Institute of Allergy and Infectious Diseases.
Footnotes
Previous Presentation: Data contained in this manuscript were previously presented in abstract form (poster session 208, abstract A-1575) at the Interscience Conference on Antimicrobial Agents and Chemotherapy, Denver, Colorado, September 10–13, 2013.
References
- 1.Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Department of Health and Human Services; Available at https://aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf (accessed 1, Oct, 2015) [Google Scholar]
- 2.Grunfeld C, Tien Phyllis. Difficulties in understanding the metabolic complications of acquired immune deficiency syndrome. Clinical Infectious Diseases. 2003;37(Suppl 2):S43–S46. doi: 10.1086/375886. [DOI] [PubMed] [Google Scholar]
- 3.Sax PE. Strategies for management and treatment of dyslipidemia in HIV/AIDS. AIDS Care. 2006 Feb;18(2):149–157. doi: 10.1080/09540120500161843. [DOI] [PubMed] [Google Scholar]
- 4.Visnegarwala F, Maldonado M, Sajja P, et al. Lipid lowering effects of statins and fibrates in the management of HIV dyslipidemias associated with antiretroviral therapy in HIV clinical practice. Journal of Infection. 2004;49:283–290. doi: 10.1016/j.jinf.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 5.Mills AM, Nelson M, Jayaweera D, et al. Once–daily darunavir/ritonavir vs. lopinavir/ritonavir in treatment–naïve, HIV–1–infected patients: 96–week analysis. AIDS. 2009;23:1679–1688. doi: 10.1097/QAD.0b013e32832d7350. [DOI] [PubMed] [Google Scholar]
- 6.Molina J, Andrade–Villanueva J, Echevarria J, et al. Once–daily atazanavir/ritonavir versus twice–daily lopinavir/ritonavir, each in combination with tenofovir and emtricitabine, for management of antiretroviral–naïve HIV–1–infected patients: 48 week efficacy and safety results of the CASTLE study. Lancet. 2008 Aug 23;372:646–655. doi: 10.1016/S0140-6736(08)61081-8. [DOI] [PubMed] [Google Scholar]
- 7.Souza SJ, Luzia LA, Santos SS, Santos SS, Rondo PH. Lipid profile of HIV-infected patients in relations to antiretroviral therapy: a review. Rev Assoc Med Bras. 2013;59:186–198. doi: 10.1016/j.ramb.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 8.Samineni D, Fichtenbaum CJ. Fenofibrate in the treatment of dyslipidemia associated with HIV infection. Expert Opin Drug Metab Toxicol. 2010;6:995–1004. doi: 10.1517/17425255.2010.504715. [DOI] [PubMed] [Google Scholar]
- 9.Duro M, Sarmento-Castro R, Almeida C, Medeiros R, Rebelo I. Lipid profile changes by high activity anti-retroviral therapy. Clinical Biochemistry. 2013;46:740–744. doi: 10.1016/j.clinbiochem.2012.12.017. [DOI] [PubMed] [Google Scholar]
- 10.Kamin DS, Grinspoon SK. Cardiovascular disease in HIV-positive patients. AIDS. 2005;19:641–652. doi: 10.1097/01.aids.0000166087.08822.bc. [DOI] [PubMed] [Google Scholar]
- 11.da Silva EF, Barbaro G. New options in the treatment of lipid disorders in HIV-infected patients. The Open AIDS Journal. 2009;3:31–37. doi: 10.2174/1874613600903010031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.National Cholesterol Education Program. Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) Final Report. National Heart, Lung, Blood Institute; Sep, 2002. pp. 1–284. Available at: http://www.nhlbi.nih.gov/guidelines/cholesterol/atp3full.pdf. Accessed July 30, 2009. [Google Scholar]
- 13.Penzak SR, Chuck SK. Management of protease inhibitor-associated hyperlipidemia. American Journal of Cardiovascular Drugs. 2002;2:91–106. doi: 10.2165/00129784-200202020-00003. [DOI] [PubMed] [Google Scholar]
- 14.Filippatos T, Milionis HJ. Treatment of hyperlipidaemia with fenofibrate and related fibrates. Expert Opinion Investigational Drugs. 2008;17(10):1599–1614. doi: 10.1517/13543784.17.10.1599. [DOI] [PubMed] [Google Scholar]
- 15.Silverberg MJ, Leyden W, Hurley L, et al. Response to newly prescribed lipid-lowering therapy in patients with and without HIV infection. Annals Internal Medicine. 2009;150:301–313. doi: 10.7326/0003-4819-150-5-200903030-00006. [DOI] [PubMed] [Google Scholar]
- 16.Norvir [package insert] North Chicago, IL: Abbott Laboratories; Feb, 2010. [Google Scholar]
- 17.Busse KH, Hadigan C, Chairez C, et al. Gemfibrozil concentrations are significantly decreased in the presence of lopinavir-ritonavir. Journal Acquired Immune Deficiency Syndrome. 2009;52:235–239. doi: 10.1097/QAI.0b013e3181b0610e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tojcic J, Benoit-Biancamano MO, Court MH, Straka RJ, Caron P, Guillemette C. In vitro glucuronidation of fenofibric acid by human UDP-glucuronosyltransferases and liver microsomes. Drug Metab Dispos. 2002;30:1280–1287. doi: 10.1124/dmd.109.029058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caldwell J. The biochemical pharmacology of fenofibrate. Cardiology. 1989;76(suppl 1):33–44. doi: 10.1159/000174545. [DOI] [PubMed] [Google Scholar]
- 20.Vaidyanathan S, Maboudian M, Warren V, et al. A study of the pharmacokinetic interactions of the direct rennin inhibitor aliskiren with metformin, pioglitazone and fenofibrate in healthy subjects. Current Medical Research and Opinion. 2008;24(8):2313–2326. doi: 10.1185/03007990802259354. [DOI] [PubMed] [Google Scholar]
- 21.Tricor [package insert] North Chicago, IL: Abbott Laboratories; Dec, 2008. [Google Scholar]
- 22.Bruce RD, Moody DE, Fang WB, Chodkowski D, Andrews L, Friedland GH. Tipranavir/ritonavir induction of buprenorphine glucuronide metabolism in HIV-negative subjects chronically receiving buprenorphine/naloxone. Am J Drug Alcohol Abuse. 2011;37:224–228. doi: 10.3109/00952990.2011.568081. [DOI] [PubMed] [Google Scholar]
- 23.Yamazaki M, Li B, Louie SW, et al. Effects of fibrates on human organic anion-transporting polypeptide 1B1-, multidrug resistance protein 2- and P-glycoprotein-mediated transport. Xenobiotica. 2005;35:737–753. doi: 10.1080/00498250500136676. [DOI] [PubMed] [Google Scholar]
- 24.Ehrhardt M, Lindenmaier H, Burhenne J, Haefeli WE, Weiss J. Influence of lipid lowering fibrates on P-glycoprotein activity in vitro. Biochem Pharmacol. 2004;67:285–292. doi: 10.1016/j.bcp.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 25.Ouellet D, Hsu A, Qian J, et al. Effect of ritonavir on the pharmacokinetics of ethinyl oestradiol in healthy female volunteers. British Journal Clinical Pharmacology. 1998;46(2):111–6. doi: 10.1046/j.1365-2125.1998.00749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van der Lee MJ, Dawood L, ter Hofstede HJ, et al. Lopinavir/ritonavir reduces lamotrigine plasma concentrations in healthy subjects. Clinical Pharmacology and Therapeutics. 2006 Aug;80:159–68. doi: 10.1016/j.clpt.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 27.Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Department of Health and Human Services; Available at https://aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf (accessed 1, Oct, 2015) [Google Scholar]
- 28.World Health Organization. Consolidated guidelines on the use of antiretroviral drugs for treating and preventing HIV infection: Recommendations for a public health approach. 2013 Jun 30; http://www.who.int/hiv/pub/guidelines/arv2013/download/en/ (accessed 1, Oct, 2015) [PubMed]
- 29.Burger DM, Huisman A, Ewijk NV, et al. The effect of atazanavir and atazanavir/ritonavir on UDP-glucuronosyltransferase using lamotrigine as a phenotypic probe. Clin Pharmacol Ther. 2008;84:698–703. doi: 10.1038/clpt.2008.106. [DOI] [PubMed] [Google Scholar]
- 30.Zhang D, Chando TJ, Everett DW, Patten CJ, Dehal SS, Humphreys WG. In vitro inhibition of UDP glucuronosyltransferases by atazanavir and other HIV protease inhibitors and the relationship of this property to in vivo bilirubin glucuronidation. Drug Metab Dispos. 2005;33:1729–1739. doi: 10.1124/dmd.105.005447. [DOI] [PubMed] [Google Scholar]
- 31.Gruber VA, Rainey PM, Moody DE, et al. Interactions between buprenorphine and the protease inhibitors darunavir-ritonavir and fosamprenavir-ritonavir. Clin Infect Dis. 2012;54:414–423. doi: 10.1093/cid/cir799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Butrans [package insert] Stamford, CT: Purdue Pharma LP; Jun, 2014. [Google Scholar]