

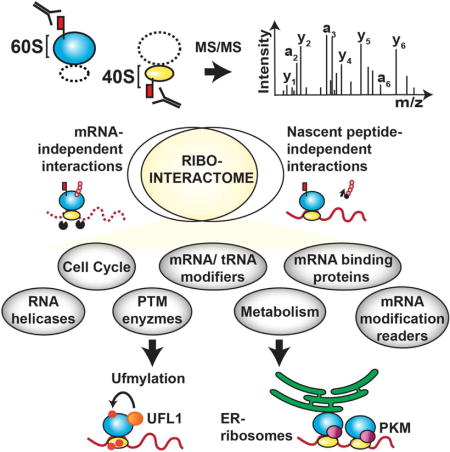
Summary
During eukaryotic evolution, ribosomes have considerably increased in size forming a surface exposed ribosomal RNA (rRNA) shell of unknown function, which may create an interface for yet uncharacterized interacting proteins. To investigate such protein interactions, we establish a ribosome affinity purification method that unexpectedly identified hundreds of ribosome associated proteins (RAPs) from categories including metabolism, cell cycle, as well as RNA and protein modifying enzymes that functionally diversify mammalian ribosomes. By further characterizing RAPs, we discover the presence of ufmylation, a metazoan-specific posttranslational modification, on ribosomes and define its direct substrates. Moreover, we show that the metabolic enzyme, pyruvate kinase muscle (PKM), interacts with sub-pools of endoplasmic reticulum (ER)-associated ribosomes, exerting a non-canonical function as an RNA binding protein in the translation of ER-destined mRNAs. Therefore, RAPs interconnect one of life’s most ancient molecular machines with diverse cellular processes, providing an additional layer of regulatory potential to protein expression.
Graphical abstract
Introduction
Although the ribosome plays a universal role in translating the genome across all kingdoms of life, mammalian ribosomes have substantially increased in size during eukaryotic evolution. In particular, ribosomes of higher eukaryotes have a unique solvent-accessible outer rRNA shell (Noeske and Cate, 2012) which may act as a platform for additional unknown interacting proteins. A few well-characterized examples suggest the importance of such ribosome interacting proteins in control of translation specificity and fidelity. For instance, the RNA binding protein (RBP) FMRP appears to bind directly to the assembled, 80S ribosome (Chen et al., 2014), and represses the translation of specific subsets of mRNAs (Darnell et al., 2011). Another example is the ubiquitin ligase Listerin which associates directly with the ribosomal large subunit as part of a quality control pathway to regulate the degradation of nascent proteins when translation is interrupted (Shao et al., 2015). Although additional ribosome interacting proteins may endow ribosomes with functional diversity and the potential for ribosome heterogeneity in subcellular space, we lack a comprehensive identification of such proteins within the complex cellular milieu of mammalian cells.
The major challenge in addressing this problem is the lack of methods to selectively isolate cytosolic mammalian ribosomes. While mass spectrometry (MS) of sucrose gradient fractions following ultracentrifugation has been attempted (Figure S1A) (Reschke et al., 2013), this approach carries many caveats. First, although this approach does enrich for ribosomes, complexes that are not bona fide components of the ribosome co-migrate in sucrose gradient fractions due to similar centrifugation properties. In fact, similar cytoplasmic lysis and centrifugation methods are used to isolate membrane fractions or centrosomes (Girard et al., 2005; Reber, 2011). Indeed, we have observed clathrin complexes and ribonucleoprotein particles such as vault complex components present within polysome fractions independently of ribosomes (Figure S1B). Second, the long durations of ultracentrifugation and sucrose gradient fractionation (4–20 hours) used may not preserve functional states of ribosomes and may cause the loss of weaker, yet biologically meaningful interactions.
Here, to determine the magnitude and the components of the mammalian ‘ribo-interactome’, we endogenously tagged both the small and large ribosomal subunits in mouse embryonic stem cells (ESCs) and performed affinity enrichment for each of the tagged ribosomal subunits to define the intersection of the two separate ribosomal subunit datasets. This has led to the identification of what we term ribosome associated proteins (RAPs), which fall under unexpected functional categories such as energy metabolism, cell cycle, and key protein and RNA modification enzymes. We further concentrate on two examples of RAPs and define their biological functions. Our findings show that UFL1 is an enzyme which leads to a metazoan specific PTM on ribosomes. Our data also reveal that PKM is a RAP found enriched at ER-ribosomes, which controls the translation of ER-destined mRNAs. These findings highlight the potential diversity in ribosome composition at the level of RAPs within key subcellular locations. Together this study identifies hundreds of RAPs with the potential to expand the functional role of the ribosome in diverse cellular processes and to define new layers of control to protein expression.
Results
A ribosome tagging method to define the ES cell ribo-interactome
To precisely purify mammalian ribosomes from cytoplasmic extracts, we aimed to tag ribosomal proteins (RPs) endogenously as tagged RPs, when overexpressed, do not efficiently incorporate into translating ribosomes and can exist in free complexes (unpublished results). To date, the only endogenously tagged RP is eL22-HA which has been used to isolate ribosome-bound mRNAs in a mouse model (Sanz et al., 2009). However, when we generated ESCs from these mice, eL22-HA is also found present in free fractions that do not contain assembled ribosomes (Figure S2A), consistent with the reported extra-ribosomal functions of eL22 (Battle et al., 2006). In order to overcome this caveat, we tagged multiple, surface accessible candidate RPs in ESCs using CRISPR/Cas9-mediated genome editing (Doudna and Charpentier, 2014). This enabled the addition of a small FLAG-tag to the large ribosomal subunit gene eL36, and the small ribosome subunit gene eS17 seamlessly at their native 3′ C-termini. Unlike eL22-HA, FLAG-tagged eL36 and eS17 RPs are not found in free, non-ribosomal pools and are incorporated into functional ribosomes (Figure S2B). To assess potential background, cells stably expressing the FLAG-tagged GFP protein at similar levels to either of the RPs were also generated (Figure 1A). We initially performed a cytoplasmic enrichment under physiological salt concentrations followed by higher salt washes and FLAG peptide elution (Figure 1B). FLAG-immunoprecipitation (IP) samples from two, distinct large and small subunit RP FLAG-tagged cells as well as FLAG-GFP cells, were analyzed by LC/MS-MS and evaluated using SAINT analysis (Mellacheruvu et al., 2013), with ribosome-interactors defined as proteins with SAINT score ≥ 0.56 (false discovery rate (FDR) <= 0.08) and a second cutoff of ≥ 4 fold change (FC) enrichment, which encompassed all of the detectable RPs that make up the two ribosome subunits (Figure 1B, Table S1).
Figure 1.

Affinity enrichment of mammalian ribosomes defines the ribo-interactome in ESCs.
A, In mouse ESCs, eL36 and eS17 are endogenously tagged with FLAG using CRISPR-Cas9 endonuclease system denoted by scissors. In addition to the endogenously FLAG tagged RPs, cells stably expressing different levels of GFP-FLAG transgenes were generated using PiggyBac transposon-mediated stable integration. GFP-FLAG transgene clone 3, expressing FLAG at similar levels to the tagged RPs, was chosen for further analyses.
B, Strategy to define the mammalian ribo-interactome. GFP-FLAG cells are used to assess the background of the ribosome affinity enrichment strategy. Cytoplasmic lysates from eL36-FLAG, eS17-FLAG, and GFP-FLAG cells are subjected to FLAG IP under similar conditions, and IPs are analyzed by LC/MS-MS. Average, normalized spectral abundance factor (NSAF) of RPs from three biological replicates of either eL36-FLAG or eS17-FLAG are shown. See Table S1.
C, Maximum SAINT probability scores and fold enrichment of eL36 and eS17 experiments are shown. SAINT probability of 0.56 corresponds to 0.08 FDR. 60S RPs are colored in blue, 40S RPs in yellow.
D, eL36 specific interactors are defined as those present in all eL36 biological replicates with at least 2 unique peptides, but not present in any of the eS17 biological replicates. The overlap between eL36 and eS17 datasets is defined as the proteins present at the intersection of at least one eL36 and one eS17 replicate with a SAINT score >=0.56. For GO biological process analysis, Benjamini–Hochberg FDR cutoff of 8% and fold enrichment >=4 are used. Examples of enriched GO categories are shown, for a full list see Table S2. The number of identified genes in each GO category is shown in comparison to the number of genes in each GO category.
The MS analysis using eS17-FLAG cells resulted in the enrichment of small and large subunits to the same degree as eL36-FLAG cells did indicating that the cytoplasmic isolation and MS are mainly covering fully assembled, translationally-competent 80S ribosomes (Figure 1B). In addition, this dataset also contains 60S and 40S exclusive interactors (Figure 1C, Table S1) including important regulators of translation previously ascribed to individual subunits. For instance, eIF6, which is identified specifically within the eL36-MS data prevents ribosomal subunit association by binding to the 60S subunit (Brina et al., 2015). Rio2 kinase, which is identified specifically by eS17-MS, is known to block the ribosomal mRNA exit channel to prevent premature translation initiation (Strunk et al., 2011).
The overlap between eL36-FLAG and eS17-FLAG datasets resulted in the identification of ~ 400 proteins that in addition to the RPs include components of the canonical translation machinery such as translation initiation and elongation factors (Figure 1D, Table S3). To characterize the representative functional features of the RAPs identified, gene ontology (GO) analysis was performed using the mouse ESC whole cell proteome as a background (Graumann et al., 2008). Surprisingly, in addition to the canonical translation machinery and protein folding functional categories, there is an enrichment of proteins controlling metabolism and cell cycle that may functionally interconnect the mammalian ribosome to diverse and important cellular processes (Figure 1D, Table S3, and see below). Moreover, this dataset contains multiple RNA helicases that can unwind secondary mRNA structures and also proteins involved in mRNA processing such as non-sense mediated decay, mRNA transport, splicing, and microRNA mediated gene silencing. Together, these findings reveal a new landscape of RAPs that either directly associate with mammalian ribosomes or indirectly via mRNA-mediated interactions.
Classification of direct, mRNA-dependent, or nascent peptide-dependent RAPs
We next systematically delineated how many of the identified RAPs (i) directly bind to the ribosome, (ii) are brought to the ribosome by interactions mediated with mRNAs, or (iii) reflect nascent peptide chains. To this end, FLAG-IPs using eL36-FLAG cells were compared to IPs that were performed after RNase digestion or puromycin treatment (Figure 2A). RNase A digestions on FLAG beads resulted in the efficient footprinting of the ribosome by digesting the mRNAs between multiple assembled, 80S subunits (Figure S3A, B). Although RNaseA was chosen as a nuclease as it largely preserved the integrity of ribosomes compared to RNase I (Figure S3A), we cannot however formally exclude that RNaseA may still partially cleave rRNA segments and disrupt interactions that are rRNA mediated. To delineate nascent peptide independent RAPs, cells were treated with puromycin, a tRNA analog that is incorporated into the C- termini of nascent peptides leading to their release from the ribosome (Pestka, 1971), at conditions previously shown in vivo to release nascent peptides (Wu et al., 2016; Yan et al., 2016). Under these conditions, terminated peptides that are puromyclated were detected in the cytoplasmic lysate but could not be detected after ribosome IP (Figure S3B). A10-plex TMT strategy was used to label peptides from untreated, RNase A digested, and puromycin treated samples, three biological replicates each, with different TMT tags (Thompson et al., 2003). For the quantification of the data, an additional peptide isolation and fragmentation event (MS3 scan) which leads to a more accurate estimate of relative protein levels than MS2-based quantification was used (Ting et al., 2011). Using this strategy, a high correlation between biological replicates (r=0.93 to 0.99) was achieved (Figure 2A, Figure S3C, Table S3).
Figure 2.

The quantitative TMT experiment to determine RNase- and puromycin-dependent RAPs.
A, Overview of the quantitative-MS experiment approach. Three biological replicates (BR) are used for each control, RNase, and puromycin-treatment. Pearson correlation coefficients for each BR within a treatment are calculated using normalized log2 TMT intensities.
B, Scatter plot of normalized log2 RNase/control ratios versus P-values. FDR and negative predictive values (NPV) are estimated by mixture modeling of test statistics (Efron, 2004). 14% of the interactions are estimated to be RNase-dependent (Figure S4). At 99% NPV, 438 interactions are estimated to be RNase-independent. Representative examples of RNase-dependent ribosome-interactions are highlighted. See Table S3.
C, Scatter plot with normalized log2 puromycin/control ratios versus P-values. Representative examples of puromycin dependent interactions are highlighted.
To classify mRNA dependent and independent RAPs, we empirically modeled the null distribution of the test statistics in the RNase treatment which allowed us to discern ~14% of the total RAPs that lose ribosome interaction upon mRNA digestion (50 proteins at FDR <0.15) from 438 RAPs that are insensitive to RNase digestion (negative predictive value (NPV) >0.99) (Figure 2B, Figure S4A). Although it is possible that proteins that lost ribosome interaction upon RNA digestion are interacting with mRNAs independent of the ribosome, they include previously established, translation-related proteins such as poly(A) binding proteins, LARP1, LARP4, and eIF2AK3 (Figure 2B). RNase-independent interactors included all detectable RPs, and it encompassed the majority of the dataset. Unlike the RNase experiment, puromycin treatment resulted in only a minor fraction of the RAPs to lose their interaction (3% compared to 14% upon RNase treatment), suggesting that nascent peptides were rarely falsely identified as RAPs in our dataset (Figure 2C, Figure S4A). This is in agreement with the N to C terminal coverage of MS-identified peptides that do not show any bias towards the N terminus (Figure S4B). In total, 4 puromycin treatment dependent proteins were identified at FDR<0.15 which include HSPA8 and DNAJC21 chaperones and proteins that are known to make functional contacts with ribosomes that are dependent on tRNAs or nascent peptides. For instance, recruitment and further interactions of NEMF to the large ribosomal subunit, which is critical for protein quality control, is dependent on its interaction with the peptidyl-tRNA (Shao et al., 2015). Therefore, these quantitative-MS experiments investigating mRNA and nascent peptide dependency permit us to gain preliminary insights into the mechanisms of potential translation regulation by the RAPs.
Landscape of direct ribosome interactors
We defined the intersection of the RNase-independent (NPV>=0.99) and puromycin-independent (NPV>=0.99) proteins as the “ribo-interactome”, which is comprised of ~430 proteins including RPs, translation initiation and elongation factors (Figure 3A). Moreover, RBPs that have known roles such as reading cis-regulatory elements in mRNAs, unwinding mRNA structures, and/or controlling mRNA stability, interact with ribosomes directly, independent of mRNAs. For instance, the ribo-interactome contains the RNA helicase DDX1 which can interact with the mammalian tRNA ligase RTCB to mediate cytoplasmic splicing of the Xbp1 mRNA (Jurkin et al., 2014; Popow et al., 2011). Another example is CNOT1/3, components of the CCR4-NOT complex that have diverse roles in mRNA metabolism (Shirai et al., 2014), which could act as anchor points on the ribosome by recruiting mRNA dependent RAPs (e.g. components of the miRNA machinery) to integrate post-transcriptional mRNA regulation with translation. This dataset also encompasses the well-characterized RBP FMRP (Chen et al., 2014; Darnell et al., 2011), loss of which leads to Fragile X Syndrome, as well as FMRP binding proteins with much less explored functions in translation. VCP and FUS are other examples of disease-related RBPs and are involved in the pathogenesis of the neurological disease amyotrophic lateral sclerosis (ALS) (Lagier-Tourenne and Cleveland, 2009). Future studies are needed to determine whether they could link ribosomes to the emerging dysfunction of translation control in ALS (Coyne et al., 2014).
Figure 3.
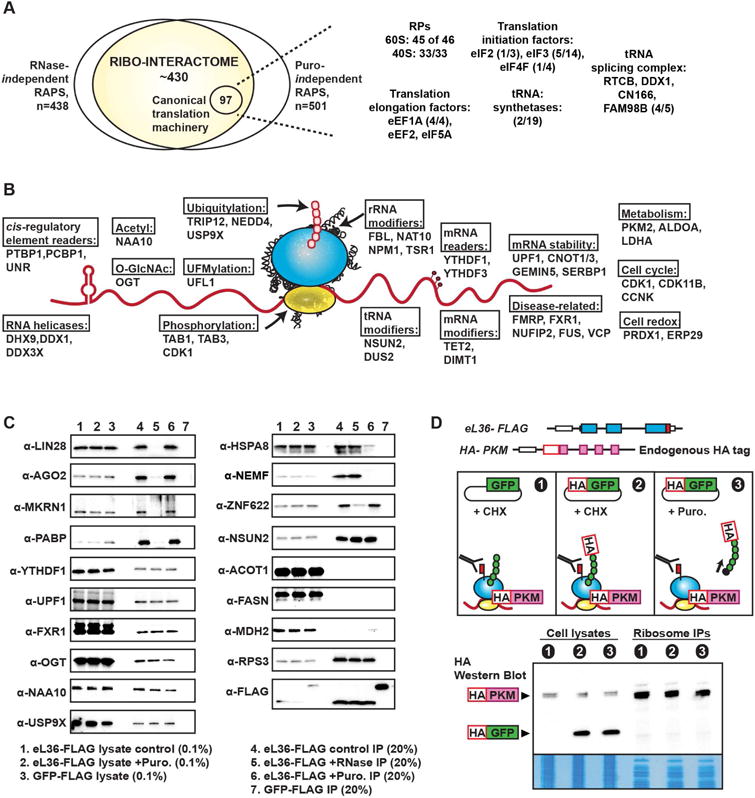
The ribo-interactome consists of diverse functional groups.
A, The ribo-interactome is defined as the intersection of RNase-independent and puromycin-independent interactions. The number of identified proteins related to canonical translation machinery in the MS experiments is presented along with the known number of factors in each class.
B, The ribosome as a hub for interactions with a multitude of proteins with diverse functions. Representative examples of direct ribosome interactors found in each functional group are presented. In the schematic, the pink circles represent the nascent peptides; red circles on the mRNA represent mRNA modifications.
C, Validation of representative examples from ribo-interactome. Western blots of the interactors from control, RNase-treated, and puromcyin-treated ribosome IP samples, along with the cytoplasmic lysates which are used as input control for these IPs.
D, PKM is endogenously tagged with HA within eL36-FLAG ES cells. Untagged GFP and HA-tagged GFP are further transfected into these cells. GFP does not interact with ribosomes, and is used as a negative control for possible ribosome interactions. GFP nascent chains are depicted by green circles. Western blots of the cell lysates and ribosome IPs are shown alongside Coomassie stained fractions. 0.01% of cytoplasmic lysates are used as input and 20% of the IPs are run in the western blot.
The ribo-interactome includes enzymes that modulate reversible, post-transcriptional mRNA modifications that are suggested to affect translation, as well as proteins that can read these modifications (Dominissini et al., 2016; Wang et al., 2015) (Figure 3B). For instance, our dataset includes two specific readers (YTHDF1 and YTHDF3) but not any of the writers of N6-methyladenosine modifications, and also includes TET2, which hydroxymethylates RNA resulting in differential translation of such modified mRNAs (Delatte et al., 2016) (Figure 3B). In addition to RNA modification enzymes, enzymes that catalyze or reverse diverse protein modifications (e.g. acetylation, O-GlcNAcylation, phosphorylation, and ubiquitylation) are direct RAPs and could modify nascent proteins and/or the translation machinery itself. Indeed, PTMs on the ribosomes are emerging as dynamic events in response to multiple stimuli and stress, although enzymes that could facilitate these modification remain largely unknown (Simsek and Barna, 2017). Therefore, the PTM enzymes such as ubiquitin ligases and deubiquitylating enzymes as well as kinases and phosphatases that directly interact with the ribosome may link translation specificity with upstream signaling pathways and contribute to ribosome heterogeneity.
Lastly, the ribo-interactome contains proteins belonging to functional categories such as cell cycle, cell redox homeostasis, and metabolism (Figure 3B). One of the most unanticipated categories of proteins within the ribo-interactome is glucose metabolism enzymes which have the potential to generate metabolic intermediates of cellular building blocks such as nucleic acids and amino acids (Shyh-Chang et al., 2013). The metabolic enzymes in this category appear to be a specific subset. For example, additional metabolism enzymes such as ACOT1, FASN, and MDH2 are not present in the ribo-interactome dataset and serve as negative controls (Figure 3C). To further validate our initial findings from the RNase A and puromcyin treated MS experiments, proteins in the categories mentioned above were examined via immunoblotting following eL36-FLAG IPs with either RNase or puromycin treatments (Figure 3C). Our findings were orthogonally validated by treating cell lysates with EDTA or RNase A and comparing the sucrose gradient fractionation profiles of the tested RAPs to those of RPs (Figure S5, S6). RAPs tested that are mRNA-dependent upon RNase A digestion no longer accumulated at the 80S, consistent with the fact that mRNAs were digested away. To further assess whether an abundant protein can be falsely detected as a RAP, the PKM protein, one of the metabolism related RAPs, was endogenously tagged at its N terminus with HA in eL36-FLAG cells (Figure 3D). To use the same antibody for detection, HA-GFP was transiently expressed at higher levels than HA-PKM in the HA-PKM; eL36-FLAG cells. Although HA-GFP could be observed within cell lysates at higher levels than HA-PKM, HA-GFP could not be detected in the ribosome IP (Figure 3D). This is an independent experiment that is consistent with the puromycin results suggesting that although nascent peptides are present at translating ribosomes, they are far less abundant compared to the RAPs and that even if proteins are highly overexpressed, they are unlikely to be falsely identified.
A new PTM at the ribosome: Ufmylation
As part of the ribo-interactome, we identified UFL1, which is the only known enzyme that determines the target specificity for the metazoan-specific PTM, ufmylation (Zhang et al., 2015). Ufmylation is a ubiquitin-like PTM in which Ufm1, an 85-amino acid (9.1 kDa) protein, is conjugated to target proteins via a single enzyme cascade (Figure 4A). Although the significance of ufmylation is underlined by its essential roles in embryonic development and erythroid differentiation, research on this modification is still in its infancy (Tatsumi et al., 2011; Yoo et al., 2014; Zhang et al., 2015). By using N-terminally HA tagged UFL1 and an antibody that detects UFL1 at its C-terminus, we find full length UFL1 present in control, RNase, and puromycin treated IPs (Figure 4B). To determine whether any RAPs are ufmylated, we blotted the eL36 ribosome IP samples with a ufmylation modification-specific antibody. In comparison to the control GFP IP, specific bands corresponding to ufmylated proteins were observed (Figure 4B). Moreover, the ufmylation signal as well as UFL1 itself is not detectable at non-ribosome containing, free fractions, but is exclusively enriched at fractions corresponding to the 60S and 80S (Figure 4C).
Figure 4.

The ufmylation enzyme UFL1 interacts with ribosomes and modifies key components of the translation machinery.
A, Schematic of the ufmylation cascade.
B, UFL1 is tagged endogenously with HA at its N terminus. The UFL1 antibody recognizes the C-terminal portion of human UFL1 protein. FLAG IPs for both control GFP-FLAG and eL36-FLAG cells are performed. Both the GFP- FLAG input and IP as well as the eL36-FLAG input and IP are blotted with HA, Ufl1, and Ufm1-specific antibodies.
C, Sucrose gradient fractionation is performed and fractions are blotted for either the Ufm1 modification or the E3 ligase enzyme, UFL1. UV signal at 260 detects RNA and indicates rRNA abundance across fractions.
D, Schematic that outlines the two-step affinity enrichment to identify ufmylated substrates at the ribosome. Fold changes (FC) of each His-Ufm1 IP compared to background IP is shown. 4-fold FC is used as a cutoff and proteins above this cutoff are marked. See Table S4. 80S human ribosome structure with the positions of uS3 (green), uS20 (orange), uL16 (dark blue), mRNA (red), E-site tRNA (dark grey), and EEF2 (black) are indicated. The ribosomal RNAs are shown in light blue (60S) or yellow (40S). PDB: 4V6X with mRNA superimposed are from PDB: 4KZZ.
Although prior studies have attempted to identify ufmylated proteins, these studies did not contain any RPs or proteins in the ribo-interactome (Tatsumi et al., 2010; Yoo et al., 2014). To selectively identify only the ufmylated RAPs, but not proteins that can recognize and bind to ufmylated proteins, His-Ufm1 was expressed in eL36-FLAG cells to perform a subsequent IP step under denaturing conditions (Figure 4D, Table S4). The LC/MS-MS analyses of the two-step purification strategy led to the identification of two small subunit RPs, uS3 and uS10, as well as a large subunit protein uL16. The translation initiation factor, eIF6, that exclusively interacts with the 60S ribosome to regulate subunit joining (and is part of our eL36-exclusive dataset (Table S1)) was also identified (Brina et al., 2015). The molecular weights of the proteins identified in the MS analysis matched the expected molecular weights of ufmylated proteins observed by blotting the ribo-interactome for ufmylation (Figure 4D). Interestingly, on the cryo-EM structure of the human ribosome (Anger et al., 2013), uS3 and uS10 small subunit RPs are immediately next to each other on the solvent exposed surface of the 40S, in close vicinity of the mRNA entry channel (Figure 4D). Identification of these small subunit RPs, even though the ufmylation signal is absent in 40S fractions implies that ufmylation of these RPs is likely to occur on assembled 80S ribosomes. uL16 is also on the same interface with uS3 and uS10 (Figure 4D), suggesting that the ufmylation of uS3, uS10, uL16 and eIF6 may work in concert to coordinate subunit joining and mRNA interactions. Future studies are required to further dissect the functional consequences of this specific modification on the ribosome.
Pyruvate Kinase: a critical metabolism regulator and a direct ribosome interactor
From the metabolism related RAPs, we chose to functionally analyze PKM, which catalyzes the last rate-limiting step in glycolysis by converting phosphoenolpyruvate (PEP) and ADP to pyruvate and ATP (Figure 5A) (Israelsen and Vander Heiden, 2015). Multiple studies have underscored PKM’s importance in cancer and cellular differentiation (Israelsen and Vander Heiden, 2015). Alternative splicing of two mutually exclusive exons of the PKM gene results in two different isoforms, PKM1 and PKM2, and PKM2 is the dominant isoform in ESCs as well as tumor cells (Shyh-Chang et al., 2013). We generated mouse ESCs that allowed inducible Cre-recombinase-mediated deletion of the PKM2 isoform-specific exon (Israelsen et al., 2013) (Figure S7A,B). Using these cells, when PKM2 levels were lowered, PKM1 levels were increased overall and the presence of PKM1 at ribosome pools was increased as well (Figure S7B), suggesting that both PKM2 and PKM1 can bind to the ribosome.
Figure 5.
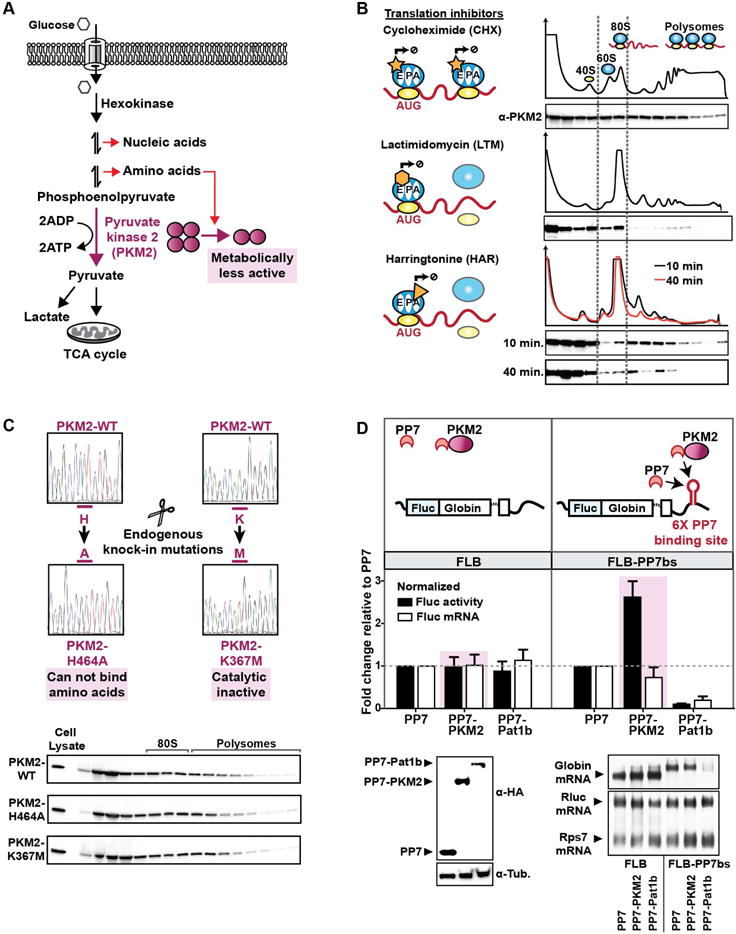
Characterization of ribosome binding by the metabolism enzyme PKM2.
A, Schematic for the glycolysis pathway.
B, Sucrose gradient fractionation for PKM2. ESCs are treated with translation elongation inhibitors that act at different stages of translation (inhibitors denoted by yellow geometric shapes). As the duration of the HAR treatment increases, the characteristic polysome UV signal decreases, since uninhibited ribosomes will ‘run-off’ the mRNA as depicted by the lighter blue shaded ribosome cartoon. Drug treatments were performed for short durations to capture immediate effects. CHX treatment was for 2 mins; LTM treatment was for 10 mins, and HAR treatments was for 10 or 40 mins. Protein levels of PKM2 are shown in each fraction.
C, Endogenous homozygous knock-in mutations are generated using the CRISPR-Cas9 endonuclease system as denoted by scissors. Sequencing chromatograms of the wild type and mutated PKM2 loci confirm mutations are in homozygosity. Sucrose gradient fractions are precipitated and blotted for PKM2.
D, Schematic representation of FLB and FLB-PP7bs reporters. Firefly luciferase activity is normalized to cotransfected Renilla luciferase control and represented relative to PP7 alone. Northern blots are performed with an exon-junction probe crossing the rabbit β-globin intron and are normalized to Renilla control. Rps7 is the loading control. The plots of luciferase activity show the mean of 6 biological replicates. The mRNA levels detected by Northern blots are the mean of 4 biological replicates. Error bars in both represent the standard deviation.
To gain further mechanistic insight into PKM binding to ribosomes, sucrose gradient fractionation experiments in the presence of specific translation inhibitors were performed. Cycloheximide (CHX) blocks the exit of uncharged tRNAs by binding to the E-site of the ribosome (Garreau de Loubresse et al., 2014) and thereby ‘freezes’ ribosomes along mRNAs in the act of translation. PKM2 is present in the free subunits, 80S, and polysome fractions under these conditions (Figure 5B). Lactimidomycin (LTM) binds to the E-site of the ribosome similarly to CHX (Garreau de Loubresse et al., 2014); however LTM will act only on the first 80S positioned at the start codon, due the presence of a bulky side group. In the presence of LTM, PKM accumulates at the 80S peak and decreases at the polysomes, revealing that PKM2 interacts with translating ribosomes. Finally, upon harringtonine (HAR) treatment, which binds and prevents entry of the charged-tRNA at the A-site (Garreau de Loubresse et al., 2014). PKM is instead depleted from the 80S fractions, suggesting that blocking the A-site prevents PKM2 interaction with the ribosome (Figure 5B). These studies suggest unexpected specificity for PKM interactions with elongating ribosomes in proximity to the A-site.
Next, to determine whether PKM2’s catalytic activity is important for its interaction with the ribosome, we generated ES cells with a homozygous PKM-K367M knock-in mutation that mutates the ADP-binding site of PKM necessary for its enzymatic activity (Le Mellay et al., 2002). K367M did not affect PKM’s interaction with the ribosome (Figure 5C). PKM has also been shown to bind amino acids, and mutating the residue H464 to alanine abrogates any amino acid binding (Chaneton et al., 2012). PKM-H464A knock-in mutations did not affect overall PKM2 protein stability and did not change its interaction with the translating ribosomes (Figure 5C). These findings demonstrate that neither PKM’s catalytic activity nor its ability to bind amino acids is critical for its interaction with the ribosome.
PKM is a translational activator that binds to specific mRNAs and regulates their translation
To examine PKM’s potential role in translation uncoupled from its role in metabolism, we used a tethered function assay that brings PKM2 in close proximity to a reporter mRNA 3′untranslated region (UTR). The PP7 coat protein was fused to the N terminus of PKM2 and was expressed alongside the FLB-PP7bs reporter, allowing PKM2 to be recruited to reporter mRNA through PP7-PP7bs interactions. When PKM2 was tethered to the FLB-PP7bs reporter, luciferase activity was increased ~2.5 fold, whereas the steady state mRNA levels did not change (Figure 5D). Importantly, this effect only occurred when PKM2 was localized to the reporter and not when the FLB reporter lacking the PP7bs was used (Figure 5D). These findings suggest that tethered PKM functions as a translation activator, unconstrained by PKM’s metabolic function. This prompted us to test whether PKM can bind to specific classes of endogenous mRNAs. Recently, metabolic enzymes, particularly glycolysis enzymes, have been found to bind mRNAs in high-throughput screens of RBPs (Castello et al., 2012; Liao et al., 2016; Matia-Gonzalez et al., 2015), although their physiological function has remained largely unknown.
To identify possible PKM2-associated RNAs systematically in vivo, we endogenously tagged the PKM2 protein with FLAG-HA employing CRISPR/Cas9 (Figure S7C) and performed FAST-iCLIP (Flynn et al., 2015) (Figure 6A). Tagging the PKM gene at either the N or C terminus with FLAG-HA did not affect PKM2 polysome association (Figure S7C) and we employed cells in which PKM2 was tagged at the C-terminus for further analysis. iCLIP identifies direct protein-RNA complexes by combining ultraviolet (UV) crosslinking, IP, and high-throughput sequencing (Huppertz et al., 2014). PKM2 iCLIP captured only the in vivo crosslinked, physiological RNA targets, such that if UV crosslinking was not used, stringent washes following two consecutive IPs resulted in almost no RNA capture (Figure S7D). PKM iCLIP analyses revealed that the two largest classes of PKM2 RNA targets are ribosomal RNAs (29% of reads) and protein coding RNAs (23% of reads) (Figure 6B, Figure S7E). Consistent with our MS analysis, the PKM2 iCLIP reads mapped to specific sites on 18S and 28S mature rRNA. Interestingly, a specific signal is observed at the tip of Helix 38 of 28S rRNA which is known as the ‘A-site finger (ASF)’ since it protrudes into the A-site and during the decoding process interacts directly with the A-site tRNA, making it a significant site for translation regulation (Budkevich et al., 2011) (Figure 6B). Thereby, the iCLIP results in conjunction with the LTM/HAR sucrose gradient fractionation experiments (Figure 5B) suggest that PKM2 binds to a specific location in the vicinity of the A-site on the ribosome.
Figure 6.

PKM2 directly binds and regulates translation of target mRNAs that are commonly translated at the ER.
A, PKM1/2 is endogenously tagged seamlessly with a C-terminal tandem FLAG-HA tag. Schematic of PKM2-FAST iCLIP experimental flow.
B, Percentage of the total iCLIP reads for various RNA classes. Positions of PKM2 crosslinks on the mature rRNA region is shown. ‘Others’ refer to U1, U2, U6 and other snoRNAs. Diagram for the A-site finger is taken from Comparative RNA Web (http://www.rna.ccbb.utexas.edu). Canonical base pairs are depicted with (-), GU wobble base pairs with (.). The nucleotide corresponding to the highest peak in the mature rRNA region, signifying the PKM2 crosslinking site on the A-site finger, is highlighted with yellow.
C, Overview of ribosome profiling workflow for control and PKM knockdown experiments.
D, Scatter plot showing the correlation between PKM2 iCLIP enrichment and translational efficiency change upon PKM depletion. Spearman coefficient (ρ) is presented.
E, Cumulative distributions of translational efficiency change upon PKM-depletion. PKM2 iCLIP targets are divided into four groups according to the degree of their iCLIP enrichment. Strong binders have lower translational efficiency in PKM-depleted cells relative to weak binders (P-value < 2.2 ×10–16 between top 5% and bottom 50% iCLIP targets, Mann-Whitney U test). See Table S5.
F, GO analysis for cellular compartment and biological process for PKM2 iCLIP targets. Adjusted P-values (Benjamini–Hochberg) are shown.
The second-most enriched class of RNAs in the PKM2 iCLIP dataset are protein coding genes. Further classification of the protein coding reads into intron, 5′UTR, CDS and 3′UTR, suggest that PKM2 is enriched at the CDS and 3′UTRs of mRNAs (Figure S7E). To understand the functional significance of PKM2-mRNA interactions, we next performed ribosome profiling (Ingolia et al., 2012) upon siRNA-mediated PKM knockdown to monitor PKM-dependent translational efficiency (TE) changes (Figure 6C, Figure S7F). Ribosome profiling which involves the deep sequencing of ribosome-protected mRNA fragments (ribosome footprints) is a means to monitor translation efficiency genome-wide (Ingolia et al., 2012). We determined translational changes of PKM2 iCLIP mRNA targets upon partial PKM2 knockdown (~70%) by performing ribosome profiling at early time points (within 36 hours of knockdown) (Figure S7F). Strikingly, there is a negative correlation (ρ = −0.327) between TE change upon PKM2 knockdown and PKM2 iCLIP enrichment scores as reflected by the fact that the strongest PKM2 mRNA binders exhibited the greatest decrease in TE upon PKM knockdown (Figure 6D, Figure 6E, Table S5). In other words, ribosome occupancy of PKM2-bound mRNAs tends to be lower in PKM2-depleted cells compared to that in control siRNA-treated cells. These results suggest that PKM2 acts as a translational activator of its direct, physiological mRNA targets.
Direct PKM2 mRNA targets are strongly enriched for genes encoding for the cellular components of the ER, and cell membrane (Figure 6F, Table S2). Furthermore, GO analysis for biological processes reveals significant enrichment for genes encoding for secretory enzymes promoting cell adhesion and enzymes involved in phospholipid and sterol synthesis for which ER is the principal production site (Holthuis and Menon, 2014) (Figure 6F). Interestingly, these mRNAs encoding for membrane and ER-localized proteins, are commonly translated by ER-bound ribosomes. Thus, PKM2 surprisingly binds key classes of mRNAs that are commonly translated at the ER.
PKM is enriched at the ER-ribosomes and localizes mRNAs to the ER
Since mRNAs that directly interact with PKM2 are enriched for putative ER translated transcripts, we next characterized the specific subsets of ribosomes that interact with PKM2. PKM-containing 80S ribosomes were isolated and analyzed by quantitative-MS experiments using SILAC (stable isotope labeling by amino acids in cell culture) optimized for ESCs (Bendall et al., 2008) (Figure 7A). The highest enriched protein specific to PKM2-containing ribosomes was PKM2 itself, highlighting that the enrichment was successful, as well as two additional metabolism enzymes aldolase and thymidylate synthase. Moreover, this analysis also revealed that PKM2-ribosomes are enriched for ER membrane proteins as well as the Sec61 translocon complex, the docking site for ER-bound ribosomes (Voorhees et al., 2014) (Figure 7A, Table S6).
Figure 7.
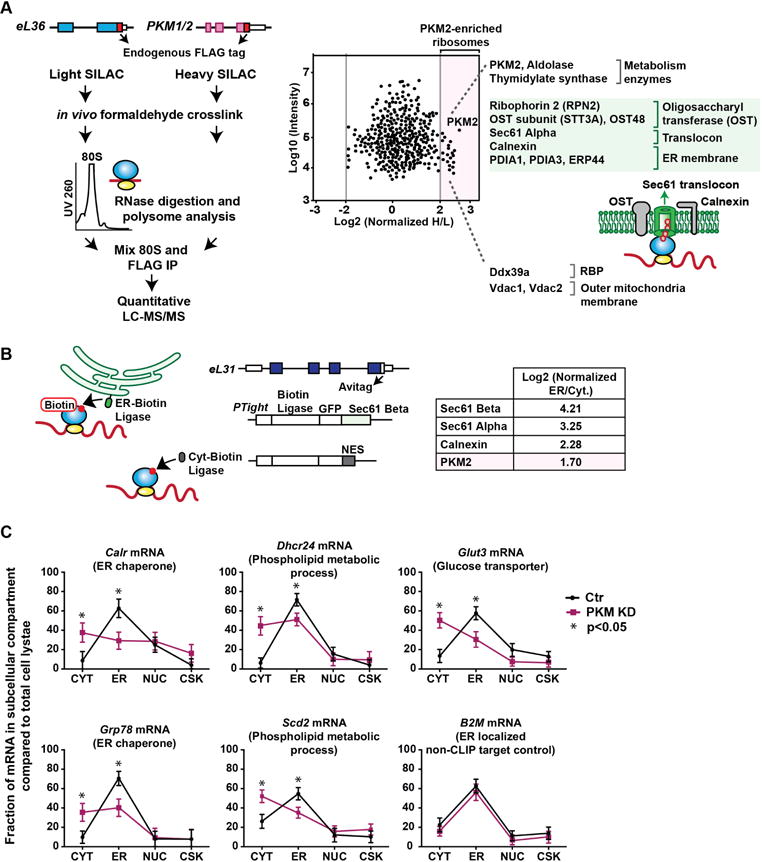
PKM2 is enriched at ER ribosomes and localizes mRNAs to the ER.
A, Quantitative-MS experiment to characterize PKM2 containing ribosomes.
Scatter plot with normalized log2 heavy/light ratios comparing eL36 and PKM2-enriched ribosomes (n=2). Mean 0.61; S.D. 0.50; cut off values for enriched proteins was 2.5 S.D. from the mean and is shown as the grey line lines. See Table S6. Green denotes ER-related components.
B, Subcellular ER-ribosome enrichment. eL31 is tagged endogenously with Avitag at the C terminus. ER-or cytoplasmic- biotin ligase is expressed from an inducible promoter. ER-biotin ligase is attached to the Sec61 Beta protein and cytoplasmic biotin ligase contains a nuclear export signal (NES). PKM2 enrichment is shown relative to known ER resident proteins.
C, Subcellular localization of PKM2 iCLIP targets. The fraction of mRNAs within subcellular fractions normalized to total mRNA are shown. Each fraction value is initially determined by normalizing to an exogenous spike-in RNA control. Data are mean and S.D. of two biological replicates. CYT, cytosol; ER, Endoplasmic reticulum; NUC, nucleus; CSK, cytoskeleton.
To test the hypothesis that PKM2 is enriched at ER-bound ribosomes, a two-component BirA proximity labeling strategy to selectively label ER-bound ribosomes for further MS analysis was employed (Figure 7B). We endogenously tagged eL31 with Avitag that is positioned close to the contact site of the ribosome with the Sec61 complex in ES cells (Voorhees et al., 2014). ES cells expressing a biotin ligase that is either localized to the ER or to the cytoplasm were generated, such that proximity of eL31 to either ligase will enable enrichment of biotin-tagged ribosomes by streptavidin IP. An analogous system, albeit with a distinct RP, has been previously employed in yeast for the purpose of ribosome profiling (Jan et al., 2014). Our current analysis revealed a marked enrichment of Sec61 components and PKM as well as additional RAPs within ER-bound ribosomes (manuscript under preparation, D.S., M.B.). These findings suggest that PKM2 interacts with sub-pools of ribosomes at the ER and reveals heterogeneous ribosomes within the subcellular space.
To understand whether PKM2 has a role in the localization of mRNAs to the ER, we further compared the localization of a subset of PKM2 iCLIP target mRNAs in the ER in comparison to other subcellular compartments upon PKM knockdown. As expected, ER-translated mRNAs such as mRNAs encoding ER-chaperones (Calx and Grp78), lipid metabolism enzymes (Dhcr24, Scd2), and glucose transporter (Glut3), were highly enriched at the ER fraction. Notably, upon PKM knockdown, PKM2 iCLIP target mRNAs were decreased at the ER fraction relative to other compartments. In contrast, a control mRNA that is localized to the ER, but is not a PKM2-CLIP target was unaffected upon PKM2 knockdown (Figure 7C). These results suggest that PKM2 may help localize its target binding mRNAs to the ER fraction.
Discussion
The mammalian ribo-interactome as evident from the directed studies of UFL1 and PKM2 yields unexpected potential regulators of translation and reveals that the ribosome is a dynamic hub of interacting proteins that may link the ribosome with diverse cellular functions and imbue regulatory potential in translating the genome. Further studies will be required to elucidate the functional significance of RAPs from diverse categories including those such as “cell redox homeostasis”, and from “cell cycle”. Recent ribosome profiling studies suggest that a special program of translational control operates during the mammalian cell cycle (Stumpf et al., 2013; Tanenbaum et al., 2015), and the cell cycle related RAPs may, at least in part, help to implement this program. Among the RAPs in the cell redox homeostasis category, PRDX1, has been suggested to act as a chaperone under oxidative stress conditions (Jang et al., 2004). Proteins in the cell redox category may represent chaperones directly associated with the mammalian ribosome that could further link protein folding to the cellular redox environment. The ribo-interactome also contains multiple classes of kinases and ubiquitin ligases. This may suggest, akin to the dynamic, multiple dynamic PTMs that make up the histone code, ribosome PTMs may similarly endow greater heterogeneity and dynamics in translation regulation upon cellular stimuli.
Our studies establish a metazoan-specific PTM ufmylation in mammalian ribosomes. Future studies are needed to elucidate whether ufmylation of critical substrates impacts ribosome subunit joining or contributes to transcript-specific translation. Interestingly, the available knockout mouse models for the enzymes of the ufmylation cascade show specific defects in erythrocyte differentiation and result in embryonic lethality (Tatsumi et al., 2011; Zhang et al., 2015). Notably, haploinsufficiency in multiple RPs result in defects in erythrocyte differentiation as a common phenotype, highlighting the sensitivity of hematopoietic cells to defects in protein production (Narla and Ebert, 2010) and raising the question of whether ufmylation of ribosomes plays a causative role in the phenotypes associated with bone marrow failure.
While metabolism enzymes have been identified in genome-wide screens aimed at identifying RBPs, their functional roles as RBPs have largely been unknown. The independent results from many integrated approaches provide complementary lines of evidence suggesting that PKM is present at sub-pools of ER-ribosomes, binds directly mRNAs translated at the ER, and act as a translation activator for its target mRNAs. In addition to PKM2, multiple other metabolism enzymes directly interact with the ribosome, and future studies will be required to determine whether these metabolism enzymes can work independently or coordinately to regulate ribosome activity. It is intriguing to consider why a glucose metabolism enzyme such as PKM2 is enriched at ER ribosomes in ESCs. ESCs are highly proliferating and it is therefore possible that PKM2 can couple metabolism to the phospholipid and ER chaperone production that is necessary for the expansion of cellular membranes associated with cellular proliferation. Similar to ESCs, cancer cells also have increased biosynthetic needs compared to differentiated adult tissues (Shyh-Chang et al., 2013), and PKM is in fact found mutated in multiple human cancers (Israelsen et al., 2013). As direct inhibitors of protein synthesis hold promise in the treatment of cancers (Bhat et al., 2015), it will be interesting to determine whether PKM mutations found in human cancers may sensitize these cells to specific translational inhibitors.
As highlighted by the example of PKM’s role in ER-ribosomes, our studies reveal that RAPs can complement and diversify the translating potential of subcellular pools of ribosomes. For example, although the ER is a critical subcellular compartment, to our knowledge there are few known examples of RBPs that can affect the translation of ER-targeted messages in mammalian cells with the exception of the translational repressor Lin28a (Cho et al., 2012). In this respect, it will be important to determine whether the translation of spatially localized mRNAs at distinct subcellular environments (e.g. cell membrane, mitochondria and ER) may be facilitated by a different set of RAPs.
Finally, the characterization of the ribo-interactome within ESCs serves as a foundation for numerous lines of additional research. For example, ESCs with the endogenously tagged ribosomes can be readily differentiated into additional cell types, to determine the selective and dynamic association of RAPs during the course of cellular differentiation. Also, the different strategies utilized here can be further applied in combination, for instance to study PTM of ribosomes at different subcellular locations. Thereby, the ribo-interactome dataset along with important functional examples presented in this study paves the way for connecting one of life’s most ancient molecular machines with more intricate control of gene expression.
Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Maria Barna (mbarna@stanford.edu).
Experimental Model and Subject Details
Mouse E14 embryonic stem cells (ESCs) (male) is a gift from Barbara Panning’s lab (UCSF) and were authenticated using q-PCR primers against ESC-specific pluripotency markers: Nanog, Oct4, and Rex1. qPCR primers used are detailed in Table S7. E14 mouse ESCs were cultured on 0.1% gelatin-coated dishes in 5% CO2-buffered incubators at 37°C using media comprised of Knockout-DMEM (Life Technologies, catalog no. 10829018), 15% Embryomax FBS (EMD Milipore, catalog no. ES-009-B), 2 mM non-essential amino acids (EMD Milipore, catalog no. TMS-001-C), 2 mM L-Glutamine (EMD Milipore, catalog no. TMS-002-C), 0.1 mM 2-mercaptoethanol (Gibco, catalog no. 21985023), and 105 U/ml LIF (EMD Millipore, catalog no. ESG1107). Cells were split every other day to have ~5×106 cells/10 cm dish and were used up to passage 35. For SILAC labeling, ESCs were grown four passages in heavy SILAC media comprised of DMEM without arginine and lysine (Gibco, catalog no. A1443101), 15% Knockout Serum Replacement (Gibco, catalog no. 10828010), 2 mM non-essential amino acids, 2 mM L-Glutamine, 0.1 mM 2-mercaptoethanol, 105 U/ml LIF, 3 μM CHIR99021 (Stemgent, catalog no. 04-0004), 1 μM PD0325901 (Stemgent, catalog no. 04-0006), 0.8 mM isotope-coded L-Lysine (13C6, 15N2, Cambridge Isotope Labs, catalog no. CNLM-291-H), 0.4 mM isotope-coded L-Arginine (13C6, 15N4, Cambridge Isotope Labs, catalog no. CNLM-539-H). Pluripotency of ESCs that have undergone SILAC labeling were analyzed using q-PCR primers against ESC-specific pluripotency markers: Nanog, Oct4, and Rex1.
Genome editing of E14 ESCs were achieved by using CRISPR/Cas9 nuclease-mediated recombination (Doudna and Charpentier, 2014). sgRNA guide sequences were cloned into the BbsI-digested expression plasmid bearing both sgRNA scaffold backbone (BB) and Cas9 nuclease, pX330-U6-Chimeric_BB-CBh-hSpCas9 (Cong et al., 2013). sgRNA guide sequences and ssODN repair templates used are detailed in Table S7. ~1×106 ESCs were plated onto a single 6-well plate, 4 hours prior to the transfection of the relevant sgRNAs cloned into the pX330-U6-Chimeric_BB-CBh-hSpCas9 plasmid and the ssODN template. 500 ng of sgRNA plasmid and 100 pmols of ssODN (~1660 ng) were transfected using 7.5 μl Lipofectamine 2000 (Thermo scientific, catalog no. 11668027). 24 hours after transfections, cells were trypsinized and plated at a dilution to obtain ~1000 cells/10 cm plate. 10 days later, single ESC colonies were picked and replica-plated into two 96 well plates. One of the two plates was used for subsequent genotyping and sequencing analyses. Primers used for genotyping are detailed in Table S7. Stable GFP-FLAG (Tg) cells were generated using a piggyBac transposon-mediated stable integration system. μ5 g of pCAG-rtTA-PB-GFP-FLAG plasmid, 0.25 μg of piggyBac transposase, and 0.5 μg of linear Puromycin resistance cassette (Clontech, catalog no. 631626) were transfected into ~1×106 ESCs that were plated on a single well of a 6-well plate, using 7.5 μl Lipofectamine 2000. Stable clones were selected with 0.5 μg/ml Puromycin dihydrochloride (Gibco, catalog no. A1113803) for 10 days. Stable GFP-FLAG (Tg) clones were initially scored by GFP median intensity and then were analyzed by FLAG western blotting along with the FLAG-tagged RPs.
ESCs for the eL22-HA; ROSA26 CAGGs CRE/WT, and PKM flox/wt; ROSA26 CRE-ERT2/wt were generated from embryonic day (E) 3.5 blastocysts from relevant mouse crosses by the Stanford transgenic facility. eL22-HA; ROSA26 CAGGs CRE/WT cells (male) ESCs were expanded on Mitomycin C-treated mouse embryonic fibroblasts (MEFs) (EMD Millipore, catalog no. PMEF-CF) and were taken off MEFs prior to experiments. PKM flox/wt; ROSA26 CRE-ERT2/wt (male) ESCs were expanded on Mitomycin C-treated MEF and were taken off MEFs prior to experiments. PKM flox/wt; ROSA26 CRE-ERT2/wt (male) ESCs were treated with 1 μM 4-hydroxytamoxifen (Sigma-Aldrich, catalog no. T176) for 24 hours to induce Cre-mediated recombination of PKM flox allele.
Methods Details
Polysome Analysis
~10×106 ESCs were passaged approximately 16 hours prior to the polysome analysis. For various drug treatments, cells were incubated with the following drugs at the indicated concentrations and incubation durations at 37 °C. For Cycloheximide treatment (CHX) (Sigma-Aldrich, catalog no. C7698-1G), ESCs were treated at 100 μg/ml for 2 minutes. For Harringtonine (Abcam, catalog no. ab141941) treatment, ESCs were treated at 2 μg/ml for 10 min or for 40 min. For Lactimidomycin (EMD Milipore, catalog no. 506291) treatment, ESCs were treated at 25 μM for 10 minutes. ESCs were lysed in buffer A (25 mM Tris-HCl pH 7.5 (Ambion, catalog no. AM9850G, and Ambion, catalog no. AM9855G), 150 mM NaCl (Ambion, catalog no. AM9759), 15 mM MgCl2 (Ambion, catalog no. AM9530G), 1 mM DTT (Ambion, catalog no. 10197777001), 8% glycerol (Sigma-Aldrich, catalog no. G5516), 1% Triton X-100 (Sigma-Aldrich, catalog no. T8787), 0.5% sodium deoxycholate (Sigma-Aldrich, catalog no. D6750), 100 μg/ml Cycloheximide (CHX) (Sigma-Aldrich, catalog no. C7698-1G), 100 U/ml SUPERase In RNAse Inhibitor (Ambion, catalog no. AM2694), 25 U/ml TurboDNAse (Ambion, catalog no. AM2238), Complete Protease Inhibitor EDTA-free (Sigma-Aldrich, catalog no. 11836170001) in nuclease-free water (Thermo Fisher Scientific, catalog no. 10977015)). For ~10 ×106 ESCs, 400 μl of buffer A was used to lyse the cells. After lysis, nuclei were removed by two consecutive centrifugations at 800g, 5 min at 4°C followed by one centrifugation at 8000g, 5 min, and one centrifugation at 20817g, 5 min. RNA concentrations were measured using Nanodrop UV spectrophotometer (Thermo Fisher Scientific) and normalized amounts of RNA were layered onto a linear sucrose gradient (10–45% sucrose (Fisher Scientific, catalog no. S5–12) (w/v), 25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 15 mM MgCl2, 1 mM DTT, 100 μg/ml CHX in nuclease-free water and centrifuged in a SW41Ti rotor (Beckman) for 2.5 h at 40,000 rpm at 4°C. Typically, 600–1000 μg RNA was used for each sucrose gradient fractionation experiment. Fractions were collected by Density Gradient Fraction System (Brandel). For RNase treatment, SUPERase In RNAse Inhibitor was omitted from buffer A. After lysis, and after centrifugations at 800g, 8000g, and 20817g, RNase A (Invitrogen, catalog no. AM2270) and RNase T1 (Invitrogen, catalog no. AM2283) were added and incubated at 25°C for 30 min. For ~ 600 μg RNA, 1 μg RNase A and 2000 U RNase T1 were used to footprint ribosomes and 180U SUPERase In was added subsequently. For EDTA treated samples, cell lysates were layered on a linear sucrose gradient (10–45% sucrose (w/v), 25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 50 mM EDTA (Ambion, catalog no. AM9260), 1 mM DTT). Polysome fractions were precipitated using Proteoextract Protein Precipitation Kit (EMD Milipore). For each 750 μl fraction, 450 μl precipitant 1 was added and incubated at −20°C for at least 1 hr. Precipitated fractions were resolved in 4–15% (Biorad, catalog no. 3450028) SDS-PAGE gels. For Western blots antibodies were diluted in PBS-0.1% Tween 20 at 1:1000 dilution either in 5% BSA (w/v) or 5% non-fat milk.
Mass spectrometry and Data Analysis
For each FLAG immunoprecipitation (IP), five ~80% confluent 15 cm plates of (~150 ×106 cells) eL36-FLAG, eS17-FLAG, or GFP-FLAG ESCs were lysed in the lysis buffer A. After lysis, nuclei were removed by two consecutive centrifugations at 800g, 5 min at 4°C followed by one centrifugation at 8000g, 5 min, and one centrifugation at 20817g, 5 min as discussed in the polysome analysis above. Protein concentrations were measured using BCA assay (Pierce, catalog no. 23228) and input protein concentrations were normalized to 15 mg/ml with the lysis buffer A. 8 mg of total protein was used for each IP and 400 μl of FLAG-Dynabeads was used and incubated with the input for 0.5 hr on rotation at 4°C.
FLAG-Dynabeads were prepared as follows: 30 μg of FLAG antibody (Sigma-Aldrich, catalog no. F3165) was covalently coupled to 1 mg of Dynabeads M-270 Epoxy beads (Life technologies, catalog no. 14301) using Dynabeads antibody coupling kit (Life technologies, catalog no. 14311D). For each IP, 150 μg FLAG M2 antibody was covalently coupled to 5 mg Dynabeads, and was resuspended in 400 μl SB buffer contained in the Dynabeads coupling kit. Even though anti-FLAG M2 agarose beads (Sigma-Aldrich, catalog no. A2220) had IP efficiencies that were ~4–6 fold higher, FLAG-Dynabeads were used to minimize background due to the agarose beads.
After 0.5 h incubation at 4°C on rotation, IP samples were first washed 3 times each for 5 min in 5 ml volume at 4°C with buffer B (25 mM Tris-HCl pH 7.5, 150 mM NaCl, 15 mM MgCl2, 1 mM DTT, 1% Triton X-100, 0.5% sodium deoxycholate, 100 μg/ml CHX). Afterwards, beads were washed 3 times for 5 min each at 4°C using buffer C (25 mM Tris-HCl pH 7.5, 300 mM NaCl, 15 mM MgCl2, 1 mM DTT, 1% Triton X-100, 0.5% sodium deoxycholate, 100 μg/ml CHX). Samples were then eluted off the anti-FLAG beads using 450 μl competitive FLAG peptide elution (25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5 mg/ml 1× FLAG peptide (Sigma-Aldrich) at 25°C for 0.5 hr. For IPs that investigated the relative abundance of a highly translated protein (HA-GFP) compared to that of a protein potentially interacting with the ribosome (HA-PKM1/2), 30 μg pCAGGs-HA-GFP plasmid were transfected into ~150 ×106 eL36-FLAG; HA-PKM ESCs using 45 μl Lipofectamine 2000 (Thermo Scientific, catalog no. 11668027), and FLAG-ribosome IP was performed as described above.
For RNase A-treated IP samples, the following conditions were adopted from a previous publication (Klass et al., 2013) and for the subsequent experiments, SuperRNase In was omitted from buffer A. After FLAG-bead incubation, beads were washed 3 times for 5 min each at 4 °C with buffer B, and then were split into two batches. For the RNAse A-treated sample, the sample was washed 3 more times for 10 min each at 25°C with buffer C containing 2 ng/μl RNase A (Invitrogen, catalog no. AM2270) for a total of 10 μg. Afterwards, beads were washed once more using buffer C containing 200 U/ml SuperRNase In. For Puromycin treated IP samples, 200 mM Puromycin dihydrochloride (Sigma-Aldrich, catalog no. P8833) was prepared in 1X D-PBS (Thermo scientific, catalog no. 14190250) the same day of the experiment and was incubated with cells at 37°C for 10 min at a final concentration of 200 μM.
For unlabeled MS experiments, each IP from eL36-FLAG, eS17-FLAG, or GFP-FLAG ESCs was dried using a speedvac, was resuspended in 30 μl SDS sample buffer with reducing agent (Alfa Aesar, catalog no. AAJ61337AC), and was incubated at 99°C for 10 minutes. Samples were run in 4–12% Bis-Tris gel (Thermo scientific, catalog no. NP0321BOX) at 120 volts for 10 min using MOPS buffer (Thermo scientific, catalog no. NP0001). For in-gel digestion of the IP samples, the protein gel was rinsed twice with HPLC water (Fisher scientific, catalog no. W5SK-1L), and fixed with 10% acetic acid (Fisher scientific, catalog no. A38500), 45% methanol (Fisher scientific, catalog no. A452SK-1) for 15 min. The gel was stained with SimplyBlue (Life technologies, catalog no. LC6060) for 0.5 h and bands were cut and incubated with 100 mM Ammonium bicarbonate (Sigma-Aldrich, catalog no. 09830) for 15 min. Gel pieces were treated with 5 mM DTT (Thermo Scientific, catalog no. 20291) in 50 mM ammonium bicarbonate at 55°C for 30 min. Afterwards, the DTT solution was discarded and gel pieces were treated with 25 mM IAA (Thermo Scientific, catalog no. 90034) in 50 mM Ammonium bicarbonate at 25°C for 30 min. Gel pieces were shrunk using 50% Acetonitrile (51101) in 50 mM ammonium bicarbonate and were dried using speedvac for 15 min. 1 μg Trypsin/Lys-C (Promega, catalog no. V5071) per gel sample was added in 0.01% ProteaseMAX (Promega, catalog no. V2071) in 50 mM ammonium bicarbonate for ~16 hrs at 37°C. Two consecutive peptide extractions were performed using the same digestion volume of 70% acetonitrile (Thermo Scientific, catalog no. 51101), 29% HPLC water, and 1% formic acid (Fisher scientific, catalog no. A117-50) at 37°C. Digested peptides were dried using speedvac and were resuspended in 8 μl of 0.1% formic acid. 2 μl of each sample was analyzed on an Orbitrap Elite mass spectrometer. Peptides were separated using a gradient of 5 to 21% acetonitrile over 90 min. MS2 spectra were searched using the Byonic (v2.12.0) algorithm against a Uniprot database derived from the mouse proteome containing its reversed complement and known contaminants. Peptide spectral matches were filtered to a 1% false discovery rate (FDR) using the target-decoy strategy combined with linear discriminant analysis. Precursor mass tolerance was set to 10 ppm and fragment mass tolerance was set to 0.4 Dalton allowing 2 miscleavages.
Normalized spectral abundance factor (NSAF)is described previously in (Florens et al., 2006), and is calculated as the number of spectral counts (SpC) identifying a protein divided by the length of the protein (L), that is divided by the sum of SpC and L ratios of all the proteins in the MS experiment. For calculation of SAINT scores, spectral counts were analyzed by SAINT (v2.5.0) (Choi et al., 2012) using the following parameters: lowmode = 1, minfold = 1, and norm = 0. Fold change (FC) is the ratio of the sum of SpC across IP experiments to the sum of SpC across background control experiments plus pseudocount of 1. For the analysis of the enrichment of Gene Ontology (GO) terms, the whole cell mESC proteome data from (Graumann et al., 2008) was used as the background and was analyzed using DAVID (david.abcc.ncifcrf.gov). P values were corrected using Benjamini–Hochberg and an initial cutoff of 0.05 was used. Then, the data were ranked by fold enrichment and a minimum 4 fold change enrichment was used as a threshold.
For the TMT experiments that compared control, RNase-treated, and Puromycin-treated IP samples, after FLAG peptide elution, RNA levels were measured using Nanodrop UV spectrophotometer and FLAG peptide elution buffer was used to normalize IPs. Samples were dried by speedvac overnight and dried samples were resuspended in 50μl of LDS sample buffer (Thermo scientific, catalog no. NP0007) with reducing agent and incubated at 60°C for 10 minutes. One half of each sample was run in 10% Bis-Tris gel (Thermo scientific, catalog no. NP0301BOX) at 120 volts for 10 min using MES buffer (Thermo scientific, catalog no. NP0002). Gel bands were cut out, destained, reduced, and alkylated. In-gel trypsin digestion was performed. Extracted peptides were labeled with TMT-10plex isobaric label reagent Set (Thermo scientific, catalog no. 90110). Labeling reactions were combined, cleaned, and dried down. Peptides were resuspended in 5% acetonitrile, 5% formic acid and half of the sample was analyzed on an Orbitrap Fusion mass spectrometer. Peptides were separated using a gradient of 6 to 28% acetonitrile in 0.125% formic acid over 180 min. Peptides were detected (MS1) and quantified (MS3) in the Orbitrap (Ting et al., 2011). Peptides were sequenced (MS2) in the ion trap. MS2 spectra were searched using the SEQUEST algorithm against a Uniprot composite database derived from the mouse proteome containing its reversed complement and known contaminants. Peptide spectral matches were filtered to a 1% false discovery rate (FDR) using the target-decoy strategy combined with linear discriminant analysis. The proteins were filtered to a <1% FDR. Proteins were quantified only from peptides with a summed SN threshold of >=200 and MS2 isolation specificity of 0.5 which was determined empirically in (Ting et al., 2011). Protein quant data is included as an excel spreadsheet. Quantitative data is provided in two forms: 1) total summed intensity of peptides assigned to each protein and 2) Log2 relative abundance.
For the comparison of PKM-FLAG enriched ribosomes to eL36-FLAG ribosomes, for each experiment, five ~80% confluent 15 cm plates of SILAC media-fed mESCs (~150 ×106 cells) were collected in D-PBS and treated with 0.05% formaldehyde (Sigma-Aldrich, catalog no. F8775) in D-PBS for 15 minutes at 25°C for 15 min on rotation. Buffer A without SuperRNase In was used to lyse the cells and RNase A was used to footprint the ribosomes as described above. For each experiment, five sucrose gradient fractionations were performed over a linear sucrose gradient of 10–45% sucrose as described above. 80S fractions from five experiments were pooled together and used as input for the FLAG IP which is described in detail above. Eluted proteins were digested overnight using the in-gel digestion protocol described above and analyzed in Orbitrap Elite mass spectrometer. Data was analyzed using MaxQuant program (v.1.2.2.5).
For the two-step enrichment of the Ufmylated ribo-interactome, 6XHis tagged Ufm1 at its N terminus was transfected into eL36-FLAG ESCs. Specifically, to each of 20×15 cm eL36-FLAG cells, 30 μg of pCAGGs-6X His Ufm1 was transfected with 45 μl Lipofectamine 2000. As a negative control, eL36-FLAG cells that were not transfected with the 6XHis-Ufm1 expression plasmid were used. After 18 hours, FLAG IP was performed as described above. The subsequent Ni-NTA pulldown under denaturing conditions were performed as follows: 9M Urea, Tris-HCl pH 8.0, 15 mM imidazole 1.5 X FLAG elution buffer without FLAG peptide was prepared and added to the FLAG elution samples to have a final concentration of 6M urea and 10 mM imidazole. The FLAG elution with 6M urea was then incubated with 60 μl of Ni-NTA agarose slurry (Thermo scientific, R90101) for 2 hours at 25°C. Ni-NTA wash buffer consists of 100 mM sodium phosphate, 10 mM imidazole, 10 mM Tris base, 1M urea, pH 8.0. Ni-NTA elution buffer contains 300 mM imidazole. His purifications were in-gel digested as described above and analyzed in Orbitrap Elite mass spectrometer. MS2 data was searched using Mascot (v2.4), and for the analysis of two experiments compared to the background, FC was calculated as described above.
iCLIP and Data Analysis
FAST-iCLIP was performed (Flynn et al., 2015) on PKM2-FLAG-HA cells by UV crosslinking cells to a total of 0.35 J cm−2. Whole-cell lysates were generated in iCLIP lysis buffer (50 mM HEPES, 200 mM NaCl, 1 mM EDTA, 10% glycerol, 0.1% NP-40, 0.2% Triton X-100, 0.5% N-lauroylsarcosine) and briefly sonicated using a probe-tip Branson sonicator to solubilize chromatin. Each iCLIP experiment was normalized for total protein amount, typically 1 mg, and partially digested with RNase I (ThermoFisher Scientific, catalog no. AM2294) for 10 min at 37°C and quenched on ice. PKM2-FLAG-HA was isolated with anti-FLAG agarose beads (Sigma-Aldrich) for 1 h at 4°C on rotation. Samples were washed sequentially in 1 ml for 5 min each at 4°C: 2× high stringency buffer (15 mM Tris-HCl, pH 7.5, 5 mM EDTA, 2.5 mM EGTA, 1% Triton X-100, 1% sodium deoxycholate, 120 mM NaCl, 25 mM KCl), 1× high salt buffer (15 mM Tris-HCl pH 7.5, 5 mM EDTA, 2.5 mM EGTA, 1% Triton X-100, 1% sodium deoxycholate, 1 μM NaCl), 1× NT2 buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM MgCl2, 0.05% NP-40). Purified PKM2-FLAG-HA was then eluted off anti-FLAG agarose beads using competitive FLAG peptide elution. Each sample was resuspended in 500 μl of FLAG elution buffer (50 mM Tris-HCl, pH 7.5, 250 mM NaCl, 0.5% NP-40, 0.1% sodium deoxycholate, 0.5 mg/ml FLAG peptide) and rotated at 4°C for 30 min. The FLAG elution was repeated once for a total of 1 ml elution. PKM2-FLAG-HA was then captured using anti-HA agarose beads (Pierce) for 1 h at 4°C on rotation. Samples were then washed as previously in the anti-FLAG agarose beads.
After the NT2 wash, HA-bound RNA-protein complexes were dephosphorylated with T4 PNK (NEB, cat# M0210) for 30 min in an Eppendorf Thermomixer at 37°C, 15s 1400rpm, 90s rest in a 30μl reaction, pH 6.5, containing 10 units of T4 PNK, 0.1 μl SUPERase-IN, and 6 μL of PEG-400 (16.7% final). After 30 min, beads were rinsed once with NT2 buffer and 3′-end ligated with T4 RNA Ligase 1 (NEB, cat# M0204) overnight in an Eppendorf Thermomixer at 16°C, 15s 1400rpm, 90s rest in a 30μL reaction containing 10 units T4 RNA Ligase, 1pmole pre-Adenylated-DNA-adapter, 0.1μL SUPERase-IN, and 6μL of PEG400 (16.7% final). The following day, samples were again rinsed with NT2 buffer and 5′ radiolabeled by adding 1μL of T4 PNK, 0.5μL g32-ATP (Perkin Elmer), 2μL 10x T4 PNK Buffer, and 0.5μL SUPERase-In, and 16μL of water for 15 minutes at 37°C. To this reaction, 1μL of 100mM DTT and 6uL of 4x LDS Buffer (ThermoFisher Scientific) was added, and samples were heated to 75°C for 10min. Released RNA-protein complexes were separated on SDS-PAGE using NuPAGE 4–12% Bis-Tris Gels (1.0mm × 12 well) at 180V for 45 min. Resolved RNP complexes were wet-transferred to nitrocelluose at 400 mA for 60 min at 4°C.
RNA was recovered and processed for library preparation as in the irCLIP protocol (Zarnegar et al., 2016). Membranes were cut into ~0.5×1mm narrow strips that easily come to rest in the bottom of a siliconized 1.5mL eppendorf tube. To each tube, 0.2 mL of Proteinase K reaction buffer (100 mM Tris, pH 7.5, 50 mM NaCl, 1 mM EDTA, 0.2% SDS) and 10μl of Proteinase K (Thermo Fisher Scientific, cat# AM2546) was added. The reaction was then incubated for 60 min at 50°C in an Eppendorf Thermomixer. Next, 200μL of saturated-phenol-chloroform, pH, 6.7 was added to each tube and incubated for 10 min at 37°C in an Eppendorf Thermomixer, 1400 rpm. Tubes were briefly centrifuged and the entire contents transferred to a 2 mL Heavy Phase Lock Gel (5Prime, cat# 2302830). After 2 min centrifugation at >13000 rpm, the aqueous layer was re-extracted with 1 mL of chloroform (invert tube 10 times to mix; do not vortex, pipet or shake) in the same 2 mL Phase Lock Gel tube and centrifuged for 2 min at >13000 rpm. The aqueous layer was then transferred to a new 2 mL Heavy Phase Lock Gel tube and extracted again with an additional 1 mL of chloroform. After 2 min centrifugation at >13000 rpm, the aqueous layer was transferred to a siliconized 1.5 mL eppendorf tube and precipitated overnight at −20°C by addition of 10 μl 5M NaCl, 3 μL Linear Polyacrylamide (Thermo Fisher Scientific, cat# AM9520) and 0.8 mL ethanol.
cDNA synthesis primers were purchased from IDT: cDNA-barcode1 (6 bp TruSeq barcode in ‘bold’):
/5phos/WWWNNNXXXXXXNNNNNTACCCTTCGCTTCACACACAAG/iSp18/GGATCC/iSp18/TACTGAACCGC. P3short (cDNA elution oligo): CTGAACCGCTCTTCCGATCT. PCR1 primers, P3tall: GCATTCCTGCTGAACCGCTCTTCCGATCT, P6tall: TTTCCCCTTGTGTGTGAAGCGAAGGGTA. PCR2 primers (PAGE purified). P3solexa: CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT, P6solexa: AATGATACGGCGACCACCGAGATCTACACTCTTTCCCCTTGTGTGTGAAGCGAAGGGTA
P6 sequencing primer (For Illumina Sequencing): CACTCTTTCCCCTTGTGTGTGAAGCGAAGGGTA
RNA fragments were pelleted at >13000 rpm for 45 min at 4°C, washed once with 1mL of ice cold 75% ethanol and air dried. Pellets were resuspended in 12 μL water. 12 μL of RNA was mixed with 1 μL of 1 μM cDNA and 1 μL of 10mM dNTPs and heated to 70°C for 10 min then rapidly cooled to 4°C. Six microliters of cDNA Master Mix (4 μL 5× SSIV Buffer, 1 μL 100mM DTT, 1 μL SSIV) was added to the annealed RNA and incubated for 30min at 55°C. cDNA:RNA hybrids were captured by addition of 5 μl of MyOne Streptavidin C1 Dynabeads (Thermo Fisher Scientific, cat# 65001) that had been rinsed and suspended in 30μl of Biotin-IP buffer (100mM Tris, pH 7.5, 1M NaCl, 1mM EDTA, 0.1% Tween), and end over end rotated for 30 min at room temperature. Beads were placed on a 96-well magnet and washed sequentially with 0.1 mL of Biotin IP buffer and PBS. Beads were resuspended in 10 μL of cDNA elution/RNA degradation buffer (8.25 μL water, 1 μL of 1 μM P3short oligo, and 0.7 5μl of 50 mM MnCl2) and placed in a thermocycler with the program: 5 min 95°C, 1 min 75°C, ramp 0.1 degree/s to 60°C forever. After 15 min, tubes were removed and mixed with 5 μL of Circligase-II reaction buffer (3.3 μL water, 1.5 μL 10x Circligase-II buffer, and 0.2 μL of Circligase-II, Epicentre, cat# CL9021K). cDNA was circularized in a thermocycler for 1.5hrs at 60°C. cDNA was captured by addition of 30 μL of Ampure XP beads (Beckman Coulter, cat# A63880), 75 μL of isopropanol and 15 min of incubation (the solution was remixed after 7.5 min). Beads were washed once with 80% ethanol, dried for 5 min and resuspended in 14 μL of water. For maximal elution, tubes were placed in a 95°C thermocycler for 2 min and immediately transferred to a 96-well magnet. The 14 μL eluate was transferred to a new 0.2mL PCR tube containing 15 μL of 2X Phusion HF-PCR Master Mix (NEB, cat# M0531), 0.5 μL of 30 μM P3/P6 PCR1 oligo mix and 0.5μl of 15X SYBR Green I (Thermo Fisher Scientific, cat# S7563). The tubes were then placed in a Stratagene MX3000P qPCR machine with the following program: 98°C 2 min, 15 cycles of 98°C 15 seconds, 65°C 30 seconds, 72°C, 30 seconds, with data acquisition set to the 72°C extension. PCR1 reactions were then subjected to one round of magnetic bead size selection by addition of 4.5 μL of isopropanol, 54 μL of Ampure XP beads and incubation for 10 min. Beads were washed once with 80% ethanol, dried for 5 min and eluted in 10 μl of water. PCR1 products were subjected to a second round of size selection by addition of 1.5μl of isopropanol, 18 μL of Ampure XP beads and incubation for 10 min. Beads were washed once with 80% ethanol, dried for 5 min and eluted in 10 μL 500 nM P3solexa/P6solexa oligo mix. 10 μl of 2X Phusion HF-PCR Master was added to each tube and placed in a thermocycler with the following program: 98°C 2 min, 3 cycles of 98°C 15 seconds, 65°C 30 seconds, 72°C, 30s seconds. Final libraries were purified by addition of 36 μL of Ampure XP beads and incubation for 5 min. Beads were washed twice with 70% ethanol, dried for 5 min and eluted in 20 μL of water. 1–2μL of libraries were quantitated by HS-DNA Bioanalyzer.
Samples were sent for deep sequencing on the Illumina NextSeq machine for single-end 75-bp cycle run. FAST-iCLIP data was processed using the FAST-iCLIP analysis pipeline (https://github.com/ChangLab/FAST-iCLIP). PCR duplicates were removed using unique molecular identifiers (UMI) in the RT primer region. Adapter and barcode sequences were trimmed, and reads were mapped step-wise to repetitive and non-repetitive genomes. Specific parameters used are as follows: -f 18 (trims 17nt from the 5′ end of the read), −l 15 (includes all reads longer than 15nt),–bm 25 (minimum MAPQ score from bowtie2 of 25 is required for repeat element mapping), –sr 0.08 (STAR mismatch-per-base ratio; 0.08 corresponds to 2 mismatches per 25 bases), and –tr 2,3 (repetitive genome) and –tn 2,3 (nonrepetitive genome) RT stop intersection (n,m; where n = replicate number and m = number of unique RT stops required per n replicates) (Dobin et al., 2013). Using the –tr/tn 2,3 parameters, a minimum of 6 RT stops are required to support any single nucleotide identified as crosslinking site. For the tRNA alignment, reference index was generated by appending CCA tail sequence to tRNA gene predictions accessed from gtRNAdb (http://gtrnadb.ucsc.edu/). All possible tRNA alignments were reported using bowtie2 -a mode, but each mapped read was counted once for calculating total proportion of tRNAs in the library.
siRNA Transfection
For PKM knockdown experiments, E14 ESCs were transfected for 36 h with either control (Dharmacon) or PKM targeting siRNAs (Dharmacon) using RNAiMAX (Invitrogen, catalog no. 13778). For ~10×106 cells, either 100 pmol PKM targeting siRNA or control siRNA were transfected using 30 μl RNAiMAX. PKM1/2 siRNAs target both PKM1 and PKM2 isoforms. The final concentrations of siRNAs were 20 nM.
Ribosome Profiling and Data Analysis
Ribosome profiling was performed as described before (Ingolia et al., 2012) with modifications. Details are described below.
Control and PKM knockdown ESCs were passaged 16 h prior to sucrose cushion purification. ESCs were treated with 100 μg ml−1 CHX for 2 minutes at 37°C. CHX-treated cells were lysed using the buffer A in the polysome analysis without SUPERase RNase Inhibitor. RNA for RNA-Seq was isolated using TRIzol (Invitrogen, catalog no. 15596-018). For ribosome profiling, after lysis, nuclei were removed by three consecutive centrifugations (800g, 5 min at 4°C). For ~ 600 μg RNA, 1 μg RNase A (Invitrogen, catalog no. AM2270) and 2000 U RNase T1 (Invitrogen, catalog no. AM2283) were used to footprint ribosomes for 30 minutes at 25°C and subsequently quenched with 180 U SUPERase In. Ribosomes were enriched by adding the lysate onto sucrose cushion buffer (33% sucrose (w/v), 25 mM Tris-HCl pH 7.5, 150 mM NaCl, 15 mM MgCl2, 1 mM DTT, 100 U/ml SUPERase In RNAse Inhibitor), and centrifuging in a TLA 120.2 rotor (Beckman) for 4 h at 70,000 rpm at 4 °C. The ribosome pellet containing the ribosome footprinted Ribo-Seq library was resuspended in TRIzol.
Library preparation was adapted from a previous protocol (Ingolia et al., 2012) and the ARTseq Ribosome Profiling Kit manual (Epicentre). In summary, total RNA and ribosome footprints were extracted using sequential TRIzol and acid-phenol:chloroform extraction. Briefly, 200 uL of chloroform was added to 1 mL of TRIzol resuspended sample, mixed, and centrifuged at 12,000g, 15 min, 4°C. The aqueous phase was removed and added to 500 uL of acid-phenol:chloroform, pH 4.5 (with IAA) (Invitrogen, catalog no. AM9722), mixed, and centrifuged at 21,000g, 5 min, RT. From this step on, nonstick RNase-free tubes were used (Invitrogen, catalog no. AM12450). The subsequent aqueous phase was removed and precipitated overnight at −80°C with 600 uL isopropanol and 1.5 uL GlycoBlue Coprecipitant (Invitrogen). The samples were then centrifuged at 21,000g, 30 min, 4°C, supernatant was removed, and the RNA pellet was washed twice with 500 uL cold 75% ethanol. Pellets were dried for 15 min, RT and resuspended in nuclease free water.
After extraction and precipitation, both ribosome footprinting and total RNA samples were depleted of rRNA using the Ribo-Zero Gold rRNA Removal Kit (H/M/R) (Illumina, catalog no. MRZG126). Briefly, magnetic beads were washed with nuclease free water and resuspended in resuspension solution with 1 uL RiboGuard RNase inhibitor. 5 ug of RNA was then probe-hybridized by incubating with Ribo-Zero rRNA Reaction Buffer and Removal Solution in a 40 uL reaction. The probe-hybridized RNA samples were then transferred to the magnetic beads and incubated at room temperature for 5 minutes. The recommended 50°C incubation was not performed. The supernatant was then removed and column purified (RNA Clean & Concentrator 5, Zymo Research, catalog no. R1016). Ribosome protected fragment samples were adjusted to 100 uL with nuclease free water and mixed with 200 uL RNA binding buffer and 450 uL 100% ethanol. Total RNA samples were adjusted to 100 uL with nuclease free water and mixed with 200 uL RNA binding buffer and 300 uL 100% ethanol. Sample was then transferred and bound to column, washed, and eluted in 12 uL nuclease free water.
Total RNA samples were then fragmented by partial alkaline hydrolysis. The samples were diluted to 100 uL with 5 mM Tris-HCl, pH 7.5 and incubated with 100 uL 2× alkaline fragmentation buffer (100 mM Na2CO3 pH 9.2, 2 mM EDTA) for 20 minutes at 95°C. The reaction was neutralized with 440 uL STOP Buffer (70 uL 3M NaOAc pH 5.5, 2 uL Glycoblue, and 370 uL nuclease free water) and isopropanol precipitated overnight at −80°C.
Ribosome protected fragments and total RNA samples were then size selected by running the samples out on a 15% TBE-Urea polyacrylamide gel. Ribosome protected fragments were size selected between 28-nt and 34-nt as marked by RNA oligonucleotides oNTI199 and oNTI265, respectively (Ingolia et al., 2011). Total RNA samples were size selected between 40–70 nt as marked by a 10 bp DNA ladder (Invitrogen, catalog no. 10821015). Gel slices were crushed and extracted at room temperature overnight in 400 uL RNA extraction buffer (300 mM NaOAc pH 5.5, 1 mM EDTA, 0.25% SDS). The eluate was then purified by acid-phenol:chloform extraction and isopropanol precipitation (see above).
Samples were then 3′dephosphorylated by denaturing at 80°C for 90 seconds and incubating with 1 uL T4 PNK (NEB, catalog no. M0201S) in a 50 uL reaction at 37°C for 1 hour. The fragmented total RNA and ribosome protected fragments were then purified using Zymo RNA Clean & Concentrator 5 columns using a protocol in which 100 uL sample, 200 uL RNA binding buffer, and 450 uL 100% ethanol were used for binding (see above). Samples were eluted with 8.5 uL nuclease free water and incubated with 1.5 uL of 0.5 ug/uL Universal miRNA Cloning Linker (NEB, catalog no. S1315S) and denatured at 80°C for 90 seconds. The denatured sample was then incubated with 1 uL T4 RNA Ligase 2, truncated (NEB, catalog no. M0242S), 2 uL 10× buffer, 1 uL SUPERase In, and 6 uL 50% PEG 8000 for 2.5 hours at room temperature. Samples were then purified using Zymo RNA Clean & Concentrator 5 columns (100 uL sample, 200 uL RNA binding buffer, 450 uL 100% ethanol). Linker ligated ribosome protected fragments and total RNA fragments were subsequently size selected on 10% TBE-Urea polyacrylamide gels and gel extracted and acid-phenol:chloroform extracted as described above.
To perform reverse transcription, samples were incubated with 2 uL of 1.25 uM RT primer (see table) and denatured for 2 min at 80°C. Reverse transcription was performed with SuperScript III (Invitrogen, catalog no. 18080-044) in a 20 uL reaction (48°C, 30 min). RNA was then hydrolyzed by adding 2.2 uL of 1N NaOH and incubating for 20 min at 98°C. Samples were then purified using Zymo RNA Clean & Concentrator 5 columns (100 uL sample, 200 uL RNA binding buffer, 300 uL 100% ethanol). RT products were size selected and gel extracted from 10% TBE Urea polyacrylamide gels as described above instead that DNA extraction buffer was used for overnight extraction (300 mM NaCl, 10 mM Tris-HCl pH 8, 1 mM EDTA, 0.1% SDS). Eluate was then isopropanol precipitated overnight at −80°C overnight.
Samples were then circularized with CircLigase (Illumina, catalog no. CL4115K) in a 20 uL reaction (15 uL cDNA, 2 uL 10x CircLigase Buffer, 1 uL 1 mM ATP, 1 uL MnCl2, 1 uL CircLigase) for 12 hours at 60°C and subsequently purified by Zymo RNA Clean & Concentrator 5 columns (100 uL sample, 200 uL RNA binding buffer, 300 uL 100% ethanol) and eluted with 12 uL nuclease free water.
1 uL of library was used for PCR amplification with Phusion High-Fidelity DNA Polymerase (Thermo Fisher, catalog no. F530S) (98°C 30s, 98°C 10s, 65°C 10s, 72°C 5s) for 10–11 cycles using the ribosome profiling library PCR forward primer and indexed reverse primers (see table).
PCR product was PAGE purified from 8% TBE polyacrylamide gels, extracted overnight using DNA extraction buffer, and isopropanol precipitated overnight at −80°C. DNA was measured and quality controlled on the Agilent 2100 Bioanalyzer (High-Sensitivity DNA) by the Stanford Protein and Nucleic Acid Facility. Libraries were sequenced by the Stanford Functional Genomics Facility on the Illumina NextSeq 500 (1×75nt).
For analysis pre-processing, the 3′ adapter sequences from reads were removed using cutadapt (Martin, 2011). The 5′ end of each read was then removed using fastx_trimmer from FASTX-Toolkit. To remove reads that aligned to rRNA, tRNA, and snRNAs, reads were first aligned to these sequences using bowtie2 (bowtie2 parameters: -L 18) and subsequently discarded. Filtered reads that did not align to rRNA/tRNA/snRNAs were then aligned to an mm9 transcriptome reference derived from UCSC knownCanonical using bowtie2 (bowtie2 parameters: –norc -L 18). Ribo-Seq reads were then parsed for uniquely aligned reads, separated into read length groups, and ribosome A site positions were determined by offsetting the distance of the 5′ end of each read to canonical start sites in each length group and adding 4 nucleotides (Ingolia et al., 2012). RNA-Seq reads were also parsed for uniquely aligned reads and were assigned to particular nucleotide positions using the above parameters. Ribo-Seq and RNA-Seq reads were then counted using UCSC mm9 knownCanonical annotations.
To remove lowly expressing genes, genes with <150 reads in the CDS of any of the Ribo-Seq or RNA-Seq libraries were removed. Translational efficiency for each gene is defined as the library size normalized counts of Ribo-Seq reads divided by normalized RNA-Seq read counts aligning to the CDS (with the first 15 codons and last 5 codons removed). Raw aligned iCLIP reads from the FAST-iCLIP analysis described above were then obtained prior to merging replicate RT stops. Alignments were assigned to the 5′ ends of reads and counted using UCSC mm9 knownCanonical annotations. The counts over the total mature transcript from each replicate were averaged to obtain the mean CLIP read count for each gene. To calculate iCLIP enrichment scores for each gene, a pseudocount of 1 was added to the mean iCLIP read count for each gene, normalized to aligned library size, and divided by the library size normalized RNA-Seq counts from the control library.
Translational efficiency changes between the PKM knockdown and control samples were calculated and plotted against PKM2 iCLIP enrichment. Spearman’s rho was calculated in R. Correlation was also analyzed by binning genes based on iCLIP enrichment scores and analyzing the resulting empirical cumulative density function of translational efficiency change between PKM1/2 knockdown and control for each group. Differences between the strong iCLIP binding (top 5 percentile) group and the weakly binding (50–100 percentile) group were quantified with the Mann-Whitney U test in R.
iCLIP GO term enrichment was performed using the set of iCLIP enriched genes, defined as genes with log2(total mature transcript iCLIP normalized read count) > 5 and log2(iCLIP enrichment) > 1, compared to a background set of genes with CDS read count >150 in mouse ESC RNA-seq and Ribo-Seq experiments. Enrichments were calculated using DAVID (david.abcc.ncifcrf.gov). 0.05 cutoff of Benjamini–Hochberg P values and a minimum 2 fold enrichment were used.
Tethered Function Assay
The following plasmids have been described previously: pCIneo-RL (p2443) (Pillai et al., 2005), pFLB (p2524), pFLB-PP7bs (p2646), pcDNA3-HA-PP7cp-Pat1b (p2634), pcDNA3-HA-PP7cp (p2211) (Ozgur et al., 2010). Tethering reporter assays were performed by transfecting 2 μg of the reporters (FLB or FLB-PP7bs) with 2 μg of PP7 fusion proteins along with the normalization control, 0.16 μg pCIneo-RL reporter, into 1×106 E14 ESCs cells using 7.5 μl Lipofectamine 2000 (Thermo Fisher). After 24 hours, cells were collected and one third of a 6 well dish of transfected E14 ESCs was used for parallel luciferase activity, western and northern blot analysis. Subsequently, total RNA was extracted from cell pellets using the GeneMatrix Universal RNA Purification Kit (EurX). For northern blot analysis, 2–10 μg of total RNA was resolved by 1.1% agarose/2% formaldehyde/MOPS gel electrophoresis using 1× MOPS running buffer and blotted over night with 8x saline-sodium citrate (SSC) buffer (1x contains 0.15 μM NaCl and 0.015 μM sodium citrate) onto Hybond-N+ Nylon membranes (Amersham, GE Healthcare). Membranes were hybridized overnight at 55°C with digoxigenin-labelled RNA probes synthesized in vitro using Sp6 polymerase (Ambion) and DIG RNA labelling mix (Roche). 500 ng RNA probe was diluted in 10 ml hybridization buffer containing 50% formamide, 5x SSC, 5x Denhard’s solution, 5 mM EDTA, 10 mM PIPES pH 7.0 at 25°C, 0.4 mg/ml torula yeast RNA (US Biological) and 1% SDS. Membranes were washed twice with 2x SSC/ 0.1% SDS for 5 minutes, and twice with 0.5x SSC/ 0.1% SDS for 20 minutes at 65°C. Alkaline phosphatase-coupled anti-digoxigenin Fab fragments and CDP-Star substrate (both Roche) were used for detection using digoxigenin-labeled RNA probes against Rluc (pCIneo-RL), rabbit β-globin and Rps7 mRNAs, which were generated by PCR using the primers listed in the Key Resource Table. The corresponding signals were quantified using ImageJ software. For luciferase assays, cells were lysed in 60 μl of 1x passive lysis buffer of the Dual-Luciferase Reporter Assay System (Promega) and frozen at −80°C. After thawing, cell debris and nuclei were removed by centrifugation for 1 min at 13,000 rpm. 20 μl of supernatant was assayed for luciferase activity in technical replicates by mixing with 50 μl of Dual-Luciferase Reporter Assay System substrates. Firefly and Renilla luciferase activities were measured on a GloMax-Multi (Promega) plate reader. Luciferase reporter activity is expressed as a ratio between Fluc and Rluc which was normalized to the ratio of Fluc to Rluc mRNA levels based on the corresponding quantified northern blot signals.
Subcellular Fractionations and qPCR
Sequential detergent extraction was used to isolate subcellular fractions as described previously (Jagannathan et al., 2011). Subcellular fractions from ~2 ×106 cells were isolated using Native Membrane Extraction Kit (Calbiochem, catalog no. 444810). To each isolation, exogenous 50 pg Luciferase mRNA was added as a control to normalize for the RNA isolation procedure and the fractions were collected in 500 μl TRIzol (Invitrogen). 1/10th of the initial lysate was used to isolate total RNA. RNA was isolated by adding 250 μl isopropanol to the cell fractions in TRIzol and incubating at 25°C for 15 min. Afterwards, fractions were centrifuged at 12000 μg at 4°C for 10 min. The pellet was washed twice with 1000 μl 75% ethanol and resuspended in RNase-free water. 0.4 μg of RNA was converted to cDNA using iScript supermix (Bio-Rad, catalog no. 1708840). cDNA was diluted ten-fold and 1 μl was used to run SYBR green detection qPCR assay using SsoAdvanced SYBR Green supermix (Bio-Rad, catalog no. 1725270) on a CFX384 machine (Bio-rad). Each fraction was normalized to the Fluc values for that fraction and was shown as a fraction of the total RNA collected from the samples before they went through the fractionation protocol. qPCR primers are detailed in Table S7.
Quantification and Statistical Analysis
For the analysis of TMT data, log2 total summed SN intensities were quantile normalized and t-statistics were calculated using voom/LIMMA method (Ritchie et al., 2015). The distribution of the statistics indicated that the theoretical null is substantially narrower. We thus performed empirical null estimation by mixture model fitting using the locfdr framework (Efron, 2004). Since we are interested in defining proteins that only lose their interaction upon treatment and call proteins that increase their interaction with the ribosome still direct ribosome interactors, a one-sided distribution was used. We used the maximum likelihood probability densities fitted by locfdr to estimate empirical p-values, false discovery rates, and negative predictive values. Controlling the FDR at 15% and using the effect size cutoff, we classified RNase-dependent or Puro-dependent proteins as those whose levels are decreased upon treatment. Controlling the Negative predictive value (NPV) at 99%, we classified RNase-independent or Puro-independent proteins as those whose levels are not decreased upon treatment.
In all figures, data is presented as mean, SD and *p < 0.05. Blinding and randomization were not used in any of the experiments. Number of independent biological replicates used for experiments are listed in the figure legends. Tests and specific p-values used are indicated in the figure legends.
Data and Software Availability
Raw and analyzed data for Ribosome profiling and iCLIP have been deposited under GEO: GSE96998. MS files will be submitted to massIVE database.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-PKM1/2 (C103A3) | Cell Signaling Technology |
Cat#3190; RRID: AB_2163695 |
| Goat polyclonal anti-PKM | Abcam | Cat#ab118499; RRID: AB_10898974 |
| Goat polyclonal anti-PKM | Santa Cruz Biotechnology |
Cat#sc-65176; RRID: AB_2163698 |
| Rabbit monoclonal anti-PKM2 (D78A4) | Cell Signaling Technology |
Cat#4053; RRID: AB_1904096 |
| Rabbit polyclonal anti-PKM1 | Sigma-Aldrich | Cat#SAB4200094; RRID: AB_10624711 |
| Rat monoclonal anti-TUBULIN (YL1/2) | Abcam | Cat#ab6160; RRID: AB_305328 |
| Mouse monoclonal anti-FLAG (M2) | Sigma-Aldrich | Cat#F3165; RRID: AB_259529 |
| Mouse monoclonal anti-HA (16B12) | Covance Research Products Inc. |
Cat#MMS-101P; RRID: AB_2314672 |
| Rabbit monoclonal anti-UPF1 (D15G16) | Cell Signaling Technology |
Cat#12040 |
| Rabbit polyclonal anti-FXR1 | Cell Signaling Technology |
Cat#4173; RRID: AB_1950347 |
| Rabbit polyclonal anti-CNOT1 | Proteintech Group | Cat#14276-1-AP; RRID: AB_10888627 |
| Rabbit polyclonal anti-DDX1 | Bethyl | Cat#A300-521A; RRID: AB_451046 |
| Rabbit polyclonal anti-RPS2/uS5 | Bethyl | Cat#A303-794; RRID: AB_11218192 |
| Rabbit polyclonal anti-RPL14/eL14 | Bethyl | Cat#A305-052; RRID: AB_2621246 |
| Rabbit polyclonal anti-RPS17/eS17 | Abcam | Cat#ab138991 |
| Rabbit polyclonal anti-NEMF | Thermo Fisher Scientific |
Cat# PA5-49768; RRID: AB_2635221 |
| Rabbit polyclonal anti-RPL7/uL30 | Bethyl | Cat# A300-740A; RRID: AB_533451 |
| Rabbit polyclonal anti-RPL36/eL36 | Abcam | Cat#ab209340 |
| Rabbit polyclonal anti-CLATHRIN1 | Abcam | Cat#ab21679; RRID: AB_2083165 |
| Mouse monoclonal anti-alpha-ADAPTIN (AC1-M11) | Abcam | Cat#ab2807; RRID: AB_2056323 |
| Rabbit polyclonal anti-MVP | Proteintech Group | Cat#16478-1-AP; RRID: AB_2147597 |
| Mouse monoclonal anti-MRP11 (3G2) | Abcam | Cat#ab150335 |
| Mouse monoclonal anti-PABP (10E10) | Santa Cruz Biotechnology |
Cat#sc-32318; RRID: AB_628097 |
| Rabbit polyclonal anti-SRSF3 | Abcam | Cat#ab125124; RRID: AB_10971360 |
| Rabbit polyclonal anti-LIN28 | Abcam | Cat#ab71415; RRID: AB_2135050 |
| Rabbit polyclonal anti-UFL1 | Bethyl | Cat#A303-456A; RRID: AB_10951658 |
| Rabbit monoclonal anti-UFM1 (EPR4264(2)) | Abcam | Cat#ab109305; RRID: AB_10864675 |
| Rabbit polyclonal anti-UFM1 | Santa Cruz Biotechnology |
Cat#sc-366565 |
| Rabbit monoclonal anti-AGO2 (C34C6) | Cell Signaling Technology |
Cat#2897; RRID: AB_2096291 |
| Rabbit polyclonal anti-MKRN1 | Bethyl | Cat#A300-990A; RRID: AB_2142814 |
| Rabbit polyclonal anti-ZNF622/ZPR | Bethyl | Cat#A304-075A; RRID: AB_2621324 |
| Rabbit polyclonal anti-YTHDF1 | Proteintech Group | Cat#17479-1-AP; RRID: AB_2217473 |
| Rabbit polyclonal anti-OGT | Abcam | Cat#ab96718; RRID: AB_10680015 |
| Rabbit polyclonal anti-NAA10 | Novus | Cat#NBP2-19461 |
| Rabbit polyclonal anti-USP9X | Bethyl | Cat#A301-351A; RRID: AB_938084 |
| Rabbit monoclonal anti-HSPA8 (D12F2) | Cell Signaling Technology |
Cat#8444; RRID: AB_10831837 |
| Rabbit polyclonal anti-NSUN2 | Proteintech Group | Cat#20854-1-AP; RRID: AB_10693629 |
| Rabbit polyclonal anti-ACOT1 | Abcam | Cat#ab100915; RRID: AB_10672166 |
| Rabbit polyclonal anti-FASN | Cell Signaling Technology |
Cat#3189; RRID: AB_10692675 |
| Rabbit polyclonal anti-MDH2 | Abcam | Cat#ab96193; RRID: AB_10679348 |
| Mouse monoclonal anti-Puromycin (12D10) | EMD Millipore | Cat#MABE343; RRID: AB_2566826 |
| Rabbit polyclonal anti-RPS3/uS3 | Bethyl | Cat#A303-840A; RRID: AB_2620191 |
| Rabbit polyclonal anti-EIF3H | Sigma-Aldrich | Cat#HPA023117; RRID: AB_1848076 |
| Rat monoclonal anti-Mouse IgG-HRP (eB144) | Rockland | Cat#18-8817-31; RRID: AB_2610850 |
| Horse anti-Mouse IgG-HRP | Cell Signaling Technology |
Cat#7076; RRID: AB_330924 |
| Mouse monoclonal anti-Rabbit IgG-HRP (eB182) | Rockland | Cat#18-8816-31; RRID: AB_2610847 |
| Goat anti-Rabbit IgG-HRP | Cell Signaling Technology |
Cat#7074; RRID: AB_2099233 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cycloheximide | Sigma-Aldrich | Cat#C7698-1G |
| Harringtonine | Abcam | Cat#ab141941 |
| Lactimidomycin | EMD Millipore | Cat#506291 |
| Puromycin dihydrochloride | Sigma-Aldrich | Cat#P8833 |
| RNase A | Invitrogen | Cat#AM2270 |
| RNase T1 | Invitrogen | Cat#AM2283 |
| LIF | EMD Millipore | Cat#P8833 |
| CHIR99021 | Stemgent | Cat#04-0004 |
| PD0325901 | Stemgent | Cat#04-0006 |
| RNA Clean and Concentrator-5 columns | Zymo Research | Cat# R1016 |
| FLAG peptide | Sigma-Aldrich | Cat#F3290 |
| Dynabeads M-270 Epoxy beads | Life technologies | Cat#14301 |
| Critical Commercial Assays | ||
| Ribo-Zero Gold rRNA Removal Kit (Human/Mouse/Rat) | Illumina | Cat# MRZG126 |
| ProteoExtract Protein Precipitation Kit | EMD Millipore | Cat#539180 |
| ProteoExtract Native Membrane Protein Extraction Kit | Calbiochem | Cat#444810 |
| Dynabeads antibody coupling kit | Life technologies | Cat#14311D |
| Dual-Luciferase Reporter Assay System | Promega | Cat#E1910 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE96998 |
| Mouse reference transcriptome mm9 knownGene | UCSC Genome Browser |
https://genome.ucsc. edu/cgi- bin/hgTables |
| Mouse mm9 knownCanonical annotation | UCSC Genome Browser |
https://genome.ucsc. edu/cgi- bin/hgTables |
| Experimental Models: Cell Lines | ||
| E14Tg2a.4 mouse ESCs | Smith et al., 1987 | N/A |
| eL36-FLAG mouse embryonic stem cells | This paper | N/A |
| GFP-FLAG (Tg) mouse embryonic stem cells | This paper | N/A |
| eS17-FLAG mouse embryonic stem cells | This paper | N/A |
| eL36-FLAG; HA-PKM mouse embryonic stem cells | This paper | N/A |
| HA-UFL1 mouse embryonic stem cells | This paper | N/A |
| FLAG HA-PKM1/2 mouse embryonic stem cells | This paper | N/A |
| PKM1/2-FLAG HA H464A mouse embryonic stem cells | This paper | N/A |
| PKM1/2-FLAG HA K367M mouse embryonic stem cells | This paper | N/A |
| PKM1/2-FLAG HA mouse embryonic stem cells | This paper | N/A |
| eL22-HA (MGI:4355967); ROSA26 CAGGs CRE/ WT mouse embryonic stem cells |
This paper | N/A |
| PKM flox/wt (MGI:5547750); ROSA26 CRE-ERT2/wt (MGI:3699244) mouse embryonic stem cells |
This paper | N/A |
| Oligonucleotides | ||
| ON-TARGETplus Non-targeting Pool | Dharmacon | Cat# D-001810-10- 05 |
| ON-TARGETplus PKM Pool | Dharmacon | Cat# L-062711-00- 0005 |
| Oligonucleotides for genome editing, for q-PCR analysis to determine pluripotency of ESCs, ribosome profiling, tethering assays, and for q-PCR analysis to determine subcellular fractions of mRNAs see Table S7 |
This paper | N/A |
| Recombinant DNA | ||
| pX330-U6-Chimeric_BB-CBh-hSpCas9 | Cong L et al., 2013 | Addgene: 42230 |
| eL36_FLAG_pX330-U6-Chimeric_BB-CBh-hSpCas9 | This paper | N/A |
| eS17_FLAG_pX330-U6-Chimeric_BB-CBh-hSpCas9 | This paper | N/A |
| PKM1/2_Nterminus_HA_pX330-U6-Chimeric_BB-CBh- hSpCas9 |
This paper | N/A |
| UFL1_ HA_pX330-U6-Chimeric_BB-CBh-hSpCas9 | This paper | N/A |
| PKM1/2_Cterminus_FLAG_HA_pX330-U6- Chimeric_BB-CBh-hSpCas9 |
This paper | N/A |
| PKM1/2_Nterminus_FLAG_HA_pX330-U6- Chimeric_BB-CBh-hSpCas9 |
This paper | N/A |
| PKM1/2_H464A_pX330-U6-Chimeric_BB-CBh- hSpCas9 |
This paper | N/A |
| PKM1/2_K367M_pX330-U6-Chimeric_BB-CBh- hSpCas9 |
This paper | N/A |
| pcDNA3-FLB | Ozgur et al., 2010 | N/A |
| pcDNA3-FLB-PP7bs | Ozgur et al., 2010 | N/A |
| pcDNA3-HA-PP7cp-Pat1b | Ozgur et al., 2010 | N/A |
| pCIneo-RL | Pillai et al, 2005 | N/A |
| pcDNA3-HA-PP7cp | Ozgur et al., 2010 | N/A |
| pcDNA3-HA-PP7cp-PKM2 | This paper | N/A |
| pCAGGs-6X His Ufm1 | This paper | N/A |
| pCAGGs-rtTA-PB-GFP-FLAG | This paper | N/A |
| pCAGGs-HA-GFP | This paper | N/A |
| Software and Algorithms | ||
| Cutadapt | Martin, 2011 |
http://cutadapt.readt hedocs.io/en/stable/ |
| SAINT v2.5.0 | Choi et al., 2012 |
http://saint- apms.sourceforge.n et/Main.html |
| FASTX-Toolkit | Cold Spring Harbor Lab |
http://hannonlab.cshl .edu/fastx_toolkit/ |
| Bowtie2 | Langmead et al, 2012 |
http://bowtie- bio.sourceforge.net/ bowtie2/index.shtml |
| FastQC | Babraham Bioinformatics |
http://www.bioinform atics.babraham.ac.u k/projects/fastqc/ |
| locfdr | Efron et al., 2015 |
https://CRAN.R- project.org/package =locfdr |
| LIMMA | Ritchie et al, 2015 |
https://bioconductor. org/packages/releas e/bioc/html/limma.ht ml |
| STAR | Dobin et al., 2013 |
https://github.com/al exdobin/STAR |
| Samtools | Li et al, 2009 |
http://samtools.sourc eforge.net/ |
| RStudio | R Studio |
https://www.rstudio.c om/ |
| FAST-iCLIP | Flynn et al, 2015 |
https://github.com/C hangLab/FAST- iCLIP |
Supplementary Material
Supplementary Figure 1. Complexes that are not bona fide components of the ribosome are present in sucrose gradient fractions due to similar centrifugation properties. Related to Figure 1.
A, Density gradient centrifugation has been used to separate ribosomes as well as membrane fractions, centrosomes, and subcellular organelles. The sucrose gradients are prepared and lysates are layered on top of the gradient. Cellular components migrate through the gradient and separate based on their density. Subsequent fractionation of sucrose gradients allows isolation of these components.
B, Representative UV absorbance at 260 nm helps monitor abundant RNAs such as ribosomal RNA within fractions. Incubating the cellular lysate with EDTA before layering the lysate on the sucrose gradient dissociates the 80S and polysomes into 40S and 60S, therefore results in the accumulation of 40S and 60S fractions and moves the ribosome subunits to earlier fractions. 40S, 60S, and 80S indicate the positions of the respective ribosomal subunits and the assembled monosome on the gradient. Distributions of the indicated proteins across the gradient are assessed by precipitating the fractions and were analyzed by Western Blotting.
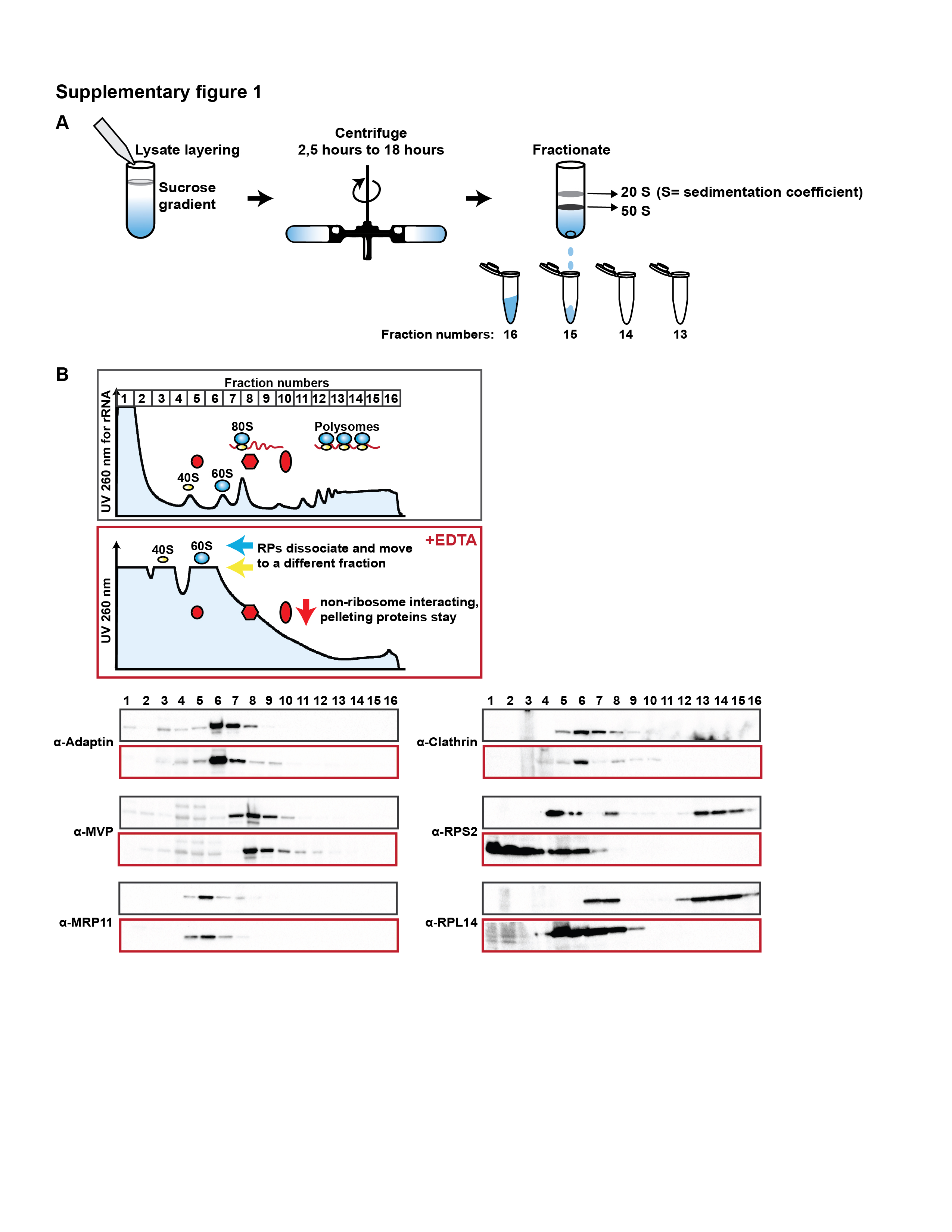{kind=link}
Table S3. TMT experiments for control, RNase, and Puromycin treated IPs. Summed signal to noise across all channels as well as the adjusted P values, FDR, and NPV values for each interaction are presented. Related to Figure 2.
Table S4. Proteins identified in the His-Ufm1 experiments in comparison to the background. Unique peptide numbers for each biological replicate along with their Fold Change (FC) are shown. Related to Figure 4D.
Table S5. Ribosome profiling for PKM knockdown compared to control and PKM2 iCLIP enrichment scores. Log2 RNA, Log2 Ribo, Log2 TE values upon PKM knockdown are shown along with iCLIP enrichment scores. Related to Figure 6D, E.
Table S6. SILAC quantitative MS for PKM-enriched ribosomes compared to control ribosomes. Mean 0.61; S.D. 0.50; cut off values for enriched proteins was 2.5 S.D. from the mean. Related to Figure 7A.
Table S7. Primers used are shown. Related to Star Methods.
Supplementary Figure 2. FLAG tagged eL36 and eS17 are incorporated into functional ribosomes. Related to Figure 1.
A, ES cells were generated from eL22-HA mice intercrossed to Cre recombinase-expressing mice resulting in deletion of the wild-type exon 4 and replacement with the HA tagged exon 4. This targeting strategy as previously described (Sanz et al., 2009) is shown. The distribution of eL22-HA across sucrose gradient fractions was analyzed by blotting for the HA antibody.
B, eL36-FLAG, eS17-FLAG ESCs and untagged ESCs were analyzed by sucrose gradient fractionation. UV absorbance at 260 nm was used to assess 40S, 60S, 80S, and polysomes traces. The UV traces of the FLAG tagged RPs shows proper ribosomal assembly demonstrating the non-perturbative nature of the small FLAG tags. Incorporation of the tagged RPs into polysomes analyzed by FLAG antibody shows that it is similar to the distribution of the endogenous untagged RPs analyzed by primary antibodies, showing that tagged RPs belong to functional ribosomes.
Supplementary Figure 3. Control, RNase-treated, and puromycin-treated ribosome IPs result in the identification of direct RAPs. Related to Figure 2.
A, RNase A treatment preserves ribosome integrity and effectively cleaves polysomes. RNase A digestion was optimized by titrating the enzyme. Unlike RNase I digestion, RNAse A resulted in complete cleavage of polysomes and a substantial increase in the 80S fraction. Shown are Western blots of the fractions using an antibody against a RP. The lowest RNase A concentration as detailed in the methods was used.
B, Schematic of the RNase A and puromycin treatments. After FLAG IP, RNase treatment is performed before FLAG peptide elution. Cytoplasmic lysates, -RNase FLAG elution as well as +RNase elution are loaded onto sucrose gradients to determine the relative ratios of different ribosome pools, such as 80S, and polysomes. Upon RNase A digestion, if IPs are eluted with FLAG peptides, polysomes are decreased, and 80S fraction is increased, consistent with RNase digestion of mRNAs resulting in ribosome footprints. For the puromycin experiment, an antibody detecting puromycin is used. 0.01% of cytoplasmic lysates are used as an input and 40% of the IPs are run in the western blot. Since puromycin is incorporated into nascent peptides, proteins with different sizes are detected within the cytoplasmic lysate.
C, Coomassie stains of control, RNase treated, and puromycin-treated ribosome IPs are shown with each biological replicate (BR).
{kind=link}
Supplementary Figure 4. Histograms of test statistics from RNase and puromycin treatment experiments and peptide coverage metaplots. Related to Figure 2.
A, Histograms and mixture models of the test statistics (t-statistic) from the RNase A or puromycintreatment experiments. The observed distribution of t-statistics are substantially wider than N(0, 1), prompting the use of empirical modeling approaches to implement a tenable null hypothesis. Green line indicates the mixture density and dashed blue lines indicate the empirical null density estimated by locfdr method. The vertical red line indicates the 15% FDR cutoff and the vertical blue line indicates the 99 % NPV cutoff.
B, Metaplot of peptide coverage in eL36-MS. Abundance-normalized coverage of peptides across 10 bins of protein length is plotted for RPs and non-RPs separately.
{kind=link}
Supplementary Figure 5. Sucrose gradient validations for the select direct and mRNA-dependent ribosome interactors using RNase A digestion. Related to Figure 3.
After cytoplasmic lysate preparation, control and RNAse A lysates are analyzed via sucrose gradient fractionation. Collected fractions are precipitated and blotted for indicated the proteins. ACOT1, FASN, MDH2 metabolism enzymes are used as negative controls.
Supplementary Figure 6. Sucrose gradient validations for the select direct and mRNA-dependent ribosome interactors using EDTA treatment. Related to Figure 3.
After cytoplasmic lysate preparation, control and EDTA lysates are analyzed via sucrose gradient fractionation. Collected fractions are precipitated and blotted for the indicated proteins.
{kind=link}
Supplementary Figure 7. Both PKM1 and PKM2 can interact with the ribosome and PKM is an RNA binding protein. Related to Figure 5 and 6.
A, Generation of conditional PKM2 knock-out mouse ESCs. PKM2flox/flox mice are crossed to mice carrying inducible Cre recombinase (Cre-ER) to produce ESCs.
B, Treatment of conditional heterozygote PKM2flox/wt mouse ESCs with 4-hydroxytamoxifen results in excision of PKM exon 10 in PKM2flox/wt cells, and Western Blot analysis showed a decrease of PKM2 protein as analyzed by an antibody that recognizes the PKM2 exclusive region. The same fractions were run on a separate gel and blotted with a PKM1 specific antibody. The * denotes non-specific bands recognized by the PKM1 specific antibody.
C, Endogenous PKM tagging does not affect its polysome localization. PKM1/2 was endogenously tagged either at the N or C terminus with FLAG-HA tag. Each tagged ESCs shows similar PKM2 distribution at t polysomes compared to the PKM2 distribution detected by primary antibodies in untagged cells, showing that the FLAG-HA tag does not affect PKM2 localization to polysomes.
D, PKM2 is an RBP. Autoradiogram of 32P-labelled RNA crosslinked to PKM2-FLAG-HA. RNAprotein complexes were seen in the purifications from PKM2-FLAG-HA cells, but not from control, untagged cells. High and low RNase I concentrations were used to confirm the signal is RNA. To validate whether the IP washing conditions after the FLAG-HA tandem IP are stringent, we used non- UV crosslinked PKM2-FLAG-HA cells. In the absence of UV crosslinking, RNA signal was not present, indicating that in vivo RNA targets that were crosslinked to PKM2 were isolated. Western blotting with HA on the right, showing that the RNA signal was above the molecular weight of PKM2.
E, Total counts of PKM2-iCLIP reads for protein-coding RNAs across introns, 5'UTRs, CDS, or 3'UTRs, normalized by total length of each annotation type. Values above the dashed line indicate enrichment.
F, PKM siRNA treated cytoplasmic lysates are blotted with antibodies that recognize PKM2 exclusively, PKM1 exclusively, and both PKM1 and PKM2.
Table S1. Biological replicates for MS experiments for the eL36-FLAG and eS17-FLAG experiments. Unique peptide numbers for each biological replicate are shown along with mean coverage in percentage. SAINT scores and Fold changes (FC) are shown. Related to Figure 1A, B.
Table S2. Enriched GO categories (biological process) for the ribo-interactome and PKM iCLIP. Fold enrichment, P-values, and the name of the associated members are shown. Related to Figure 1C and Figure 6F.
Highlights.
A mammalian ribosome affinity approach reveals ribosome associated proteins (RAPs)
A multitude of RAPS link the ribosome to diverse cellular and molecular functions
Ribosomes are modified by metazoan specific ufmylation
A metabolic enzyme, PKM, is at ER-ribosomes and translates ER destined mRNAs
Acknowledgments
We thank Josh Elias, Stanford and Randall K. Mann, Stanford for advice on mass spectrometry approaches. We thank Ryan Kunz and Rachel B. Rodrigues (Thermo Fisher Scientific Center for Multiplexed Proteomics at Harvard Medical School) for the TMT experiment. We thank Tom Cech for naming RAPs. We thank Georg Stoecklin at DKFZ and ZMBH, Germany for kindly sharing the plasmids used in PKM tethering experiments. We thank Khanh Ngo for the initial optimization of sucrose gradient-mass spectrometry experiments. We thank Davide Ruggero, UCSF for his critical comments on the manuscript. This research was supported by the New York Stem Cell Foundation (M.B), NIH Director’s New Innovator Award, 7DP2OD00850902 (M.B.), R21HD086730 (M.B), Alfred P. Sloan Research Fellowship (M.B), Mallinckrodt Foundation Award (M.B.), Pew Scholars Award (M.B.) and P50-HG007735 (H.Y.C), R01-HG004361 (H.Y.C), and R01-ES023168 (H.Y.C; R.A.F is supported by NIH 1F30CA189514-01 and Stanford Medical Scientist Training Program; G.C.T. is supported by The Paul and Daisy Soros Fellowships for New Americans and Stanford Medical Scientist Training Program; K.L. is a Layton family fellow of the of the Damon Runyon Cancer Research Foundation; A.F.X. is supported by the Stanford Medical Scientist Training Program; G.W.B. is supported by the Benchmark Stanford Graduate Fellowship; D.S.B. is a Philip O’Bryan Montgomery, Jr., MD fellow of the Damon Runyon Cancer Research Foundation and Postdoctoral Fellow of American Heart Association. M.B. is a New York Stem Cell Foundation Robertson Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
M.B. and D.S.B. conceived and M.B. supervised the project; D.S.B. and M.B. designed the experiments and D.S.B performed experiments; G.C.T. performed the ribosome profiling experiment and analyzed the data and performed polysome gradient experiments; H.Y.C. and R.A.F. designed the iCLIP experiment and analyzed the resulting data; G.W.B. analyzed data for both of the ribosome profiling and iCLIP experiments; K.L. performed the assays for the tethered function experiment; A.F.X. generated His-Ufn1 plasmid; D.S.B. performed the rest of the experiments. M.B. and D.S.B wrote the manuscript with input from all the co-authors.
References
- Battle DJ, Kasim M, Yong J, Lotti F, Lau CK, Mouaikel J, Zhang Z, Han K, Wan L, Dreyfuss G. The SMN complex: an assembly machine for RNPs. Cold Spring Harb Symp Quant Biol. 2006;71:313–320. doi: 10.1101/sqb.2006.71.001. [DOI] [PubMed] [Google Scholar]
- Bendall SC, Hughes C, Stewart MH, Doble B, Bhatia M, Lajoie GA. Prevention of amino acid conversion in SILAC experiments with embryonic stem cells. Mol Cell Proteomics. 2008;7:1587–1597. doi: 10.1074/mcp.M800113-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, Topisirovic I. Targeting the translation machinery in cancer. Nat Rev Drug Discov. 2015;14:261–278. doi: 10.1038/nrd4505. [DOI] [PubMed] [Google Scholar]
- Brina D, Miluzio A, Ricciardi S, Biffo S. eIF6 anti-association activity is required for ribosome biogenesis, translational control and tumor progression. Biochim Biophys Acta. 2015;1849:830–835. doi: 10.1016/j.bbagrm.2014.09.010. [DOI] [PubMed] [Google Scholar]
- Budkevich T, Giesebrecht J, Altman RB, Munro JB, Mielke T, Nierhaus KH, Blanchard SC, Spahn CM. Structure and dynamics of the mammalian ribosomal pretranslocation complex. Mol Cell. 2011;44:214–224. doi: 10.1016/j.molcel.2011.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen E, Sharma MR, Shi X, Agrawal RK, Joseph S. Fragile X mental retardation protein regulates translation by binding directly to the ribosome. Mol Cell. 2014;54:407–417. doi: 10.1016/j.molcel.2014.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H, Liu G, Mellacheruvu D, Tyers M, Gingras AC, Nesvizhskii AI. Analyzing protein-protein interactions from affinity purification-mass spectrometry data with SAINT. Curr Protoc Bioinformatics Chapter. 2012;8 doi: 10.1002/0471250953.bi0815s39. Unit8 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne AN, Siddegowda BB, Estes PS, Johannesmeyer J, Kovalik T, Daniel SG, Pearson A, Bowser R, Zarnescu DC. Futsch/MAP1B mRNA is a translational target of TDP-43 and is neuroprotective in a Drosophila model of amyotrophic lateral sclerosis. J Neurosci. 2014;34:15962–15974. doi: 10.1523/JNEUROSCI.2526-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S, Peer E, Kol N, Ben-Haim MS, Dai Q, Di Segni A, Salmon-Divon M, Clark WC, et al. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature. 2016;530:441–446. doi: 10.1038/nature16998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Efron B. Large-Scale Simultaneous Hypothesis Testing: The Choice of a Null Hypothesis. Journal of the American Statistical Association. 2004;99:96–104. [Google Scholar]
- Florens L, Carozza MJ, Swanson SK, Fournier M, Coleman MK, Workman JL, Washburn MP. Analyzing chromatin remodeling complexes using shotgun proteomics and normalized spectral abundance factors. Methods. 2006;40:303–311. doi: 10.1016/j.ymeth.2006.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn RA, Martin L, Spitale RC, Do BT, Sagan SM, Zarnegar B, Qu K, Khavari PA, Quake SR, Sarnow P, et al. Dissecting noncoding and pathogen RNA-protein interactomes. RNA. 2015;21:135–143. doi: 10.1261/rna.047803.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garreau de Loubresse N, Prokhorova I, Holtkamp W, Rodnina MV, Yusupova G, Yusupov M. Structural basis for the inhibition of the eukaryotic ribosome. Nature. 2014;513:517–522. doi: 10.1038/nature13737. [DOI] [PubMed] [Google Scholar]
- Girard M, Allaire PD, Blondeau F, McPherson PS. Isolation of clathrin-coated vesicles by differential and density gradient centrifugation. Curr Protoc Cell Biol Chapter. 2005;3 doi: 10.1002/0471143030.cb0313s26. Unit 3 13. [DOI] [PubMed] [Google Scholar]
- Graumann J, Hubner NC, Kim JB, Ko K, Moser M, Kumar C, Cox J, Scholer H, Mann M. Stable isotope labeling by amino acids in cell culture (SILAC) and proteome quantitation of mouse embryonic stem cells to a depth of 5,111 proteins. Mol Cell Proteomics. 2008;7:672–683. doi: 10.1074/mcp.M700460-MCP200. [DOI] [PubMed] [Google Scholar]
- Holthuis JC, Menon AK. Lipid landscapes and pipelines in membrane homeostasis. Nature. 2014;510:48–57. doi: 10.1038/nature13474. [DOI] [PubMed] [Google Scholar]
- Huppertz I, Attig J, D’Ambrogio A, Easton LE, Sibley CR, Sugimoto Y, Tajnik M, Konig J, Ule J. iCLIP: protein-RNA interactions at nucleotide resolution. Methods. 2014;65:274–287. doi: 10.1016/j.ymeth.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc. 2012;7:1534–1550. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, Li J, Yu Y, Sasaki M, Horner JW, et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell. 2013;155:397–409. doi: 10.1016/j.cell.2013.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israelsen WJ, Vander Heiden MG. Pyruvate kinase: Function, regulation and role in cancer. Semin Cell Dev Biol. 2015;43:43–51. doi: 10.1016/j.semcdb.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagannathan S, Nwosu C, Nicchitta CV. Analyzing mRNA localization to the endoplasmic reticulum via cell fractionation. Methods Mol Biol. 2011;714:301–321. doi: 10.1007/978-1-61779-005-8_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan CH, Williams CC, Weissman JS. Principles of ER cotranslational translocation revealed by proximity-specific ribosome profiling. Science. 2014;346:1257521. doi: 10.1126/science.1257521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkin J, Henkel T, Nielsen AF, Minnich M, Popow J, Kaufmann T, Heindl K, Hoffmann T, Busslinger M, Martinez J. The mammalian tRNA ligase complex mediates splicing of XBP1 mRNA and controls antibody secretion in plasma cells. EMBO J. 2014;33:2922–2936. doi: 10.15252/embj.201490332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klass DM, Scheibe M, Butter F, Hogan GJ, Mann M, Brown PO. Quantitative proteomic analysis reveals concurrent RNA-protein interactions and identifies new RNA-binding proteins in Saccharomyces cerevisiae. Genome Res. 2013;23:1028–1038. doi: 10.1101/gr.153031.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Cleveland DW. Rethinking ALS: the FUS about TDP-43. Cell. 2009;136:1001–1004. doi: 10.1016/j.cell.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet Journal. 2011;17:10–12. [Google Scholar]
- Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY, et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat Methods. 2013;10:730–736. doi: 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narla A, Ebert BL. Ribosomopathies: human disorders of ribosome dysfunction. Blood. 2010;115:3196–3205. doi: 10.1182/blood-2009-10-178129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noeske J, Cate JH. Structural basis for protein synthesis: snapshots of the ribosome in motion. Curr Opin Struct Biol. 2012;22:743–749. doi: 10.1016/j.sbi.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozgur S, Chekulaeva M, Stoecklin G. Human Pat1b connects deadenylation with mRNA decapping and controls the assembly of processing bodies. Mol Cell Biol. 2010;30:4308–4323. doi: 10.1128/MCB.00429-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestka S. Inhibitors of ribosome functions. Annu Rev Microbiol. 1971;25:487–562. doi: 10.1146/annurev.mi.25.100171.002415. [DOI] [PubMed] [Google Scholar]
- Pillai RS, Bhattacharyya SN, Artus CG, Zoller T, Cougot N, Basyuk E, Bertrand E, Filipowicz W. Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science. 2005;309:1573–1576. doi: 10.1126/science.1115079. [DOI] [PubMed] [Google Scholar]
- Popow J, Englert M, Weitzer S, Schleiffer A, Mierzwa B, Mechtler K, Trowitzsch S, Will CL, Luhrmann R, Soll D, et al. HSPC117 is the essential subunit of a human tRNA splicing ligase complex. Science. 2011;331:760–764. doi: 10.1126/science.1197847. [DOI] [PubMed] [Google Scholar]
- Reber S. Isolation of centrosomes from cultured cells. Methods Mol Biol. 2011;777:107–116. doi: 10.1007/978-1-61779-252-6_8. [DOI] [PubMed] [Google Scholar]
- Reschke M, Clohessy JG, Seitzer N, Goldstein DP, Breitkopf SB, Schmolze DB, Ala U, Asara JM, Beck AH, Pandolfi PP. Characterization and analysis of the composition and dynamics of the mammalian riboproteome. Cell Rep. 2013;4:1276–1287. doi: 10.1016/j.celrep.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz E, Yang L, Su T, Morris DR, McKnight GS, Amieux PS. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci U S A. 2009;106:13939–13944. doi: 10.1073/pnas.0907143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S, Brown A, Santhanam B, Hegde RS. Structure and assembly pathway of the ribosome quality control complex. Mol Cell. 2015;57:433–444. doi: 10.1016/j.molcel.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai YT, Suzuki T, Morita M, Takahashi A, Yamamoto T. Multifunctional roles of the mammalian CCR4-NOT complex in physiological phenomena. Front Genet. 2014;5:286. doi: 10.3389/fgene.2014.00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyh-Chang N, Daley GQ, Cantley LC. Stem cell metabolism in tissue development and aging. Development. 2013;140:2535–2547. doi: 10.1242/dev.091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek D, Barna M. An emerging role for the ribosome as a nexus for post-translational modifications. Curr Opin Cell Biol. 2017;45:92–101. doi: 10.1016/j.ceb.2017.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strunk BS, Loucks CR, Su M, Vashisth H, Cheng S, Schilling J, Brooks CL, 3rd, Karbstein K, Skiniotis G. Ribosome assembly factors prevent premature translation initiation by 40S assembly intermediates. Science. 2011;333:1449–1453. doi: 10.1126/science.1208245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsumi K, Yamamoto-Mukai H, Shimizu R, Waguri S, Sou YS, Sakamoto A, Taya C, Shitara H, Hara T, Chung CH, et al. The Ufm1-activating enzyme Uba5 is indispensable for erythroid differentiation in mice. Nat Commun. 2011;2:181. doi: 10.1038/ncomms1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A, Schafer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK, Hamon C. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem. 2003;75:1895–1904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- Ting L, Rad R, Gygi SP, Haas W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat Methods. 2011;8:937–940. doi: 10.1038/nmeth.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorhees RM, Fernandez IS, Scheres SH, Hegde RS. Structure of the mammalian ribosome-Sec61 complex to 3.4 A resolution. Cell. 2014;157:1632–1643. doi: 10.1016/j.cell.2014.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, He C. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell. 2015;161:1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Eliscovich C, Yoon YJ, Singer RH. Translation dynamics of single mRNAs in live cells and neurons. Science. 2016;352:1430–1435. doi: 10.1126/science.aaf1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Hoek TA, Vale RD, Tanenbaum ME. Dynamics of Translation of Single mRNA Molecules In Vivo. Cell. 2016;165:976–989. doi: 10.1016/j.cell.2016.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarnegar BJ, Flynn RA, Shen Y, Do BT, Chang HY, Khavari PA. irCLIP platform for efficient characterization of protein-RNA interactions. Nat Methods. 2016;13:489–492. doi: 10.1038/nmeth.3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Zhu X, Zhang Y, Cai Y, Chen J, Sivaprakasam S, Gurav A, Pi W, Makala L, Wu J, et al. RCAD/Ufl1, a Ufm1 E3 ligase, is essential for hematopoietic stem cell function and murine hematopoiesis. Cell Death Differ. 2015;22:1922–1934. doi: 10.1038/cdd.2015.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Complexes that are not bona fide components of the ribosome are present in sucrose gradient fractions due to similar centrifugation properties. Related to Figure 1.
A, Density gradient centrifugation has been used to separate ribosomes as well as membrane fractions, centrosomes, and subcellular organelles. The sucrose gradients are prepared and lysates are layered on top of the gradient. Cellular components migrate through the gradient and separate based on their density. Subsequent fractionation of sucrose gradients allows isolation of these components.
B, Representative UV absorbance at 260 nm helps monitor abundant RNAs such as ribosomal RNA within fractions. Incubating the cellular lysate with EDTA before layering the lysate on the sucrose gradient dissociates the 80S and polysomes into 40S and 60S, therefore results in the accumulation of 40S and 60S fractions and moves the ribosome subunits to earlier fractions. 40S, 60S, and 80S indicate the positions of the respective ribosomal subunits and the assembled monosome on the gradient. Distributions of the indicated proteins across the gradient are assessed by precipitating the fractions and were analyzed by Western Blotting.
Table S3. TMT experiments for control, RNase, and Puromycin treated IPs. Summed signal to noise across all channels as well as the adjusted P values, FDR, and NPV values for each interaction are presented. Related to Figure 2.
Table S4. Proteins identified in the His-Ufm1 experiments in comparison to the background. Unique peptide numbers for each biological replicate along with their Fold Change (FC) are shown. Related to Figure 4D.
Table S5. Ribosome profiling for PKM knockdown compared to control and PKM2 iCLIP enrichment scores. Log2 RNA, Log2 Ribo, Log2 TE values upon PKM knockdown are shown along with iCLIP enrichment scores. Related to Figure 6D, E.
Table S6. SILAC quantitative MS for PKM-enriched ribosomes compared to control ribosomes. Mean 0.61; S.D. 0.50; cut off values for enriched proteins was 2.5 S.D. from the mean. Related to Figure 7A.
Table S7. Primers used are shown. Related to Star Methods.
Supplementary Figure 2. FLAG tagged eL36 and eS17 are incorporated into functional ribosomes. Related to Figure 1.
A, ES cells were generated from eL22-HA mice intercrossed to Cre recombinase-expressing mice resulting in deletion of the wild-type exon 4 and replacement with the HA tagged exon 4. This targeting strategy as previously described (Sanz et al., 2009) is shown. The distribution of eL22-HA across sucrose gradient fractions was analyzed by blotting for the HA antibody.
B, eL36-FLAG, eS17-FLAG ESCs and untagged ESCs were analyzed by sucrose gradient fractionation. UV absorbance at 260 nm was used to assess 40S, 60S, 80S, and polysomes traces. The UV traces of the FLAG tagged RPs shows proper ribosomal assembly demonstrating the non-perturbative nature of the small FLAG tags. Incorporation of the tagged RPs into polysomes analyzed by FLAG antibody shows that it is similar to the distribution of the endogenous untagged RPs analyzed by primary antibodies, showing that tagged RPs belong to functional ribosomes.
Supplementary Figure 3. Control, RNase-treated, and puromycin-treated ribosome IPs result in the identification of direct RAPs. Related to Figure 2.
A, RNase A treatment preserves ribosome integrity and effectively cleaves polysomes. RNase A digestion was optimized by titrating the enzyme. Unlike RNase I digestion, RNAse A resulted in complete cleavage of polysomes and a substantial increase in the 80S fraction. Shown are Western blots of the fractions using an antibody against a RP. The lowest RNase A concentration as detailed in the methods was used.
B, Schematic of the RNase A and puromycin treatments. After FLAG IP, RNase treatment is performed before FLAG peptide elution. Cytoplasmic lysates, -RNase FLAG elution as well as +RNase elution are loaded onto sucrose gradients to determine the relative ratios of different ribosome pools, such as 80S, and polysomes. Upon RNase A digestion, if IPs are eluted with FLAG peptides, polysomes are decreased, and 80S fraction is increased, consistent with RNase digestion of mRNAs resulting in ribosome footprints. For the puromycin experiment, an antibody detecting puromycin is used. 0.01% of cytoplasmic lysates are used as an input and 40% of the IPs are run in the western blot. Since puromycin is incorporated into nascent peptides, proteins with different sizes are detected within the cytoplasmic lysate.
C, Coomassie stains of control, RNase treated, and puromycin-treated ribosome IPs are shown with each biological replicate (BR).
Supplementary Figure 4. Histograms of test statistics from RNase and puromycin treatment experiments and peptide coverage metaplots. Related to Figure 2.
A, Histograms and mixture models of the test statistics (t-statistic) from the RNase A or puromycintreatment experiments. The observed distribution of t-statistics are substantially wider than N(0, 1), prompting the use of empirical modeling approaches to implement a tenable null hypothesis. Green line indicates the mixture density and dashed blue lines indicate the empirical null density estimated by locfdr method. The vertical red line indicates the 15% FDR cutoff and the vertical blue line indicates the 99 % NPV cutoff.
B, Metaplot of peptide coverage in eL36-MS. Abundance-normalized coverage of peptides across 10 bins of protein length is plotted for RPs and non-RPs separately.
Supplementary Figure 5. Sucrose gradient validations for the select direct and mRNA-dependent ribosome interactors using RNase A digestion. Related to Figure 3.
After cytoplasmic lysate preparation, control and RNAse A lysates are analyzed via sucrose gradient fractionation. Collected fractions are precipitated and blotted for indicated the proteins. ACOT1, FASN, MDH2 metabolism enzymes are used as negative controls.
Supplementary Figure 6. Sucrose gradient validations for the select direct and mRNA-dependent ribosome interactors using EDTA treatment. Related to Figure 3.
After cytoplasmic lysate preparation, control and EDTA lysates are analyzed via sucrose gradient fractionation. Collected fractions are precipitated and blotted for the indicated proteins.
Supplementary Figure 7. Both PKM1 and PKM2 can interact with the ribosome and PKM is an RNA binding protein. Related to Figure 5 and 6.
A, Generation of conditional PKM2 knock-out mouse ESCs. PKM2flox/flox mice are crossed to mice carrying inducible Cre recombinase (Cre-ER) to produce ESCs.
B, Treatment of conditional heterozygote PKM2flox/wt mouse ESCs with 4-hydroxytamoxifen results in excision of PKM exon 10 in PKM2flox/wt cells, and Western Blot analysis showed a decrease of PKM2 protein as analyzed by an antibody that recognizes the PKM2 exclusive region. The same fractions were run on a separate gel and blotted with a PKM1 specific antibody. The * denotes non-specific bands recognized by the PKM1 specific antibody.
C, Endogenous PKM tagging does not affect its polysome localization. PKM1/2 was endogenously tagged either at the N or C terminus with FLAG-HA tag. Each tagged ESCs shows similar PKM2 distribution at t polysomes compared to the PKM2 distribution detected by primary antibodies in untagged cells, showing that the FLAG-HA tag does not affect PKM2 localization to polysomes.
D, PKM2 is an RBP. Autoradiogram of 32P-labelled RNA crosslinked to PKM2-FLAG-HA. RNAprotein complexes were seen in the purifications from PKM2-FLAG-HA cells, but not from control, untagged cells. High and low RNase I concentrations were used to confirm the signal is RNA. To validate whether the IP washing conditions after the FLAG-HA tandem IP are stringent, we used non- UV crosslinked PKM2-FLAG-HA cells. In the absence of UV crosslinking, RNA signal was not present, indicating that in vivo RNA targets that were crosslinked to PKM2 were isolated. Western blotting with HA on the right, showing that the RNA signal was above the molecular weight of PKM2.
E, Total counts of PKM2-iCLIP reads for protein-coding RNAs across introns, 5'UTRs, CDS, or 3'UTRs, normalized by total length of each annotation type. Values above the dashed line indicate enrichment.
F, PKM siRNA treated cytoplasmic lysates are blotted with antibodies that recognize PKM2 exclusively, PKM1 exclusively, and both PKM1 and PKM2.
Table S1. Biological replicates for MS experiments for the eL36-FLAG and eS17-FLAG experiments. Unique peptide numbers for each biological replicate are shown along with mean coverage in percentage. SAINT scores and Fold changes (FC) are shown. Related to Figure 1A, B.
Table S2. Enriched GO categories (biological process) for the ribo-interactome and PKM iCLIP. Fold enrichment, P-values, and the name of the associated members are shown. Related to Figure 1C and Figure 6F.