

Abstract
Mammalian protein O-mannosylation, initiated by attachment of α-mannopyranose to Ser or Thr residues, comprise a group of post-translational modifications (PTMs) involved in muscle and brain development. Recent advances in glycoproteomics methodology and the “Simple Cell” strategy have enabled rapid identification of glycoproteins and specific glycosylation sites. Despite the enormous progress made, the biological impact of the mammalian O-mannosyl glycoproteome remains largely unknown to date. Tools are still needed to investigate the structure, role, and abundance of O-mannosyl glycans. Although O-mannosyl branching has been shown to be of relevance in integrin-dependent cell migration, and also plays a role in demyelinating diseases, such as multiple sclerosis, a broader understanding of the biological roles of branched O-mannosyl glycans is lacking in part due to the paucity of detection tools. In this work, a glycopeptide vaccine construct was synthesized and used to generate antibodies against branched O-mannosyl glycans. Glycopeptide microarray screening revealed high selectivity of the induced antibodies for branched glycan core structures presented on different peptide backbones, with no cross-reactivity observed with related linear glycans. For comparison, microarray screening of the mannose-binding lectin concanavalin A(ConA), which is commonly used in glycoproteomics workflows to enrich tryptic O-mannosyl peptides, showed that the ConA lectin did not recognize branched O-mannosyl glycans. The binding preference of ConA for short linear O-mannosyl glycanswas rationalized in terms of molecular structure using crystallographic data augmented by molecular modeling. The contrast between the ConA binding specificity and that of the new antibodies indicates a novel role for the antibodies in studies of protein O-mannosylation.
Keywords: antibodies, carbohydrates, glycopeptides, lectins, vaccines
Introduction
Protein O-mannosylation comprises a group of modifications involved in important functions in brain and muscle glycobiology.[1] In mammals, the protein O-mannosyltransferase 1 and 2 (POMT1, POMT2)are responsible for the transfer of an initial mannose (Man) monosaccharide to Ser or Thr residues on protein targets.[2] Subsequent addition of N-acetylglucosamine (GlcNAc) at the 2-position of the core Man residue by the enzyme protein O-mannose-β-1,2-N-acetylglucosaminyl transferase 1(POMGnT1) results in the formation of linear m1 core structures (GlcNAcβ1,2Manα).[3] Addition of a second GlcNAc at the mannose 6-position by β-1,6-N-acetylglucosaminyl transferase (GnT-Vb/GnT-IX) forms the branched m2 core glycan (GlcNAcβ1,2[GlcNAcβ1,6]Manα).[4] The core ml and m2 structures typically undergo further extension forming more complex O-glycanstructures.[5] For instance, the O-mannosyl core m1 glycans in the protein α-Dystroglycan(α-DG) have been found to be modified with sialic acid (Sia, neuraminic acid, Neu5Ac)attached to a lactosamine (3Galβ1,4GlcNAcβ)extension at the 4-position of the GlcNAc residues, resulting in a sialylated tetrasaccharide epitope (Neu5Acα2,3Galβ1,4GlcNAcβ1,2Manα).[6] Other O-mannose core m1 and m2 elongated glycan epitopes found on α-DG contain fucosyl-N-acetyllactosamine, or a sulfated trisaccharide (GlcA[3S]β1,3Galβ1,4GlcNAcβ) known as the human natural killer-1 (HNK-1) epitope.[7] The HNK-1epitopemodulates the function of the receptor protein-tyrosine phosphatase, known as RPTPβ.[4a] Increased HNK-1 epitope presentation by core m2 branching results in RPTPβ phosphatase inhibition, causing increased levels of integrin-dependent cell migration.[4a,8] Furthermore, RPTPβ is important for recovery from demyelinating diseases, such as multiple sclerosis and spinal cord injury, and inhibition by HNK-1 core m2 branching is believed to block the remyelination process.[9] A recent study has also showed that core m2 branching is involved in increased prostate cancer tumor growth and metastasis.™ A third O-mannosyl core structure, the core m3 glycan (GlcNAcβ1,4Man), was recently discovered.[11] In α-DG, a GalNAc residue may elongate the core m3 glycan (GalNAcβ1,3GlcNAcβ1,4Man). This core can be further extended by phosphorylation of the terminal GalNAc, addition of two ribitol-5-phosphate groups, and linking to repeating disaccharide units (-3Xylα1,3GlcAβ1-).[11,12] The GalNAc elongated core m3 glycans and ribitol-5-phosphate linkages are necessary for formation of the proteoglycan structures on α-DG, and are essential for stabilization of the dystrophin–glycoprotein complex, which acts as a transmembrane linker between the extracellular matrix and intracellular cytoskeleton.[6a,11,12b,13] Impaired glycosylation of α-DG has been shown to result in severe congenital muscular dystrophies.[14]
α-DG was the first O-mannosylated protein found in mammals, however, a study of O-glycans in total rabbit brain indicated that many more unidentified proteins carry O-mannosyl glycans, and led to the conclusion that about 30% of the released O-glycans were mannosylated.[15] The high abundance of protein O-mannosyl glycans in brain was also shown in a study of the mice brain proteome.[16] Subsequently, a handful of additional mammalian glycoproteins were identified containing O-mannosyl modifications, including the transmembrane protein CD24, amonoclonal IgG2 light chain from Chinese hamster ovary cells, phosphacan, tenascin-R, neurofascin 186, neurocan, brevican, versican, RPTPβ, and RPTPζ.[4a,17] Recently, the analytical “SimpleCell” strategy combined with glycopeptide enrichment using the lectin concanavalin A(ConA) and LC-MS identification, showed that many proteins from cultured breast cancer cells are modified with protein O-mannosylation.[18] In particular, a large family of cadherin cell membrane receptors were found to be major carriers of O-mannosyl glycans. Concurrently, LC-MS in combination with α-mannosidase treatment identified mono-O-mannosyl glycans on T-cadherin from rabbit muscle and on recombinant epithelial E-cadherin from human embryonic kidney cells.[19] O-mannosylation of E-cadherin has been shown to be crucial for the function of cadherin mediated cell–cell adhesion.[19b] A recent study revealed that in gastric carcinomas, decreased O-mannosylation unexpectedly affects the N-glycosylation pattern of E-cadherin, thereby causing its dysregulation.[20] A monoclonal antibody (RKU-1-3-5) specific for O-linked α-mannose was later developed and applied in immunostaining of murine brain glycoproteins,[21] leading to the observation that mono-O-mannosyl glycans are prevalent in inhibitory GABAergic neurons and perineuronal nets. In the same study, using ConA glycopeptide enrichment, α-mannosidase treatment, and LC-MS-based identification of (glyco)peptides, numerous murine brain glycoproteins were found containing mono-O-mannosyl glycans, and among them, members of the cadherin and plexin superfamilies and the perineural net protein neurocan.
In spite of the clear importance of O-mannosyl glycosylation, the tools available are still insufficient for the specific detection, enrichment, and identification of proteins containing these modifications. A previous study generated antibodies that were directed against a linear core m1 Galβ1,4GlcNAcβ1,2-Manα-Thr glycopeptide.[22] The induced antibodies showed very high specificity against the antigen structure, but retained some specificity for the peptide, making them unsuitable for general detection of O-mannosyl glycans presented on diverse peptide backbones.
In the present work, we aimed to generate antibodies directed against branched core m2 (GlcNAcβ1,2[GlcNAcβ1,6]Manα-Thr) O-mannosyl structures for applications in glycoprotein and glycopeptide detection. A synthetic vaccine construct was designed carrying two core m2 glycans on a tetrapeptide, with glycine residues flanking the carbohydrate epitopes. The glycopeptide was conjugated through a C-terminal squarate linker to a mutant diphtheria toxin carrier protein (CRM197). An efficient synthetic strategy was developed to prepare the branched core m2 glycosylated Fmoc-amino acid building block (Fmoc = 9-fluorenylmethoxycarbonyl) that was applied in Fmoc-solid-phase peptide synthesis (Fmoc-SPPS) to generate various glycopeptides. To evaluate the potential to use the induced core m2 antibodies in glycoprotein/glycopeptide detection, detailed characterization of the specificity was undertaken employing microarray screening of a library of 39 O-mannosyl glycopeptides. The glycopeptides included short, extended, linear, and branched core glycans presented on a variety of peptides. The glycopeptide microarray was also employed to evaluate the binding specificity of ConA to the assorted O-mannosyl glycan structures. Computational carbohydrate grafting[23] was performed on the O-mannosyl glycan–ConA crystal structure complexes (taken from PDB ID 1TEI) to gain a structural understanding of the observed specificities.
Results and Discussion
Synthesis
A limited number of chemical and chemoenzymatic preparations of linear core m1 and core m3 O-mannosyl glycosylated amino acids and glycopeptides have been reported previously.[12a,21,22,24] The first synthesis of core m2 structures is described here. A retrosynthetic analysis was carried out for the design of a straightforward route to the peracetylated GlcNAcβ1,2[GlcNAcβ1,6]Manα-Thr Fmoc-SPPS amino acid building block 14, which was later incorporated into the glycopeptide synthesis for the conjugation to CRM197 or for glycopeptide immobilization on microarray slides (Figure 1). We focused on a convergent coupling approach using a p-methoxybenzyl- and NHTroc-protected (NHTroc = N-trichloroethoxycarbonyl) thioglycoside donor 3 that could extend the free 2- and 6-hydroxyl groups of a dimethoxybutane acetal protected Manα-Thr acceptor 10 (disconnection A, Figure 1). For preparation of the orthogonally protected Manα-Thr amino acid 10, the trichloroactamidate method was selected (disconnection B, Figure 1).[25]
Figure 1.
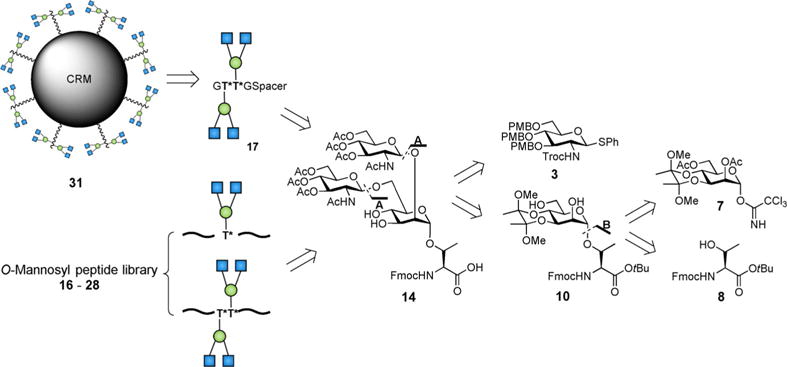
Disconnection approach for synthesis of core m2 branched O-mannosyl glycoconjugates, glycopeptides, and amino acid building blocks.
The chemical synthesis commenced with preparation of the thioglycoside disaccharide donor 3. Here, p-methoxybenzyl protection was selected instead of the more common benzyl protection to circumvent a later hydrogenation step, which would be problematic in the presence of Fmoc groups on the amino acid. Because the Troc group can be removed under mild conditions in later steps and is a participating β-directing group[26] (desired for the coupling with the mannose acceptor 10), the known GlcN3 thioglycoside 1[27] was converted to the desired GlcN-Troc-protected donor 3. After p-methoxybenzyl protection of 1, the resulting compound 2 was subjected to azide zinc reduction, and Troc-protection to give 3 (Scheme 1). For preparation of the Manα-Thr amino acid 10, the 2,6-di-O-acetyl-3,4-O-dimethoxybutane-α-D-mannopyranosyl trichloroa-cetamidate donor 7 first needed to be prepared. Starting from the known allyl 3,4-O-(2′,3′-dimethoxybutane-2′,3′-diyl)-α-D-mannopyranoside 4,[28] 2,6-acetyl protection, anomeric deallylation with Wilkinson’s catalyst, and reaction with trichloroacetonitrile and 1,8-diazabicyclo[5.4.0lundec-7-ene (DBU) was performed. The obtained trichloroacetamidate donor 7 was then coupled to the Fmoc-Thr-O-tBu amino acid 8[29] by trimethylsilyltriflate (TMSOTf) activation in diethyl ether to give 9. Selective removal of the acetyl protecting groups on the mannose 2- and 6-positions using NaOMe in methanol at pH 9.5 yielded the amino acid acceptor 10. By NIS/TfOH[30] (NIS = N-iodosuccinimide; TfOH = trifluoromethanesulfonic acid) activation at −50°C, glycosylation took place providing the GlcNAcβ1,2[GlcNAcβ1,6]Manα-Thr trisaccharide amino acid 11 in good yield. The trisaccharide amino acid 11 was converted into a building block compatible with Fmoc-SPPS by three protecting group manipulation steps. Initially, the p-methoxybenzyl groups were removed by ceric ammonium nitrate (CAN) followed by acetylation. Subsequently the N-Troc groups were removed by reductive elimination followed by acetylation to give 13. Simultaneous removal of the glycan dimethoxybutane acetal and the threonine tert-butyl ester (tBu) was followed to give the desired Fmoc-SPPS-glycosylated amino acid l4. The mannose 3,4 hydroxyl groups of 14 were left unprotected during peptide synthesis (Scheme 1).
Scheme 1.
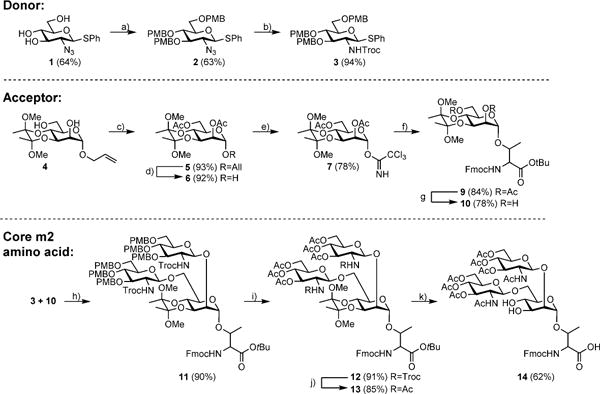
Synthesis of the trisaccharide core m2 building block 14: a) NaH, 4-methoxybenzyl chloride, DMF, RT 2 h; b) Zn, 1,4-dioxane/AcOH 10:1, RT 24 h; NaHCO3, 1,4-dioxane/H2O 2:1, TrocCl, RT 24 h; c) Pyridine/Ac2O, RT.24 h; d) Wilkinson’s catalyst, Tol/EtOAc/H2O 20:10:1, reflux 24 h; I2, THF/H2O 4:1, RT 2 h; e) trichloroacetonitrile, DBU, dichloromethane, 0°C 3 h; f) Fmoc-Thr acceptor 5, TMSOTf, molecular sieves, Et2O, RT 30 min; g) NaOMe in MeOH, pH 9.5, RT 24 h; NaHCO3, 1,4-dioxane/H2O 1:1, Fmoc-OSu, RT 2 h; h) NIS, TfOH, dichloromethane, −50°C 4 h; i) CAN, MeCN/H2O 9:1, RT 30 min;Pyridine/Ac2O, RT 24 h; j) Zn, AcOH, RT 4 days; Pyridine/Ac2O, RT 24 h; k) TFA/dichloromethane 3:1, RT 24 h.
The obtained GlcNAcβ1,2[GlcNAcβ1,6]Manα-Thr Fmoc–SPPS amino acid 14 and the previously described GlcNAcβ1-2Manα-Thr Fmoc–SPPS amino acid 15[22] were employed in the synthesis of the glycopeptides 16–28. Starting from preloaded Fmoc-Gly-, Fmoc-Ala-, Fmoc-Asp(tBu)-, or Fmoc-Arg(Pbf) (Pbf= 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl) trityl resin (12.5 μmol), coupling of the standard Fmoc-amino acids (8equiv) was carried out using HBTU/HOBt (HBTU = 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate; HOBt=1-hydroxybenzotriazole) and the glycosylated Fmoc-Thr building blocks 14 and 15 (1.5equiv) were coupled using HATU/HOAt (HATU=(7-aza-benzotriazol-1-yl)tetramethyluronium hexafluorophosphate; HOAt=1-hydroxy-7-azabenzo-triazole).[31] The glycosylated amino acids were pre-activated in a smaller volume of solvent and manually added to the resin. Furthermore, the reaction time of the glycosylated amino acids was extended (to 8 h instead of 40 min). After full assembly of the glycopeptides on the resin, and for later microarray surface immobilization, peptides 20–28 were terminated by coupling of a triethylene glycol spacer.[32] Detachment from the resin was performed by applying a mixture of TFA/TIPS/H2O (90:5:5) (TFA = trifluoroacetic acid; TIPS = triisopropylsilane). Peptides 16–19 were instead capped at the N-terminal with Ac2O, HOBt, and N,N-diisopropylethylamine (DIPEA) followed by cleavage from the resin, which allowed a spacer[33] to be connected to the C-terminal for later conjugation with an immune carrier or for immobilization on a microarray surface. Insertion of a linker at the C-terminal instead of the N-terminal avoids immunogenic response to a C-terminal amide or free carboxylic acid in close proximity to the glycosylation sites. After a desalting step on C-18 cartridges, the O-acetyl groups on all generated glycopeptides (16–28) were removed by treatment with catalytic amounts of NaOMe in methanolat pH 9.5. Deprotected glycopeptides 16–28 were finally purified by preparative HPLC. For vaccine construction and enzyme-linked immuosorbent assay (ELISA) antibody titer determination, glycopeptide–protein conjugates were prepared. The CRM197 immune carrier mutant originating from diphtheria toxin has successfully been used for immune stimulation of different glycoconjugates and was therefore selected for conjugation to peptide 17 and later immunization. The glycopeptide 17, which contained a spacer with a free amine at the C-terminal, was conjugated to diphtheria toxin CRM197 and bovine serum albumin (BSA) by an initial coupling with diethylsquarate in EtOH/H2O (1:1) at pH 8 to give the squaric acid monoamide 29. The coupling to BSA and CRM197 was performed in aqueous Na2HPO4 buffer at pH 9.5 to give the glycopeptide–BSA 30 and CRM197 31 conjugates (Scheme 2).
Scheme 2.
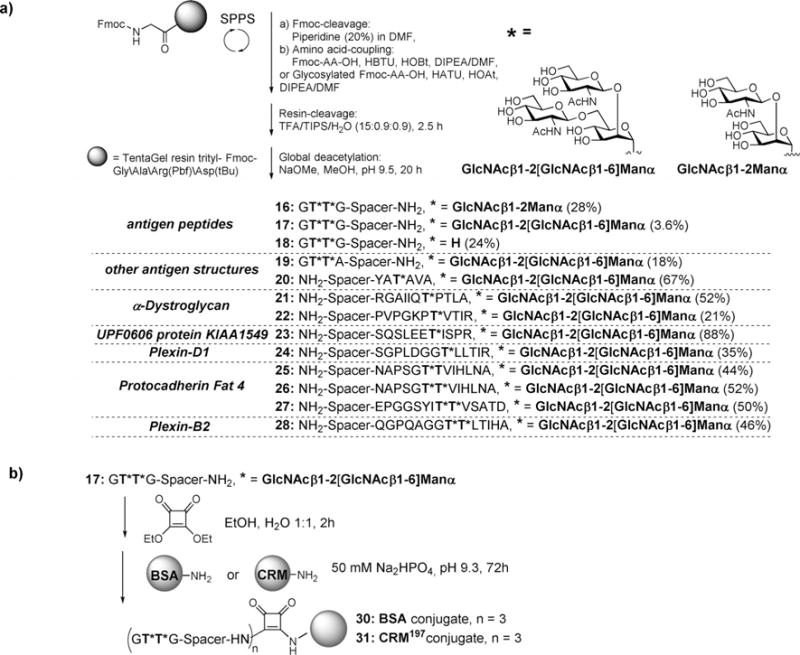
Synthesis of glycopeptides 16–28, BSA-conjugate 30 and CRM-conjugate 31.
Immunization
For antibody induction against the GlcNAcβ1,2[GlcNAcβ1,6]Manα glycopeptide, two rabbits (26559 and 26560) were immunized with the diphtheria toxin mutant CRM197 vaccine 31 together with a lipopolysaccharide (LPS) cocktail (containing LPS, Span 80, Tween 80, mineral oil and double distilled water) as adjuvant. Four booster immunizations were carried out over 8 weeks. Analysis of sera from the pre-immune, first bleed, and final bleed by ELISA coated with the GlcNAcβ1,2[GlcNAcβ1,6]-Manα glycopeptide–BSA conjugate 30 showed that strong immune responses were directed against the antigen structure (Figure S1 in the Supporting Information).
Microarray analysis and molecular modeling
The binding specificities of the antibody sera were evaluated by microarray analysis, against glycopeptides 16–28 and the previously synthesized O-mannosyl glycopeptides 32–57[21,22,24e] (Table 1). The induced antibodies showed high selectivity towards both the glycopeptide antigen structure 17 (microarray nr: 38) and all other peptides presenting the branched core m2 GlcNAcβ1,2[GlcNAcβ1,6]Manα trisaccharide 19–28 (microarray nr: 15–23, 39, respectively), regardless of whether the glycans were presented as mono- or divalent structures on the peptide backbone. Notably, neither the non-glycosylated peptide 18 (microarray nr:33) nor the linear core m1 GlcNAcβ1,2Manα disaccharide antigen peptide 16 (microarray nr: 37) were recognized by the induced antibody sera. Furthermore, the antibodies did not cross-react with the other linear core m1 disaccharides (microarray nr:5–14), the core m1 tri-saccharide (microarray nr:1–4), or α-mannose monosaccharide (microarray nr: 24–32) modified peptides, such as sequences from α-DG, Plexin, and Prothocadherin. Altogether, we could conclude that new antibodies were generated, which recognized branched core m2 structures with very high selectivity that should serve as useful tools for O-mannosyl glycan detection (Figure 2 and Table 1).
Table 1.
List of O-mannosyl peptides spotted on the microarray slides.
| Microarray peptide Nr | Compound Nr | Origin | Sequence | Glycan |
|---|---|---|---|---|
| 1 | 32 | α-Dystroglycan | SpPVPGKPT*VTIR | Galβ1–4GlcNAcβ1–4Glc2Manα |
| 2 | 33 | α-Dystroglycan | SpRGAIIQT*PTLG | |
| 3 | 34 | antigen sequence | SpGT*G | |
| 4 | 35 | antigen sequence | SpYAT*AVA | |
| 5 | 36 | α-Dystroglycan | SpPVPGKPT*VTIR | GlcNAcβ1–2Manα |
| 6 | 37 | α-Dystroglycan | SpRGAIIQT*PTLG | |
| 7 | 38 | antigen sequence | SpGT*G | |
| 8 | 39 | antigen sequence | SpYAT*AVA | |
| 9 | 40 | UPF0606 protein KIAA1549 | SpSQSLEET*ISPR | |
| 10 | 41 | Plexin-D1 | SpSGPLDGGT*LLTIR | |
| 11 | 42 | Protocadherin Fat 4 | SpNAPSGT*TVIHLNA | |
| 12 | 43 | Protocadherin Fat 4 | SpNAPSGT*T*VIHLNA | GlcNAcβ1–2Manα ×2 |
| 13 | 44 | Plexin-B2 | SpQGPQAGGT*T*LTIHG | |
| 14 | 45 | Protocadherin Fat 4 | SpEPGGSYIT*T*VSATD | |
| 15 | 22 | α-Dystroglycan | SpPVPGKPT*VTIR | GlcNAcβ1–2[GlcNAcβ1–6] Manα |
| 16 | 21 | α-Dystroglycan | SpRGAIIQT*PTLG | |
| 17 | 20 | antigen sequence | SpYAT*AVA | |
| 18 | 23 | UPF0606 protein KIAA1549 | SpSQSLEET*ISPR | |
| 19 | 24 | Plexin-D1 | SpSGPLDGGT*LLTIR | |
| 20 | 25 | Protocadherin Fat 4 | SpNAPSGT*TVIHLNA | |
| 21 | 26 | Protocadherin Fat 4 | SpNAPSGT*T*VIHLNA | GlcNAcβ1–2[GlcNAcβ1–6] Manα x2 |
| 22 | 28 | Plexin-B2 | SpQGPQAGGT*T*LTIHG | |
| 23 | 27 | Protocadherin Fat 4 | SpEPGGSYIT*T*VSATD | |
| 24 | 46 | antigen sequence | YAT*AV | Manα |
| 25 | 47 | α-Dystroglycan | SpPVPGKPT*VTIR | |
| 26 | 48 | α-Dystroglycan | SpRGAIIQT*PTLG | |
| 27 | 49 | UPF0606 protein KIAA1549 | SpSQSLEET*ISPR | |
| 28 | 50 | Plexin-D1 | SpSGPLDGGT*LLTIR | |
| 29 | 51 | Protocadherin Fat 4 | SpNAPSGT*TVIHLNA | |
| 30 | 52 | Protocadherin Fat 4 | SpNAPSGT*T*VIHLNA | Manα × 2 |
| 31 | 53 | Plexin-B2 | SpQGPQAGGT*T*LTIHG | |
| 32 | 54 | Protocadherin Fat 4 | SpEPGGSYIT*T*VSATD | |
| 33 | 18 | antigen sequence | GTTGSp | non-glycan |
| 34 | 55 | antigen sequence | SpGTG | |
| 35 | 55 | antigen sequence | SpYATAVA | |
| 36 | 57 | antigen sequence | YATAV | |
| 37 | 16 | antigen sequence | GT*T*GSp | GlcNAcβ1–2Manα |
| 38 | 17 | antigen sequence | GT*T*GSp | GlcNAcβ1–2[GlcNAcβ1–6] Manα |
| 39 | 19 | antigen sequence | GT*T*ASp |
Figure 2.
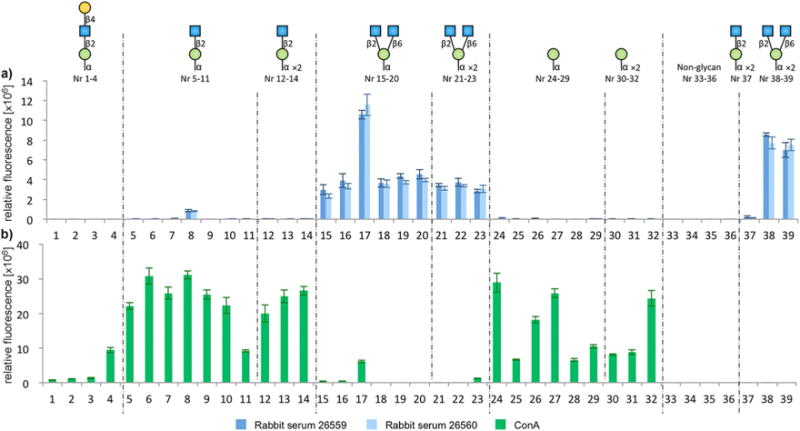
Microarray analysis and evaluation of binding to spotted peptides 16–28 and 32–57 with a) the core m2 polyclonal rabbit sera 26559 and 26560 and b) Biotin–ConA. The x-axis shows the microarray peptide spotting ID numbers (details in Table 1) and y-axis shows the relative fluorescence, readout by a) a Cy5 secondary anti-rabbit antibody or b) Cy5-streptavidin.
The concanavalin A (ConA) lectin that is commonly used for enrichment of tryptic O-mannosyl peptides was also evaluated on the glycopeptide microarray. Interestingly, the ConA recognition pattern of the O-mannosyl glycopeptide structures contrasted that of the antibody sera. ConA showed high reactivity to all α-mannose monosaccharide peptides (microarray nr: 24–32) and to all linear core m1 GlcNAcβ1,2Manα disaccharide peptides (microarray nr: 5–14), except the diglycosylated antigen peptide structure 16 (microarray nr:37). Unlike the antibody sera, with the exception of glycans conjugated to YAT*AVA (microarray nr: 4 and 17), no ConA binding recognition was observed for branched core m2 (microarray nr:15, 16, 18–23, 38, 39) and extended linear core m1 (microarray nr: 1–3) trisaccharide peptides (Figure 2 and Table 1). Notably, glycan presentation on the YAT*AVA sequence consistently resulted in enhanced binding recognition, both in the case of ConA and the core m2 antibody sera (Figure 2 and Table 1).
The observed selectivity of ConA towards the O-mannosyl glycopeptide array was rationalized by molecular modeling based on an existing co-crystal structure of ConA (PDB ID 1TEI)[34] bound to a pentasaccharide ligand (GlcNAcβ1,2Manα1,3[GlcNAcβ1,2Manα1,6]Man) that contains three Man residues (core, 3-arm, and 6-arm) with GlcNAc residues β1,2-linked to the 3- and 6-arms. The Man residues in the glycans in the microarray could potentially align in any of the three Man orientations in the co-crystal structure, thus each Man binding mode was examined for each arrayed glycan to determine its ability to rationalize the ConA data (Figure 3).
Figure 3.
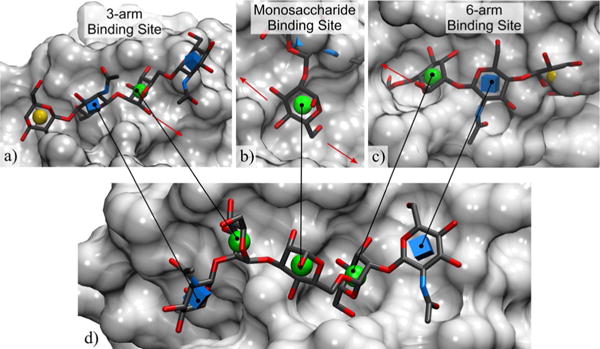
Structure-based rationalization of the observed binding specificity of ConA towards the arrayed glycans; a) only the weaker 3-arm binding site tolerates linear core m1 trisaccharides and branched glycans, which do not display affinity for ConA on the array; b) the core Man does not tolerate substitution at the 2-position of the bound Man; c) the 6-arm binding site does not tolerate substitution at the 4-position of the bound GlcNAc, nor at the 6-position of the Man, which is consistent with the binding specificity of ConA amongst the arrayed glycans; d) the co-complex of GlcNAcβ1-2Manα1-3[GlcNAcβ1-2Manα1-6]Manα with ConA from PDB ID 1TEI[34] shows the relative position of the 3-arm,6-arm, and monosaccharide binding sites. Glycans represented in 3D-SNFG icon mode,[37] the protein surface is in light grey, red arrows indicate the position of the neighboring binding site(s).
Using computational carbohydrate grafting,[23b,36] putative bound structures for each of the arrayed O-mannosyl glycans were generated by adding the relevant glycan branches to the pertinent components (core, 3-arm, and 6-arm Man) of the crystallographic ligand, while attempting to resolve any steric overlaps by adjusting the glycosidic linkages. The Gly-Spec webtool[35] (www.glycam.org/gr) was used to perform this analysis. The predictions from grafting to the 6-arm were consistent with the selectivity of ConA; namely, that substitution at the 2-position of Man by GlcNAc would be tolerated, whereas extension of the GlcNAcβ1,2Man disaccharide by Gal or modification at the 6-position of the core Man by GlcNAc would not be (Figure 3). Thus, the linear glycans (array 5–14) that show affinity for ConA were predicted to be able to bind in either the 6-arm or 3-arm alignments, whereas the monosaccharide glycans (array 24–32) could potentially bind in any of the three Man positions. In contrast, the linear core m1 trisaccharides (array 1–4) and branched glycans (array 15, 16, 18–23, 38, 39) that do not display affinity for ConA could only be tolerated in the 3-arm site (Figure 3). Grafting to the core Man residue indicated that the binding site could not tolerate substitution at the 2-position of that residue, thus only glycopeptides 24–32 could bind through this site (Figure 3). This latter observation is notable since studies based on N-linked glycans binding to ConA lead to the conclusion that interactions within the 3-arm site are weaker than those within the 6-arm.[36] The binding observed for the glycopeptide array elements 4 and 17 could be a combination of weak binding through the 3-arm site and the consistently observed enhancing effect of the YAT*AVA peptide sequence.
Conclusion
In anumber of cases, O-mannosyl core m2 branching has been shown to be important in brain glycobiology, cell migration, and tumor cell growth and metastasis. In this study, core m2 glycans, glycopeptides and core m2 glycoconjugates were synthesized for the first time, and were used to generate glycan specific antibodies and to evaluate the immune response. Immunizations with the core m2 glycoconjugate vaccine 31 provided evidence that a strong immune response was directed to the glycopeptide antigen structure 17. Evaluation of the binding specificity of the antisera using glycopeptide microarrays showed highly specific recognition of the branched core m2 O-mannosyl glycans on different peptide backbones, without cross-reactivity with linear core m1 or monosaccharide Manα-modified peptides. We aim to further characterize and apply the new antibodies as tools in future immunostaining experiments to explore functions of O-mannosyl branching in brain biology and tumor progression. For comparison, the Man-specific ConA lectin was also evaluated for binding to the glycopeptide array. The data indicated that ConA recognized short core m1 and monosaccharide Manα-modified peptides, but showed little, if any, binding to branched core m2 or extended linear core m1 trisaccharides. A computational examination of the ConA binding modes was able to provide a structural rationalization for the array data. In conclusion, the specificity of the new antibodies for branched O-mannosyl ligands stands in contrast to the binding specificity of ConA, indicating a novel role for the antibodies in studies of protein O-mannosylation.
Supplementary Material
Acknowledgments
This work was supported by Deutsche Forschungs Gemein-schaft grant number WE 4751/2-1 (to U.W.) and Sonderfor-schungsbereich 1036, project 11 (to S.S.). S.S. is a member of the excellence cluster CellNetworks. R.J.W. thanks the National Institutes of Health (U01 CA207824 and P41 GM103390) for support. Financial support by the Ministerium für Innovation, Wissenschaft und Forschung des Landes Nordrhein-Westfalen, the Senatsverwaltung für Wirtschaft, Technologie und Forschung des Landes Berlin, and the Bundesministerium für Bildung und Forschung is gratefully acknowledged (ISAS, U.W.).
Footnotes
Supporting information and full experimental details for this article can be found under http://dx.doi.org/10.1002/chem.201605627.
References
- 1.a) Lommel M, Stahl S. Glycobiology. 2009;19:816–828. doi: 10.1093/glycob/cwp066. [DOI] [PubMed] [Google Scholar]; b) Endo T. Glycoconjugate J. 2004;21:3–7. doi: 10.1023/B:GLYC.0000043740.26062.2c. [DOI] [PubMed] [Google Scholar]; c) Dobson CM, Hempel SJ, Stalnaker SH, Stuart R, Wells L. Cell Mol Life Sci. 2013;70:2849–2857. doi: 10.1007/s00018-012-1193-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manya H, Chiba A, Yoshida A, Wang X, Chiba Y, Jigami Y, Margolis RU, Endo T. Proc Natl Acad Sci USA. 2004;101:500–505. doi: 10.1073/pnas.0307228101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Takahashi S, Sasaki T, Manya H, Chiba Y, Yoshida A, Mizuno M, Ishida HK, Ito F, Inazu T, Kotani N, Takasaki S, Takeuchi M, Endo T. Glycobiology. 2001;11:37–45. doi: 10.1093/glycob/11.1.37. [DOI] [PubMed] [Google Scholar]; b) Zhang W, Betel D, Schachter H. Biochem J. 2002;361:153–162. doi: 10.1042/0264-6021:3610153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Abbott KL, Matthews RT, Pierce M. J Biol Chem. 2008;283:33026–33035. doi: 10.1074/jbc.M803646200. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Inamori KI, Endo T, Gu J, Matsuo I, Ito Y, Fujii S, Iwasaki H, Narimatsu H, Miyoshi E, Honke K, Taniguchi N. J Biol Chem. 2004;279:2337–2340. doi: 10.1074/jbc.C300480200. [DOI] [PubMed] [Google Scholar]; c) Lee JK, Matthews RT, Lim JM, Swanier K, Wells L, Pierce JM. J Biol Chem. 2012;287:28526–28536. doi: 10.1074/jbc.M112.367565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Praissman JL, Wells L. Biochemistry. 2014;53:3066–3078. doi: 10.1021/bi500153y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Michele DE, Campbell KP. J Biol Chem. 2003;278:15457–15460. doi: 10.1074/jbc.R200031200. [DOI] [PubMed] [Google Scholar]; b) Henry MD, Campbell KP. Curr Opin Cell Biol. 1999;11:602–607. doi: 10.1016/s0955-0674(99)00024-1. [DOI] [PubMed] [Google Scholar]
- 7.a) Yuen CT, Chai W, Loveless RW, Lawson AM, Margolis RU, Feizi T. J Biol Chem. 1997;272:8924–8931. doi: 10.1074/jbc.272.14.8924. [DOI] [PubMed] [Google Scholar]; b) Smalheiser NR, Haslam SM, Sutton-Smith M, Morris HR, Dell A. J Biol Chem. 1998;273:23698–23703. doi: 10.1074/jbc.273.37.23698. [DOI] [PubMed] [Google Scholar]
- 8.a) Abbott KL, Troupe K, Lee I, Pierce M. Exp Cell Res. 2006;312:2837–2850. doi: 10.1016/j.yexcr.2006.05.022. [DOI] [PubMed] [Google Scholar]; b) Lee I, Guo HB, Kamar M, Abbott K, Troupe K, Lee JK, Alvarez-Manilla G, Pierce M. J Neurochem. 2006;97:947–956. doi: 10.1111/j.1471-4159.2006.03785.x. [DOI] [PubMed] [Google Scholar]
- 9.Kanekiyo K, Inamori K-i, Kitazume S, Sato K, Maeda J, Higuchi M, Kizuka Y, Korekane H, Matsuo I, Konke K, Taniguchi N. J Neurosci. 2013;33:10037–10047. doi: 10.1523/JNEUROSCI.3137-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lange T, Ullrich S, Mueller I, Nentwich MF, Stuebke K, Feldhaus S, Knies C, Hellwinkel OJC, Vessella RL, Abramjuk C, Anders M, Schroeder-Schwarz J, Schlomm T, Huland H, Sauter G, Schumacher U. Clin Cancer Res. 2012;18:1364–1373. doi: 10.1158/1078-0432.CCR-11-2900. [DOI] [PubMed] [Google Scholar]
- 11.Yoshida-Moriguchi T, Yu L, Stalnaker SH, Davis S, Kunz S, Madson M, Oldstone MBA, Schachter H, Wells L, Campbell KP. Science. 2010;327:88–92. doi: 10.1126/science.1180512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Mo KF, Fang T, Stalnaker SH, Kirby PS, Liu M, Wells L, Pierce M, Live DH, Boons GJ. J Am Chem Soc. 2011;133:14418–14430. doi: 10.1021/ja205473q. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kanagawa M, Kobayashi K, Tajiri M, Manya H, Kuga A, Yamaguchi Y, Akasaka-Manya K, Furukawa JI, Mizuno M, Kawakami H, Shinohara Y, Wada Y, Endo T, Toda T. Cell Rep. 2016;14:2209–2223. doi: 10.1016/j.celrep.2016.02.017. [DOI] [PubMed] [Google Scholar]
- 13.Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- 14.Live D, Wells L, Boons GJ. ChemBioChem. 2013;14:2392–2402. doi: 10.1002/cbic.201300417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chai W, Yuen CT, Kogelberg H, Carruthers RA, Margolis RU, Feizi T, Lawson AM. Eur J Biochem. 1999;263:879–888. doi: 10.1046/j.1432-1327.1999.00572.x. [DOI] [PubMed] [Google Scholar]
- 16.Breloy I, Pacharra S, Aust C, Hanisch FG. Biol Chem. 2012;393:709–717. doi: 10.1515/hsz-2012-0214. [DOI] [PubMed] [Google Scholar]
- 17.a) Bleckmann C, Geyer H, Lieberoth A, Splittstoesser F, Liu Y, Feizi T, Schachner M, Kleene R, Reinhold V, Geyer R. Biol Chem. 2009;390:627–645. doi: 10.1515/BC.2009.044. [DOI] [PubMed] [Google Scholar]; b) Martinez T, Pace D, Brady L, Gerhart M, Balland A. J Chromatogr A. 2007;1156:183–187. doi: 10.1016/j.chroma.2007.04.050. [DOI] [PubMed] [Google Scholar]; c) Yahiro K, Wada A, Yamasaki E, Nakayama M, Nishi Y, Hisatsune J, Morinaga N, Sap J, Noda M, Moss J, Hirayama T. J Biol Chem. 2004;279:51013–51021. doi: 10.1074/jbc.M406473200. [DOI] [PubMed] [Google Scholar]; d) Wing DR, Rademacher T, Schmitz B, Schachner M, Dweck RA. Biochem Soc Trans. 1992;20:386–390. doi: 10.1042/bst0200386. [DOI] [PubMed] [Google Scholar]; e) Pacharra S, Hanisch FG, Breloy I. J Proteome Res. 2012;11:3955–3964. doi: 10.1021/pr200996y. [DOI] [PubMed] [Google Scholar]; f) Pacharra S, Hanisch FG, Muehlenhoff M, Faissner A, Rauch U, Breloy I. J Proteome Res. 2013;12:1764–1771. doi: 10.1021/pr3011028. [DOI] [PubMed] [Google Scholar]; g) Dwyer CA, Baker E, Hu H, Matthews RT. Neuroscience. 2012;220:47–61. doi: 10.1016/j.neuroscience.2012.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Trinidad JC, Schoepfer R, Burlingame AL, Medzihradszky KF. Mol Cell Proteomics. 2013;12:3474–3488. doi: 10.1074/mcp.M113.030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vester-Christensen MB, Halim A, Joshi HJ, Steentoft C, Bennett EP, Levery SB, Vakhrushev SY, Clausen H. Proc Natl Acad Sci USA. 2013;110:21018–21023. doi: 10.1073/pnas.1313446110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Winterhalter PR, Lommel M, Ruppert T, Strahl S. FEBS Lett. 2013;587:3715–3721. doi: 10.1016/j.febslet.2013.09.041. [DOI] [PubMed] [Google Scholar]; b) Lommel M, Winterhalter PR, Willer T, Dahlhoff M, Schneider MR, Bartels MF, Renner-Mueller I, Ruppert T, Wolf E, Strahl S. Proc Natl Acad Sci USA. 2013;110:21024–21029. doi: 10.1073/pnas.1316753110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carvalho S, Oliveira T, Bartels MF, Miyoshi E, Pierce M, Taniguchi N, Carneiro F, Seruca R, Reis CA, Strahl S, Pinho SS. Oncotarget. 2016;7:65231–65246. doi: 10.18632/oncotarget.11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bartels MF, Winterhalter PR, Yu J, Liu Y, Lommel M, Möhrlen F, Hu H, Feizi T, Westerlind U, Ruppert T, Strahl S. PLoS One. 2016;11(11):e0166119. doi: 10.1371/journal.pone.0166119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu J, Westerlind U. ChemBioChem. 2014;15:939–945. doi: 10.1002/cbic.201300537. [DOI] [PubMed] [Google Scholar]
- 23.a) Tessier MB, Grant OC, Heimburg-Molinaro J, Smith D, Jadey S, Gulick AM, Glushka J, Deutscher SL, Rittenhouse-Olson K, Woods RJ. PLoS ONE. 2013;8:e54874. doi: 10.1371/journal.pone.0054874. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Grant OC, Smith HMK, Firsova D, Fadda E, Woods RJ. Glycobiology. 2014;24:17–25. doi: 10.1093/glycob/cwt083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.a) Sardzik R, Green AP, Laurent N, Both P, Fontana C, Voglmeir J, Weissenborn MJ, Haddoub R, Grassi P, Haslam SM, Widmalm G, Flitsch SL. J Am Chem Soc. 2012;734:4521–4524. doi: 10.1021/ja211861m. [DOI] [PubMed] [Google Scholar]; b) Zhang Y, Meng C, Jin L, Chen X, Wang F, Cao H. Chem Commun. 2015;51:11654–11657. doi: 10.1039/c5cc02913a. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Seifert J, Ogawa T, Ito Y. Tetrahedron Lett. 1999;40:6803–6807. [Google Scholar]; d) Matsuo I, Isomura M, Ajisaka K. Tetrahedron Lett. 1999;40:5047–5050. [Google Scholar]; e) Yu J, Schorlemer M, Gomez Toledo A, Pett C, Sihlbom C, Larson G, Westerlind U, Nilsson J. Chem Eur J. 2016;22:1114–1124. doi: 10.1002/chem.201503659. [DOI] [PubMed] [Google Scholar]; f) Kawahira K, Tanaka H, Ueki A, Nakahara Y, Hojo H, Nakahara Y. Tetrahedron. 2009;65:8143–8153. [Google Scholar]
- 25.Schmidt RR. Angew Chem Int Ed. 1986;25:212–235. [Google Scholar]; Angew Chem. 1986;98:213–236. [Google Scholar]
- 26.a) Ellervik U, Magnusson G. Carbohydr Res. 1996;280:251–260. doi: 10.1016/0008-6215(95)00318-5. [DOI] [PubMed] [Google Scholar]; b) Zhang Z, Ollman IR, Ye XS, Wischnat R, Basov T, Wong CH. J Am Chem Soc. 1999;121:734–753. [Google Scholar]
- 27.Schmidt RR, Behrendt M, Toepfer A. Synlett. 1990:694–696. [Google Scholar]
- 28.Marwood RD, Riley AM, Jenkins DJ, Potter BVL. J Chem Soc Perkin Trans. 2000;1:1935–1947. [Google Scholar]
- 29.a) Kunz H. In: Preparative Carbohydrate Chemistry. Hanessian S, editor. Marcel Dekker; New York: 1997. p. 265. [Google Scholar]; b) Paquet A. Can J Chem. 1982;60:976. [Google Scholar]
- 30.a) Konradsson P, Udodong UE, Fraser-Reid B. Tetrahedron Lett. 1990;31:4313–4316. [Google Scholar]; b) Veeneman GH, van Boom JH. Tetrahedron Lett. 1990;31:275–278. [Google Scholar]
- 31.a) Carpino L, El-Faham A, Minor CA, Albericio F. J Chem Soc Chem Commun. 1994:201–203. [Google Scholar]; b) Carpino LA. J Am Chem Soc. 1993;115:4397–4398. [Google Scholar]; c) Dourtoglou V, Gross B, Lambropoulou V, Zioudrou C. Synthesis. 1984:572–574. [Google Scholar]
- 32.a) Westerlind U, Schroder H, Hobel A, Gaidzik N, Kaiser A, Niemeyer Christof M, Schmitt E, Waldmann H, Kunz H. Angew Chem Int Ed. 2009;48:8263–8267. doi: 10.1002/anie.200902963. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2009;121:8413–8417. [Google Scholar]; b) Keil S, Claus C, Dippold W, Kunz H. Angew Chem Int Ed. 2001;40:366–369. doi: 10.1002/1521-3773(20010119)40:2<366::AID-ANIE366>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2001;113:379–382. [Google Scholar]
- 33.Trester-Zedlitz M, Kamada K, Burley SK, Fenyö D, Chait BT, Muir TW. J Am Chem Soc. 2003;125:2416–2425. doi: 10.1021/ja026917a. [DOI] [PubMed] [Google Scholar]
- 34.Moothoo DN, Naismith JH. Glycobiology. 1998;8:173–181. doi: 10.1093/glycob/8.2.173. [DOI] [PubMed] [Google Scholar]
- 35.Grant OC, Xue X, Ra D, Khatamian A, Foley BL, Woods RJ. Glycobiology. 2016;26:1027–1028. doi: 10.1093/glycob/cww094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grant OC, Tessier MB, Meche L, Mahal LK, Foley BL, Woods RJ. Glycobiology. 2016;26:772–783. doi: 10.1093/glycob/cww020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thieker DF, Hadden JA, Schulten K, Woods RJ. Glycobiology. 2016;26:786–787. doi: 10.1093/glycob/cww076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.