

Abstract
Toward improving synthetic efficiency, organic chemists have turned to bioinspired organocascade or domino processes that generate multiple bonds and stereocenters in a single operation. However, despite the great importance of substituted cyclopentanes, given their prevalence in complex natural products and pharmaceutical agents, the rapid, enantioselective assembly of these carbocycles lags behind cyclohexanes. Herein, we describe a novel Michael-aldol-β-lactonization organocascade process for the synthesis of complex cyclopentanes utilizing chiral α,β-unsaturated acylammonium intermediates, readily generated by activation of commodity unsaturated acid chlorides with chiral isothiourea catalysts. This efficient methodology enables the construction of two C-C bonds, one C-O bond, two rings, and three contiguous stereogenic centers delivering complex cyclopentanes with high levels of relative and absolute stereocontrol. Our results suggest that unsaturated acyl ammonium intermediates have broad utility for the design of organocascade and multicomponent processes with the latter demonstrated by a Michael-Michael-aldol-β-lactonization.
Synthetic transformations that rapidly assemble complexity are actively being pursued given the importance of these processes for improvements in synthetic efficiency. In this regard, domino,1,2 tandem,3 and most recently organocascade4,5 processes have emerged as some of the most useful strategies for quickly generating structural complexity.6,7 Such methods for the construction of 6-membered carbocycles are numerous and include a range of classical methods with the Robinson annulation,8 the cationic polyene olefin cyclization,9 and the venerable Diels-Alder reaction10 serving as benchmarks. By contrast the construction of 5-membered carbocycles falls short of such diverse and widely used methods.11 While several elegant strategies exist for 5-membered carbocycle synthesis or annulation including the Pauson-Khand reaction,12 trimethylenemethane [3+2] cycloaddition,13 photochemical olefin-arene cycloaddition,14 and the Nazarov cyclization,15 most of these methods deliver cyclopentanes possessing no more than two stereogenic centers and most are not enantioselective. Organocatalytic methods for cyclopentane synthesis have begun to emerge, however many of these methods are quite limited in scope proceeding best with arene-substituted substrates.16 To address the challenge of efficient enantioselective construction of stereochemically rich cyclopentanes, a common motif in bioactive natural products17 and pharmaceuticals (Fig. 1a), we sought to design an organocascade process that could rapidly assemble these carbocycles from readily available materials.
Figure 1. Natural products and pharmaceutical agents bearing complex cyclopentanes and accessible cyclopentyl systems employing the described NCMAL process.

a, Bioactive natural products and drugs including polycyclics possessing highly substituted cyclopentane cores (red). b, Natural products possessing β-lactone-fused cyclopentanes (red). c, Several diverse cyclopentane scaffolds accessible by the described nucleophile-catalyzed, Michael-aldol-β-lactonization (NCMAL) process.
Building on our interest in tertiary amine mediated nucleophilic catalysis,18,19 we were attracted to the potential utility of chiral α,β-unsaturated acylammonium intermediates 3 given the presence of three disparate reactive sites that could be revealed sequentially to induce tandem bond-forming events (Fig. 2a). Within nucleophilic catalysis, chiral acylammonium intermediate 4, bearing one electrophilic site, has proven exceedingly useful for kinetic resolution of alcohols.20,21,22,23 The chiral ammonium enolate 5,24 bearing a nucleophilic site and a latent electrophilic site, is a highly versatile intermediate for a variety of transformations including aldol-β-lactonizations,25,26,27 α-halogenation,28 and hetero Diels-Alder reactions (Fig. 2b).29 Another more recent member in this family of chiral intermediates is the ammonium dienolate 6 derived from γ-deprotonation of an α,β-unsaturated acylammonium intermediate.30 We anticipated generation of the unsaturated acylammonium 3 through catalytic, asymmetric activation of acid chlorides by a chiral amine nucleophile (Fig. 2c). Following Michael addition by an appropriate nucleophile (Nuc1), a chiral ammonium enolate 7 is revealed enabling α-substitution with an electrophile (E) to deliver the acylammonium 8, and finally regeneration of the amine catalyst by acyl substitution with a second nucleophile (Nuc2) would deliver the triply functionalized adduct 9 (Fig. 2c). If the reaction partners are tethered (dashed lines), a bicyclic adduct 9 would result. A seminal report by the Fu group in 2006 demonstrated the potential of unsaturated acylammonium catalysis employing an unsaturated acyl fluoride and a chiral 4-pyrrolidinopyridine derivative to deliver a net [3+2] cycloaddition with a silylated indene with moderate enantioselectivities.31 The fluoride released during acyl substitution was required to increase the reactivity of the allylsilane nucleophile. This remained the only report employing this chiral intermediate until very recently when the Smith group reported the use of unsaturated, mixed anhydrides in combination with 1,3-dicarbonyl compounds to provide aryl-substituted enol lactones through an enantioselective, tandem Michael-enol-lactonization process.32 It should be noted that in both of these previous reports, the full potential of the latent, triply-reactive, unsaturated acylammonium 3 was not realized through formation of three new bonds. Herein, we describe the development of a novel organocascade process involving a nucleophile-catalyzed, Michael-aldol-β-lactonization (NCMAL) sequence that rapidly generates stereochemically complex cyclopentanes from commodity unsaturated acid chlorides including both fused and bridged cyclopentyl systems (Fig. 1c). As a consequence of the terminating lactonization step, the NCMAL delivers β-lactone-fused cyclopentanes, a structural motif found in the natural products spongiolactone and vibralactone A (Fig. 1b) in addition to a growing family of activity-based probes for proteome profiling.33 The NCMAL process was rendered enantioselective by use of chiral isothiourea nucleophilic catalysts34,35 leading to the asymmetric synthesis of cyclopentanes with up to three contiguous stereocenters including quaternary carbons.
Figure 2. Generation and reactivity of the chiral unsaturated acylammonium 3 with contrast to related chiral intermediates 4–6 and a proposed catalytic cycle leading to bicyclic-β-lactones 14.

a, Proposed synthesis of α,β-unsaturated acylammonium intermediate 3 from acid chlorides 1 and nucleophilic amines 2. b, Related chiral intermediates include the acylammonium intermediate 4, ammonium enolate 5, and ammonium dienolate 6. c, The latent, triple reactivity of the α,β-unsaturated acylammonium 3 first leads to an ammonium enolate 7 following Michael addition. The latter nucleophilic intermediate leads to α-substitution delivering acyl ammonium 8 and final acyl substitution leads to overall functionalizaton of three carbons of an α,β-unsaturated acid halide 1. d, Proposed catalytic cycle employing a latent, triply-reactive species 10, such as the keto anion 11, possessing two nucleophilic and one electrophilic site, capable of reaction with unsaturated acyl ammonium 3 to form three bonds resulting in cyclopentanes 14 bearing a fused β-lactone through a Michael-aldol-lactonization organocascade process.
The envisioned catalytic cycle for the organocascade process would be initiated by an intermolecular Michael of a tethered, triply-reactive reagent 11, such as anionic ketone 10, to the α,β-unsaturated acyl ammonium intermediate 3. The key, chiral intermediate 3 would be derived from simple substitution of a chiral amine catalyst (NR3) 2 with an unsaturated acid chloride 1 (Fig. 2d). Generation of the versatile ammonium enolate 12 enables engagement of the pendant ketone in an intramolecular aldol-β-lactonization templated by a Li(I) cation to deliver a stereoselective aldol reaction leading to formation of cyclopentane 13. Based on our previous studies, high anti diastereoselectivity was anticipated for β-substituted acid chlorides since this alleviates developing A1,3-strain during the aldol-lactonization step (cf. 12).26 A final β-lactonization would deliver the fused β-lactone ring and regenerate the nucleophilic amine catalyst.
Results and Discussion
We first set out to identify a suitable Brønsted base to generate a reactive Michael donor from the latent anionic reagent, keto malonate 10a. Our initial reaction conditions involved generation of the malonate anion with various Brønsted bases in the presence of a nucleophilic catalyst followed by slow addition of the acid chloride to the mixture. Use of the nucleophile alone or the strong Brønsted base, DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) did not provide the desired bicyclic-β-lactone 14a (Table 1, entries 1, 2). However, the synergistic action of DBU and a Lewis acid (LiClO4, entry 3) afforded the desired product in 68% yield.36 To ensure complete enolate formation, stronger lithium bases were studied and found to provide further improvement in yields (entries 4–6) up to 75% yield. Use of more weakly coordinating counterions such as sodium and potassium led to drastically lower yields (entries 7, 8), while magnesium afforded a comparable yield (entry 9). A brief screening of nucleophiles,37 including 9-azajulolidine, revealed that 4-pyrrolidinopyridine (4-PPY) and LiHMDS was an optimal combination for the NCMAL process (entries 10, 11). As evidence for the requirement of the nucleophile, a control reaction without 4-PPY did not afford any β-lactone (entry 12).
Table 1.
Optimization of the nucleophile-catalyzed, Michael-aldol-β-lactonization (NCMAL) process.
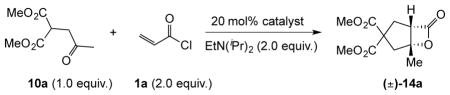
| ||||
|---|---|---|---|---|
| entry | base | catalyst | solvent | % yield* |
| 1 | -- | DMAP | CH2Cl2 | <5 |
| 2 | DBU | DMAP | CH2Cl2 | <5 |
| 3 | LiClO4+DBU | DMAP | CH2Cl2 | 68 |
| 4 | LDA | DMAP | THF/CH2Cl2 | 73 |
| 5 | tBuLi | DMAP | THF/CH2Cl2 | 72 |
| 6 | LiHMDS | DMAP | THF/CH2Cl2 | 75 |
| 7 | NaHMDS | DMAP | THF/CH2Cl2 | 23 |
| 8 | KHMDS | DMAP | THF/CH2Cl2 | <5 |
| 9 | iPrMgCl | DMAP | THF/CH2Cl2 | 75 |
| 10 | LiHMDS | 4-PPY | THF/CH2Cl2 | 84 |
| 11 | LiHMDS | 9-AJ† | THF/CH2Cl2 | 77 |
| 12 | LiHMDS | -- | THF/CH2Cl2 | <5 |
Refers to isolated yields. <5% yield indicates that β-lactone was not detected by TLC, 1H NMR (500 MHz), or FT-IR.
9-azajulolidine (9-AJ).
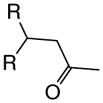
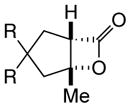
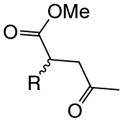
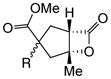
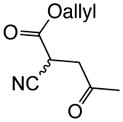
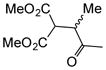
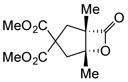
With suitable conditions for the NCMAL process in hand, we probed variation of the Michael donors (Table 2a, entries 1–9). Successful Michael donors bore two electron-withdrawing groups, an important feature pointing to the requirement of a soft enolate for the Michael addition. These substrates were obtained in a single step by alkylation of the corresponding malonates with chloroacetone (Supplementary, S3–S11). As depicted in Table 2, a variety of keto diesters, diketones, and dinitriles (entries 1–5) participate in the NCMAL process to provide the desired cyclopentanes 14b–f in 60–84% yields. When a β-keto ester or α-cyano esters were used as Michael donors (Table 2, entries 6–8), cyclopentanes 14g,h were obtained in 65–73% yields with poor diastereoselectivity (dr 1–1.7:1) as expected, however this is inconsequential given that these diastereomers would ultimately converge to a single diastereomer following decarboxylation (vide infra). Introduction of an α-keto stereocenter, as in ketomalonate 10j, also led to a mixture of diastereomers 14j (dr 2:1) in 81% yield.
Table 2.
Scope of the nucleophile catalyzed, Michael-aldol-β-lactonization (NCMAL) organocascade process.
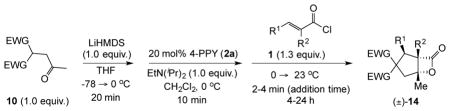
| |||||||
|---|---|---|---|---|---|---|---|
| a Varying the Michael donor with acryloyl chloride 1a | b Varying the Michael acceptor with acetodimethyl malonate 10a*,† | ||||||
|
| |||||||
| entry | starting material | bicyclic-β-lactone | % yield (dr)* | entry | starting material | bicyclic-β-lactone | % yield (dr)*,† |
| 1 |
 10b-d |
|
(±)-14b: R = tBu, 73 | 10 |
 1b–c |
|
(±)-14k: R =
Me 94 (>19:1)‡ |
| 2 | (±)-14c: R = allyl, 84 | 11 | (±)-14l: R =
CO2Et 91 (>19:1) |
||||
| 3 | (±)-14d: R = Bn, 63 | 12 |
 1d–f |
 (±)-14m–o |
(±)-14m: R = H, 90 (>19:1)‡ | ||
| 4 |
 10e–f |
|
(±)-14e: R = C(O)Me, 64 | 13 | (±)-14n: R = NO2, 83 (>19:1) | ||
| 5 | (±)-14f: R = CN, 60 | 14 | (±)-14o: R =
OMe 87 (>19:1) |
||||
| 6 |
 (±)-10g–h |
|
(±)-14g: R =
C(O)Me 73 (1:1) |
15 |
1g |
 (±)-14p |
64 (>19:1) |
| 7 | (±)-14h: R =
CN 65 (1.7:1) |
||||||
| 8 |
 (±)-10i |
 (±)-14i |
60 (1.16:1) | 16 |
 1h |
 (±)-14q |
78 |
| 9 |
 (±)-10j |
 (±)-14j |
84 (2:1)† | 17 |
 1i |
 (±)-14r |
60 (>19:1)‡ |
All yields refer to isolated yields.
Diasteromeric ratio determined by 1H NMR.
Relative stereochemistry verified by X-ray analysis. DMAP = 4-dimethyl amino pyridine,
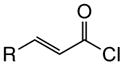
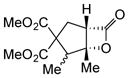
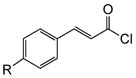
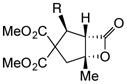
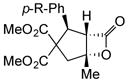
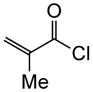
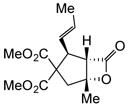
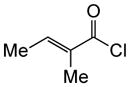
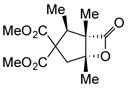
Variation of the Michael acceptors through the study of a diverse array of commercially available α,β-unsaturated acid chlorides was undertaken next. Under the optimized NCMAL conditions, cyclopentanes were readily accessed bearing up to three contiguous stereocenters with excellent relative stereochemical control (Table 2b, entries 10–17). β-Substituted acid chlorides uniformly gave high diastereoselectivity, due to A1,3-strain (cf. Fig. 2d).38 The breadth of β-substituted acid chlorides proved to be fruitful allowing for installation of various functional groups leading to β-alkyl substituted cyclopentane 14k (entry 10) and ester substituted cylopentane 14l (entry 11). Aryl-substituted acid chlorides with varied electronic properties (entries 12–14) proceeded in 83–90% yields. Use of sorbic chloride (1g, entry 15) provided a 64% yield of cyclopentane 14p but allowed for the installation of an alkene moiety on the cyclopentane providing a robust functional handle for subsequent manipulations and diversification. An α-substituted acid chloride 1h (entry 16) was also reactive under standard conditions and afforded cyclopentane 14q in 78% yield bearing two contiguous quaternary stereocenters, one of which is an all carbon quaternary center. Finally, a highly congested cyclopentane 14r was accessible using α,β-dimethyl acryloyl chloride 1i (entry 17) in 60% yield as a single diastereomer.
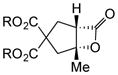
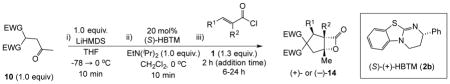
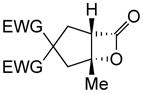
Toward applying this complexity-generating process to bioactive natural product synthesis, we next explored an enantioselective variant of the NCMAL process. Building on our previously described aldol-lactonization process,26 the chiral isothiourea, homobenzotetramisole (HBTM) developed by Birman39 was found to be the optimal catalyst to promote an enantioselective NCMAL (Table 3). Employing the aforementioned optimized conditions for the racemic NCMAL, HBTM uniformly delivered the cyclopentanes (+)-14a, (+)-14c, and (+)-14d in 59–74% yields and 93–96% ee (Table 3, entries 1–4). The bis-allyl malonate ester 10d was of particular interest for enabling further functionalization or removal of an electron-withdrawing group from the cyclopentane adducts (+)-14c and (+)-14d through mild Pd(0)-catalyzed decarboxylative transformations (entry 3).40 The absolute configuration of β-lactone (+)-14a was confirmed by X-ray analysis of a derivative following ring opening with p-bromobenzylamine (Supplementary, Fig. S3). The diketone substrate 10e was also well tolerated in the asymmetric process, leading to diketo cyclopentanes (+)-14e in 61% yield and 95% ee (Table 3, entry 5). The practicality of the NCMAL process was demonstrated by a gram-scale reaction using dimethyl keto malonate 10a as Michael donor delivered (+)-14a with comparable results (74% yield, 93% ee, Table 3, entry 2).
Table 3.
Enantioselective nucleophile catalyzed, Michael–aldol-β-lactonization (NCMAL) organocascade.
| ||||||
|---|---|---|---|---|---|---|
| entry | Michael donor (EWG) | acid chloride | bicyclic-β-lactone | % yield (dr)* | % ee | |
| 1† | 10a (CO2Me) |
 1a |
|
(+)-14a§ | 72 | 97 |
| 2†,‡ | 10a | (+)-14a | 74 | 93 | ||
| 3† | 10c (CO2allyl) | (+)-14c | 73 | 95 | ||
| 4† | 10d (CO2Bn) | (+)-14d | 59 | 98 | ||
| 5† | 10e (C(O)Me) | (+)-14e | 61 | 95 | ||
| 6 | 10a |
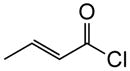 1b |
|
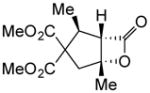
(+)-14k | 80 (>19:1) | 94 |
| 7† | 10a |
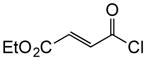 1c |
|
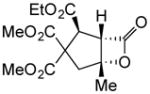
(+)-14l | 95 (>19:1) | 90 |
| 8† | 10a | (−)-14l∫ | 90 (>19:1) | 89 | ||
| 9 | 10a |
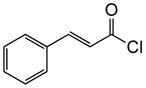 1d |
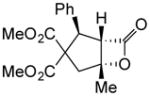
|
(+)-14m | 80 (>19:1) | 99 |
| 10 | 10a |
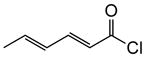 1g |
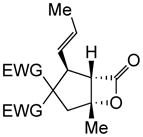
|
(+)-14p§ | 62 (>19:1) | 99 |
| 11 | 10a | (−)-14p∫ | 60 (>19:1) | 99 | ||
| 12 | 10c | (+)-14s | 55 (>19:1) | 94 | ||
| 13 | 10a |
 1h |
|
(+)-14q | 80 (>19:1) | 99 |
All yields refer to isolated yields, enantiomeric excess was determined by chiral GC or HPLC, diastereomeric ratios were determined by 1H NMR (500 MHz), and addition of acid chloride was over a 2 h period. Reaction times varied from 6 to 24 h (see Supplementary for reaction details).
Acid chlorides were added over 4 min.
Reaction was performed on gram scale.
Absolute stereochemistry was confirmed by X-ray analysis of (+)-14p and a derivative of (+)-14a (Supplementary, Fig. S6 and Fig. S3, respectively). (R)-HBTM was employed as catalyst.
To probe whether the initial Michael addition could proceed in an enantioselective manner, we next studied β-substituted acid chlorides. Indeed, use of (S)-HBTM with acid chlorides 1b–c and 1d,g gave cyclopentanes (+)-14k–l and (+)-14m,p in 89–99% ee as single diastereomers in 62–95% yields (Table 3, entries 6–12) including those bearing ethyl ester and alkenyl substituents suitable for further functionalization. The relative and absolute stereochemistry of the alkenyl substituted cyclopentane (+)-14p was confirmed by X-ray analysis (Supplementary, Fig. S6). When β-substituted acid chlorides were studied, an important difference in reaction outcome was noted under standard conditions. Optimization studies revealed that extending the addition times of the acid chloride once again led to high conversions and enantioselectivities. A competitive, racemic background pathway with the NCMAL, in the case of β-substituted acid chlorides, is proposed for this difference (Supplementary, Table S1). Under these optimized conditions, use of (R)-HBTM as catalyst provided the enantiomeric β-lactones (−)-14l and (−)-14p in 90% yield, 89% ee and 60% yield, 99% ee, respectively (Table 3, entries 8 and 11). When applied to sorbic chloride (1g), the bis-allyl keto malonate 10c provided cyclopentane (+)-14s in 55% yield and 94% ee as a single diastereomer. α-Methacrylolyl chloride (1h) was also a viable substrate delivering the sterically congested cyclopentane (+)-14q bearing two contiguous quaternary carbons, one being an all carbon quaternary center in 80% yield and 99% ee (entry 13).
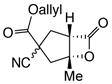
To demonstrate the utility of the described organocascade process for ring annulation leading to more complex molecular architectures, a collection of polycyclic carbocycles with fused or bridged topology was targeted using monocyclic Michael donors. Cyclopentanone 10k was readily combined with acryloyl chloride (1a) to give the tricyclic 5,5,4-bicyclic system 14t as a single diastereomer in 91% yield (Fig. 3a). In contrast, cyclohexanone 10l afforded the corresponding tricyclic products 14u in 70% yield as a 1:1 mixture of diastereomers likely owing to the lower energy difference between cis- and trans-fused 5,6-bicyclic systems. Cyclohexanedione 10m also participated as a Michael donor with acryloyl chloride (1a) to give the tricyclic cyclopentane 14v possessing a bridged topology, albeit in 25% yield (Fig. 3b). The relative stereochemistry was confirmed by single crystal X-ray analysis (inset, Fig. 3b; Supplementary, Fig. S9) and the bridged cyclopentane in 14v is reminiscent of one substructure of the gibberellin family of terpenoids (cf. Fig. 1a).41 The tetralone-derived malonate 10n also participated in the NCMAL, however the presumed intermediate β-lactone 15, possessing a benzylic C-O bond, underwent facile decarboxylation to deliver the cyclopentene 16. Following hydrogenation and Krapcho decarboxylation, the monoester 17 was obtained, demonstrating removal of an activating group in the Michael donor. Furthermore, the monoester 17 resembles a previously described steroidal intermediate42 (cf. Fig. 1b). A milder method was also sought for removal of one of the activating groups required for competent, soft Michael donors. The Pd(0)-mediated decarboxylation of the allyl ester substituted cyclopentane 14i was explored.43 Mild conditions were identified that led to reductive decarboxylation of the mixture of diastereomeric cyclopentanes 14i at 77 °C which importantly left the β-lactone intact and converged to a single diastereomer of the cyano-substituted cyclopentane 14x bearing an additional stereocenter (Fig. 3d). The relative stereochemistry of 14x was verified by X-ray analysis (inset, Fig. 3d; Supplementary Fig. S10). To the best of our knowledge, this is the first example of ester decarboxylation in the presence of a β-lactone (Fig. 3d).
Figure 3. Rapid molecular complexity generation, structural modifications of cyclopentane products, and extensions of both accessible Michael donors and unsaturated acyl ammonium intermediates.

[All NCMAL reactions were performed under standard reaction conditions shown in Table 2 unless noted otherwise.] a, Monocyclic Michael donors with acrylolyl chloride deliver tricyclic 5,5- and 5,6-fused cyclopentyl systems 14t and 14u; b, bridged tricylic cyclopentanes; and c, Truncated steroid intermediates through bis-decarboxylation; d, Mild Pd(0)-mediated reductive decarboxylations leads to cyano substituted cyclopentane 14x. e, Application of aldehyde-containing Michael donors. f, In situ generation of tosyl anhydrides delivers tricyclic 5,5,4-systems from starting carboxylic acids. Relative stereochemistry determined by X-ray analysis (Supplementary, Fig. S8).
Extension of the NCMAL from ketone substrates to the corresponding aldehyde malonates was expected to be challenging given the potential for side reactions of the reactive aldehyde under the basic conditions of the NCMAL. However, we were prompted to develop a strategy toward these types of bicyclic-β-lactones given the structure of vibralactone A (Fig. 1b). We envisioned that upon deprotonation of the malonate moiety of aldehyde 10o, equilibrium would be established between the lithiated malonate anion 18 and the cyclopropyl lithium alkoxide 19 which could mask the reactivity of the aldehyde.44 Indeed, metallation of aldehyde malonate 10o and addition of acryloyl chloride (1a) in the presence of 20 mol% 4-PPY led to the desired cyclopentane 14y in 60% yield.
We also demonstrated that in situ activated carboxylic acids could be utilized in the NCMAL greatly expanding the variety of accessible unsaturated acyl ammonium intermediates 3. Commercially available cyclopentene acid 18a was activated in situ with p-toluenesulfonyl chloride to the corresponding tosyl anhydride 18b and without isolation, slow addition of this intermediate to the anion of keto malonate 10a afforded the tricyclic 5,5,4-cyclopentyl system 14z in 56% yield as a single diastereomer.
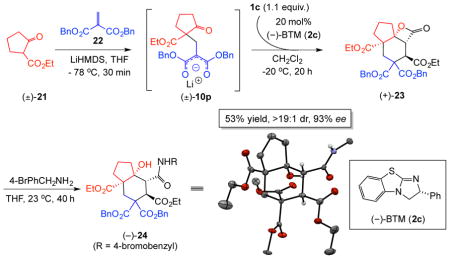
Towards combining the efficiency of the described organocascade with a multicomponent reaction,45 we sought to perform an initial Michael reaction to obtain a competent Michael donor for the subsequent Michael-aldol-lactonization to rapidly achieve molecular complexity in a highly atom-economic manner. We envisioned that in situ generation of the ketomalonate anion 10p through a Michael addition would initiate the cascade process and ultimately lead to a cyclohexyl fused-β-lactone 22. Following extensive optimization, a three-component process was realized from β-ketoester 21, fumaroyl chloride (1c), and dibenzyl 2-methylenemalonate (22) to provide the 6,5,4-tricyclic cyclohexane (+)-23 in 53% yield and 93% ee generating four contiguous stereocenters, three new C-C bonds, one C-O bond, and two new rings in a highly stereoselective manner with (−)-BTM (2c) as nucleophilic promoter. The relative and absolute configuration was confirmed following ring opening of the β-lactone with p-bromobenzylamine to afford amide (−)-23 (inset, Supplementary Fig. S11). This three-component reaction highlights the potential of incorporating α,β-unsaturated acylammonium intermediates 3 into the design of multicomponent, organocascade processes.
The complexity-generating, nucleophile-catalyzed, Michael-aldol-β-lactonization process described herein delivers high synthetic efficiency for complex cyclopentane synthesis by formation of three new bonds, up to three stereogenic centers, and two rings in a single operation from simple starting materials. Furthermore, the simple yet powerful asymmetric activation mode of commercially available, unsaturated acid chlorides utilized to its full potential for the first time by reaction at all three positions, provides a new paradigm for the design of additional organocascade processes. This tandem reaction was rendered highly enantioselective with the application of chiral isothiourea catalysts. Extensions to polycyclic carbon frameworks and multicomponent processes such as the Michael-Michael-aldol-lactonization demonstrate the potential of this concept for more sophisticated reaction design aimed at synthesis of natural products and pharmaceutical agents containing complex cyclopentanes. The β-lactones present in these systems are versatile platforms for further transformations including dyotropic and Beckman rearrangements,26 ring expansions,46 enabling further elaboration of the cyclopentane products.47 The combination of readily accessible catalysts ((R) and (S)-HBTM; soon available from Aldrich),48 unsaturated acid chlorides (Sigma-Aldrich: 13 variously substituted, MatrixScientific: 37 β-aryl-substituted unsaturated, acid chlorides), and Michael donors (TCI, diethyl acetonylmalonate) in conjunction with in situ activated carboxylic acids enables an attractive and practical strategy for the expeditious synthesis of optically active cyclopentanes with correspondence to natural products and pharmaceutical agents. We anticipate that the described latent, triple reactivity of the unsaturated acyl ammonium intermediate will lead to additional, novel synthetic methods in the area of organocatalysis. Studies of additional transformations exploiting this underexplored activation mode of commodity unsaturated acid chlorides and synthetic applications of these methods are underway.
Methods
Gram-scale NCMAL leading to tricyclic-β-lactone (+)-14a
To an oven-dried, 250 mL round-bottomed flask equipped with a magnetic stir bar was added dimethyl 2-(2-oxopropyl) malonate (10a, 1.0 g, 5.32 mmol, 1.0 equiv.) along with THF (17 mL) and the mixture was cooled to −78 °C. With vigorous stirring, LiHMDS (5.32 mL of a 1.0 M solution in THF, 5.32 mmol, 1.0 equiv.) was added dropwise via gas tight syringe. After complete addition, the reaction was stirred for 10 min at −78 °C and then warmed to 0 °C by switching the dry ice/acetone bath to an ice/water bath. Stirring was continued for an additional 10 min at this temperature, and then CH2Cl2 (60 mL), (S)-HBTM (283 mg, 1.06 mmol, dissolved in 7 mL CH2Cl2, 0.20 equiv.), and EtN(iPr)2 (0.93 mL, 5.32 mmol, 1.0 equiv.) were added sequentially via gas-tight syringe. The reaction was allowed to stir for an additional 10 min at 0 °C before acryloyl chloride (1a, 3.30 mL of a 1.612 M solution in CH2Cl2, 6.91 mmol, 1.30 equiv.) was added via microliter syringe dropwise over ~2 min. After complete addition, the ice bath was removed and the reaction was stirred for 6 h at ambient temperature (23 °C). The reaction was then cooled to 0 °C and silica gel (2 g) was added and stirred at 0 °C for 10 min. Then the ice/water bath was removed and the reaction stirred at ambient temperature (23 °C) for 20 min. The mixture was then diluted with hexanes (4.0 mL), filtered through a short silica gel pad (~2 g of silica gel), and rinsed with EtOAc (3 × 4 mL). The filtrate was then concentrated by rotary evaporation, and the crude mixture analyzed by 1H NMR which indicated a >19:1 dr. Purification by an automated flash chromatography system (gradient of EtOAc/hexanes) afforded a single diastereomer of bicyclic-β-lactone (+)-14a (0.9497 g, 74%) as a colorless oil: Enantiomeric excess was determined by chiral GC analysis in comparison with authentic racemic material; tminor = 251.6 min, tmajor = 262.6 min; 93% ee. Spectral data matched that previously reported26 and absolute stereochemistry was assigned following β-lactone ring opening with p-bromobenzylamine as described in the Supplementary.
Procedure for multicomponent Michael-Michael-aldol-β-lactonization delivering cyclohexane (+)-23
An oven-dried, 100-mL round-bottomed flask was charged with a solution of LiHMDS (1.80 mL of 1.0 M solution in THF, 1.80 mmol, 1.2 equiv,) in THF (2.5 mL) and cooled to −78 °C. Following slow addition of a solution of cyclopentanone 21 (234 mg, 1.50 mmol, 1.0 equiv.) in THF (2.5 mL), the reaction mixture was warmed to −20 °C and stirred for 30 min. A solution of diester 21 (1.80 mL of 1.0 M solution in benzene, 1.80 mmol, 1.2 equiv) in THF (2.5 mL) was added dropwise. After 1 h at −20 °C, a solution of (−)-BTM (75.7 mg, 0.30 mmol, 20 mol%) and EtN(iPr)2 (194 mg, 1.50 mmol, 1.0 equiv) in CH2Cl2 (2.5 mL) was added. Then a solution of acid chloride 1c (366 mg, 2.25 mmol, 1.5 equiv.) in CH2Cl2 (5.0 mL) was added at −20 °C over 5 h by a syringe pump. The reaction temperature was maintained at −20 °C throughout the addition of 1b and then the reaction was stirred at this temperature for 15 h. Upon completion (as judged by TLC), the reaction was concentrated by rotary evaporation and the product was purified by an automated flash chromatography system (gradient of EtOAc/hexanes) to afford a single diastereomer of tricyclic-β-lactone (+)-23 (457 mg, 53% yield) as a yellow, viscous liquid. Complete characterization data is provided in the Supplementary. The relative and absolute stereochemistry was confirmed by X-ray analysis (Supplementary Fig. S11) following β-lactone ring opening with p-bromobenzylamine.
Supplementary Material
Acknowledgments
The work was supported by NSF (CHE-0809747) and the Welch Foundation (A-1280) along with partial support from NIH (5R37GM052964). We thank Natalie Harvey for synthesis of HBTM and assistance with early studies of the NCMAL. Dr. Nattamai Bhuvanesh (Center for X-ray Analysis, TAMU) and Dr. Bill Russell (Laboratory for Biological Mass Spectrometry, TAMU) secured structural and mass data, respectively.
Footnotes
Author Contributions
G.L. initiated the studies of the α,β-unsaturated acyl ammonium intermediate from acid chlorides. D. R., G.L., and M.E.S. were involved in the design of experiments for exploration of the NCMAL. D.R. and K.N.V. conceived and developed the three-component NCMAL process. G.L., M.E.S., K.N.V., and R.L.M. performed the experiments. D.R., G.L., and M.E.S. composed the manuscript with input from all authors.
References
- 1.Tietze LF, Brasche G, Gericke KM. Domino reactions in organic synthesis. Wiley-VCH; Weinheim: 2006. [Google Scholar]
- 2.Pellissier H. Stereocontrolled domino reactions. Chem Rev. 2013;113:442–524. doi: 10.1021/cr300271k. [DOI] [PubMed] [Google Scholar]
- 3.Zhou J. Recent Advances in multicatalyst promoted asymmetric tandem reactions. Chem Asian J. 2010;5:422–434. doi: 10.1002/asia.200900458. [DOI] [PubMed] [Google Scholar]
- 4.Grondal C, Jeanty M, Enders D. Organocatalytic cascade reactions as a new tool in total synthesis. Nat Chem. 2010;2:167–178. doi: 10.1038/nchem.539. [DOI] [PubMed] [Google Scholar]
- 5.Jones SB, Simmons B, Mastracchio A, MacMillan DWC. Collective synthesis of natural products by means of organocascade catalysis. Nature. 2011;475:183–188. doi: 10.1038/nature10232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coulthard G, Erb W, Aggarwal VK. Stereocontrolled organocatalytic synthesis of prostaglandin PGF2α in seven steps. Nature. 2012;489:278–281. doi: 10.1038/nature11411. [DOI] [PubMed] [Google Scholar]
- 7.Enders D, Huttl MR, Grondal C, Raabe G. Control of four stereocentres in a triple cascade organocatalytic reaction. Nature. 2006;441:861–863. doi: 10.1038/nature04820. [DOI] [PubMed] [Google Scholar]
- 8.Surhone LM, Tennoe MT. In: Robinson Annulation. Henssonow SF, editor. Betascript Publishing; 2011. [Google Scholar]
- 9.Sakakura A, Ukai A, Ishihara K. Enantioselective halocyclization of polyprenoids induced by nucleophilic phosphoramidites. Nature. 2007;445:900–3. doi: 10.1038/nature05553. [DOI] [PubMed] [Google Scholar]
- 10.Nicolaou KC, Synder SA, Montagnon T Vassilikogiannakis. The Diels-Alder reaction in total synthesis. Angew Chem Int Ed. 2002;41:1668–1698. doi: 10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 11.Heasley B. Stereocontrolled preparation of fully substituted cyclopentanes: Relevance to total synthesis. Eur J Org Chem. 2009:1447–1489. [Google Scholar]
- 12.Jorg H. The Pauson-Khand reaction in the synthesis of pharmacologically active compounds. Curr Org Chem. 2010;14:1139–1152. [Google Scholar]
- 13.Trost BM, Chan DMT. New conjunctive reagents. 2-Acetoxymethyl-3-allyltrimethylsilane for methylenecyclopentane annulations catalyzed by palladium(0) J Am Chem Soc. 1979;101:6429–6432. [Google Scholar]
- 14.Streit U, Bochet CG. The arene–alkene photocycloaddition. Beilstein J Org Chem. 2011;7:525–542. doi: 10.3762/bjoc.7.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frontier AJ, Collison C. The Nazarov cyclization in organic synthesis. Recent advances. Tetrahedron. 2005;61:7577–7606. [Google Scholar]
- 16.Moyano A, Ramon Rios R. Asymmetric organocatalytic cyclization and cycloaddition reactions. Chem Rev. 2011;111:4703–4832. doi: 10.1021/cr100348t. [DOI] [PubMed] [Google Scholar]
- 17.Barrero AF, Quilez del Moral JF, Herrador MM, Rodriguez H, Morales MCP. Cyclopentane sesquiterpenes from fungi: ocurrence-bioactivity, biosynthesis and chemical synthesis. Curr Org Chem. 2009;18:1164–1181. [Google Scholar]
- 18.Cortez GS, Tennyson R, Romo D. Intramolecular nucleophile catalyzed aldol-lactonization (NCAL) reactions: catalytic, asymmetric synthesis of bicyclic β-lactones. J Am Chem Soc. 2001;123:7945–7946. doi: 10.1021/ja016134+. [DOI] [PubMed] [Google Scholar]
- 19.France S, Guerin DJ, Miller SJ, Lectka T. Nucleophile chiral amines as catalysts in asymmetric synthesis. Chem Rev. 2003;103:2985–3012. doi: 10.1021/cr020061a. [DOI] [PubMed] [Google Scholar]
- 20.Fu GC. Asymmetric catalysis with “planar-chiral” derivatives of 4-(dimethylamino)pyridine. Acc Chem Res. 2004;37:542–547. doi: 10.1021/ar030051b. [DOI] [PubMed] [Google Scholar]
- 21.Birman VB, Uffman EW, Jiang H, Li X, Kilbane CJ. 2,3-Dihydroimidazo[1,2-a]pyridines: a new class of enantioselective acyl transfer catalysts and their use in kinetic resolution of alcohols. J Am Chem Soc. 2004;126:12226–12227. doi: 10.1021/ja0491477. [DOI] [PubMed] [Google Scholar]
- 22.Kobayashi M, Okamoto S. Unexpected reactivity of annulated 3H-benzothiazol-2-ylideneamines as acyl transfer catalyst. Tetrahedron Lett. 2006;47:4347–4350. [Google Scholar]
- 23.Wagner AJ, Rychnovsky SD. Determination of Absolute Configuration of Secondary Alcohols Using Thin-Layer Chromatography. J Org Chem. 2013;78:4594–4598. doi: 10.1021/jo400432q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaunt MJ, Johansson CCC. Recent developments in the use of catalytic asymmetric ammonium enolates in chemical synthesis. Chem Rev. 2007;107:5596–5605. doi: 10.1021/cr0683764. [DOI] [PubMed] [Google Scholar]
- 25.Wynberg H, Staring EG. Asymmetric synthesis of (S)- and (R)-malic acid from ketene chloral. J Am Chem Soc. 1982;104:166–168. [Google Scholar]
- 26.Leverett CA, Purohit VC, Romo D. Enantioselective, organocatalyzed intramolecular aldol-lactonizations with ketoacids leading to bicyclic and tricyclic β-lactones and topology morphing transformations. Angew Chem Int Ed. 2010;122:9669–9673. doi: 10.1002/anie.201004671. [DOI] [PubMed] [Google Scholar]
- 27.Nelson SC, Zhu C, Shen X. Catalytic asymmetric acyl halide-aldehyde cyclocondensation reactions of substituted ketenes. J Am Chem Soc. 2004;126:14–15. doi: 10.1021/ja0391208. [DOI] [PubMed] [Google Scholar]
- 28.Lectka T, et al. Catalytic, asymmetric α-chlorination of acid halides. J Am Chem Soc. 2004;126:4245–4255. doi: 10.1021/ja039046t. [DOI] [PubMed] [Google Scholar]
- 29.Abraham CJ, Paull DH, Scerba MT, Grebinski JW, Leckta T. Catalytic, enantioselective bifunctional inverse electron demand hetero-Diels-Alder reactions of ketene enolates and o-benzoquinone diimides. J Am Chem Soc. 2006;128:13370–13371. doi: 10.1021/ja065754d. [DOI] [PubMed] [Google Scholar]
- 30.Tiseni PS, Peters R. Catalytic asymmetric formation of δ-lactones by [4+2] cycloaddition of zwitterionic dienolates generated from α,β-unsaturated acid chlorides. Angew Chem Int Ed. 2007;46:5325–5328. doi: 10.1002/anie.200700859. [DOI] [PubMed] [Google Scholar]
- 31.Bappert E, Muller P, Fu GC. Asymmetric [3 + 2] annulations catalyzed by a planar-chiral derivative of DMAP. Chem Commun. 2006;42:2604–2606. doi: 10.1039/b603172b. [DOI] [PubMed] [Google Scholar]
- 32.Robinson E, Fallan C, Simal C, Slawin A, Smith AD. Anhydrides as α,β-unsaturated acyl ammonium precursors: Isothiourea-promoted catalytic asymmetric annulation processes. Chem Sci. 2013;4:2193–2200. [Google Scholar]
- 33.Böttcher T, Sieber SA. β-Lactams and β-lactones as activity-based probes in chemical biology. Med Chem Commun. 2012;3:408–417. [Google Scholar]
- 34.Taylor JE, Bull SD, Williams JMJ. Amidines, isothioureas, and guanidines as nucleophilic catalysts. Chem Soc Rev 2012. 2012;41:2109–2121. doi: 10.1039/c2cs15288f. [DOI] [PubMed] [Google Scholar]
- 35.Yang X, Birman VB. Homobenzotetramisole-catalyzed kinetic resolution of α-Aryl-, α-Aryloxy-, and α-Arylthioalkanoic acids. Adv Synth Catal. 2009;351:2301–2304. doi: 10.1002/adsc.200900451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blanchette MA, et al. Horner-Wadsworth-Emmons reaction: use of lithium chloride and an amine for base-sensitive compounds. Tetrahedron Lett. 1984;25:2183–2186. [Google Scholar]
- 37.Rycke ND, Couty F, David ORP. Increasing the reactivity of nitrogen catalysts. Chem Eur J. 2011;17:12852–12871. doi: 10.1002/chem.201101755. [DOI] [PubMed] [Google Scholar]
- 38.Liu G, Shirley ME, Romo D. A diastereoselective, nucleophile-promoted aldol-lcatonization of ketoacids leading to bicyclic-β-lactones. J Org Chem. 2012;77:2496–2500. doi: 10.1021/jo202252y. [DOI] [PubMed] [Google Scholar]
- 39.Birman VB, Li X. Homobenzotetramisole: An effective catalyst for kinetic resolution of aryl-cycloalkanols. Org Lett. 2008;6:1115–1118. doi: 10.1021/ol703119n. [DOI] [PubMed] [Google Scholar]
- 40.Tsuji H, Yamada T, Minami I, Yuhara M, Nisar M, Shimizu I. Palladium-catalyzed decarboxylation-allylation of allylic esters of α-substituted β-keto carboxylic, malonic, cyanoacetic, and nitroacetic acid. J Org Chem. 1997;52:2988–2995. [Google Scholar]
- 41.Lang A. Gibberellins: structure & mechanism. Ann Rev Plant Physiol. 1970;21:537–570. [Google Scholar]
- 42.Wilds AL, Harnik M, Shimizu RZ, Tyner DA. Methods for total synthesis of steroids. XVII Δ14–16-Keto steroid approach to ring d H Introduction of 17-caboxy group Synthesis of 14α,17β and 14β,17α isomers of rac-Estra-5(10),6,8-triene-17-carboxylic acid. J Am Chem Soc. 1965;88:799–804. doi: 10.1021/ja00956a036. [DOI] [PubMed] [Google Scholar]
- 43.Minami I, Nisar M, Yuhara M, Shimizu I, Tsuji M. New methods for the syntheses of α,β-unsaturated ketones, aldehydes, and nitriles by the palladium-catalyzed reactions of allyl β-oxo esters, allyl 1-alkenyl carbonates, and allyl α-cyano esters. Synthesis. 1987:992–999. [Google Scholar]
- 44.Candish L, Lupton DW. N-Heterocyclic carbene-catalyzed Ireland-Coates Claisen rearrangement: synthesis of functionalized β-lactones. J Am Chem Soc. 2013;135:58–61. doi: 10.1021/ja310449k. [DOI] [PubMed] [Google Scholar]
- 45.Seayad J, List B. Multicomponent Reactions. Wiley-VCH; 2005. pp. 277–299. [Google Scholar]
- 46.Zhang W, Romo D. Transformation of fused bicyclic and tricyclic β-lactones to fused γ-lactones and 3(2H)-furanones via ring expansions and O-H insertions. Org Lett. 2007;9:2111–2114. doi: 10.1021/jo7012934. [DOI] [PubMed] [Google Scholar]
- 47.Wang Y, Tennyson R, Romo D. β-Lactones: Intermediates for natural product total synthesis and new transformations. Heterocycles. 2004;64:605–658. [Google Scholar]
- 48.Ranieri B, Robles O, Romo D. Concise synthesis of the isothiourea organocatalysts homobenzotetramisole and derivatives. J Org Chem. 2013;78:XXX–XXX. doi: 10.1021/jo400603n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.