

Abstract
Mononuclear nonheme iron complexes that serve as structural and functional mimics of the thiol dioxygenases (TDOs), cysteine dioxygenase (CDO) and cysteamine dioxygenase (ADO), have been prepared and characterized with crystallographic, spectroscopic, kinetic, and computational methods. The high-spin Fe(II) complexes feature the facially-coordinating tris(4,5-diphenyl-1-methylimidazol-2-yl)phosphine (Ph2TIP) ligand that replicates the three histidine (3His) triad of the TDO active sites. Further coordination with bidentate L-cysteine ethyl ester (CysOEt) or cysteamine (CysAm) anions yielded five-coordinate (5C) complexes that resemble the substrate-bound forms of CDO and ADO, respectively. Detailed electronic-structure descriptions of the [Fe(Ph2TIP)(LS,N)]BPh4 complexes, where LS,N = CysOEt (1) or CysAm (2), were generated through a combination of spectroscopic techniques [electronic absorption, magnetic circular dichroism (MCD)] and density functional theory (DFT). Complexes 1 and 2 decompose in the presence of O2 to yield the corresponding sulfinic acid (RSO2H) products, thereby emulating the reactivity of the TDO enzymes and related complexes. Rate constants and activation parameters for the dioxygenation reactions were measured and interpreted with the aid of DFT calculations for O2-bound intermediates. Treatment of the TDO models with nitric oxide (NO) – a well-established surrogate of O2 – led to a mixture of high-spin and low-spin {FeNO}7 species at low temperature (−70 °C), as indicated by electron paramagnetic resonance (EPR) spectroscopy. At room temperature, these Fe/NO adducts convert to a common species with EPR and infrared (IR) features typical of cationic dinitrosyl iron complexes (DNICs). To complement these results, parallel spectroscopic, computational, and O2/NO reactivity studies were carried out using previously-reported TDO models that feature an anionic hydrotris(3-phenyl-5-methyl-pyrazolyl)borate (Ph,MeTp−) ligand. Though the O2 reactivities of the Ph2TIP- and Ph,MeTp-based complexes are quite similar, the supporting ligand perturbs the energies of Fe 3d-based molecular orbitals and modulates Fe-S bond covalency, suggesting possible rationales for the presence of neutral 3His coordination in CDO and ADO.
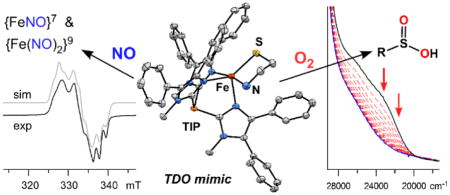
Graphical Abstract
Synthetic models of the thiol dioxygenases (TDOs) have been prepared using a tris(imidazolyl)phosphine ligand that reproduces the atypical three histidine triad of the enzyme active sites. Geometric- and electronic- structure descriptions were obtained with crystallographic, spectroscopic, and computational methods. Dioxygen reactivity experiments confirmed that the complexes behave as functional TDO models, and insights into the O2 activation mechanism were derived from kinetic studies and DFT. Exposure to NO yields multiple mono- and dinitrosyl iron complexes.
1. Introduction
The catabolism of a diverse array of cellular compounds depends upon mononuclear nonheme iron dioxygenases (MNIDs) that incorporate both atoms of O2 into the product(s).1–2 The best-known MNIDs are bacterial enzymes that catalyze the oxidative cleavage of aromatic and aliphatic C-C bonds and play a central role in the degradation of environmental pollutants (i.e., biodegradation).3–5 MNIDs are also involved in human metabolism, as exemplified by cysteine dioxygenase (CDO) and cysteamine dioxygenase (ADO).6–8 Both enzymes are thiol dioxygenases (TDOs) that convert an alkylthiol (RSH) to the corresponding sulfinic acid (RSO2H) using O2 (Scheme 1). CDO regulates the cellular concentration of L-cysteine (Cys) by performing the first step in the catabolism of this amino acid that leads to the formation of either taurine or sulfate as the end product.9–10 Insufficient levels of CDO activity cause exogenous Cys to accumulate to deleterious levels, and this metabolic imbalance is associated with several neurological disorders, including Parkinson’s and Alzheimer’s diseases,11–14 as well as autoimmune disorders.15–16 ADO is required for the degradation of coenzyme A in mammals, and is the major contributor to the production of hypo(taurine) in the brain where levels of CDO are low.6,17
Scheme 1.
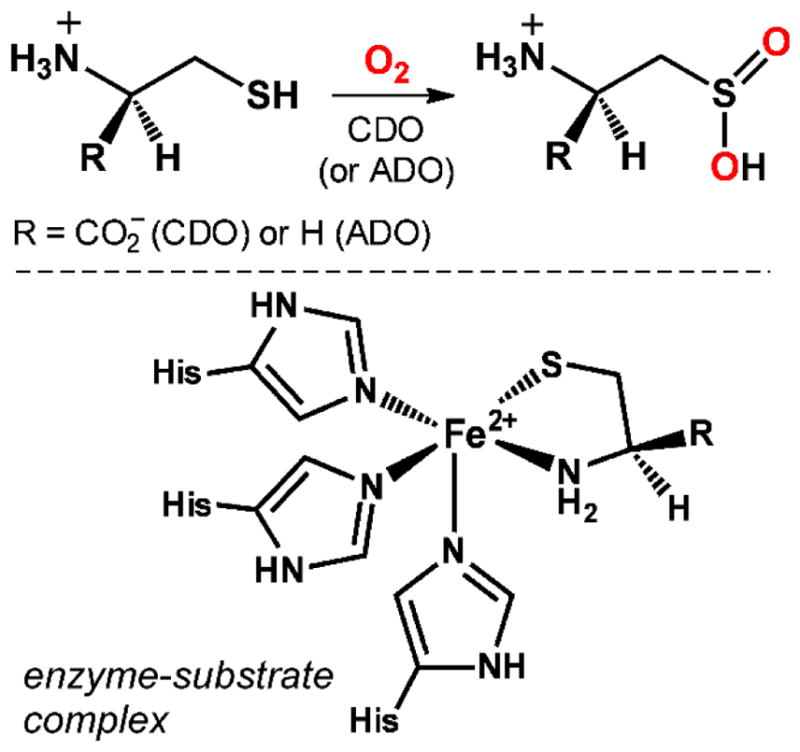
Beyond its medical importance, CDO has attracted attention due to its atypical active-site structure. The overwhelming majority of MNIDs feature a high-spin Fe(II) center bound by one Asp (or Glu) and two His residues in a facial orientation (i.e., the 2-His-1-carboxylate (2H1C) facial triad).18–19 In contrast, crystal structures of CDO have revealed a mononuclear Fe site with a neutral 3-histidine (3His) facial triad (Scheme 1).20–21 The X-ray crystallographic data confirmed that Cys binds directly to Fe in a bidentate manner via its thiolate and amine donors, yielding a five-coordinate (5C) Fe(II) site capable of O2 binding.22 While a crystal structure of ADO is currently lacking, sequence analysis suggests that it features a similar active-site structure.17 Since the publication of the first CDO structure in 2006, the 3His triad has also been observed in three microbial MNIDs: β-diketone dioxygenase (Dke1),23–25 gentisate dioxygenase (GDO),26 and salicylate dioxygenase (SDO).27 The catalytic implications of this alteration in the first-sphere coordination sphere (3His vs. 2H1C triad) have been widely discussed, but no consensus explanation has emerged.28
The first biomimetic models of CDO, prepared by Goldberg and coworkers, were based on 2,6-bis(imino)pyridine frameworks that provide three neutral N-donors in a meridional arrangement.29–32 These complexes feature an exogenous or pendant aryl thiolate ligand that converts to a sulfonato (RSO3−) donor upon exposure to O2. The same group has utilized the pentadentate N4S chelate, N3PyS, to prepare TDO models in which the aryl thiolate is appended to a tripodal scaffold.33 Reaction of the resulting Fe(II) complex, [Fe(N3PyS)(MeCN)]+, with O2 leads to double-oxygenation of the thiolate donor, thereby mimicking the thiolate-to-sulfinate conversion catalyzed by CDO. Treatment of the same complex with nitric oxide (NO) generates a stable {FeNO}7 species that exhibits spectroscopic properties similar to those observed for the low-spin (S = 1/2) NO adduct of substrate-bound CDO.34–35 Recently, Limberg and coworkers have prepared structural and functional CDO and ADO mimics supported by the hydrotris(3-phenyl-5-methylpyrazolyl)borate (Ph,MeTp−) ligand and featuring bidentate L-cysteine ethyl ester (CysOEt) or cysteamine (CysAm) anions, respectively.36–38 These 5C Fe(II) complexes react with O2 to give the S-dioxygenated products, as confirmed by experiments involving isotopically-labeled O2. De Visser and others have explored the O2-activation mechanisms of CDO and select TDO models using computational methods.39–42
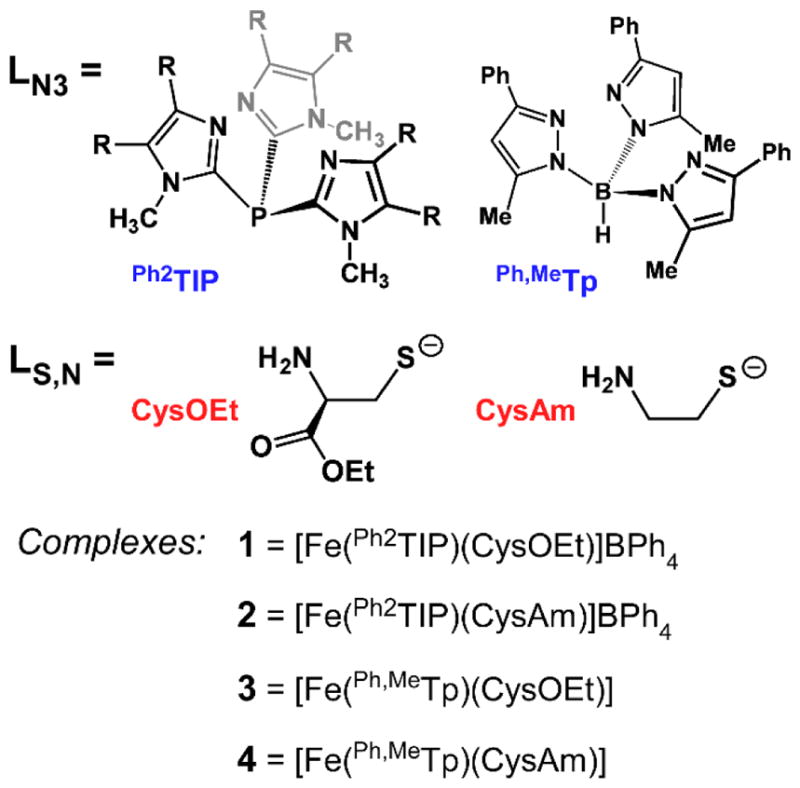
While these previous biomimetic studies of CDO and ADO have yielded many exciting results, the models reported to date lack either the facial geometry or neutral charge of the 3His triad. As part of our efforts to prepare faithful Dke1 and GDO models, our group has found that easily-prepared tris(imidazolyl)phosphine (TIP) ligands nicely replicate the fac arrangement of three imidazole donors found in the active sites.43–46 Here, we report the synthesis and structural characterization of two TDO models: [Fe(Ph2TIP)(CysOEt)]BPh4 (1) and [Fe(Ph2TIP)(CysAm)]BPh4 (2), where Ph2TIP is tris(4,5-diphenyl-1-methylimidazol-2-yl)phosphine (Scheme 2). These complexes are related to the TDO mimics of Limberg and coworkers (3 and 4 in Scheme 2); the critical difference is that 1 and 2 feature a neutral TIP scaffold, whereas 3 and 4 employ a monoanionic Ph,MeTp− ligand. Our previous studies of Dke1 models indicated that TIP ligands accurately reproduce the coordination environment and donor strength of the 3His triad, whereas the properties of Tp− ligands align better with the 2H1C triad.43 Since the two sets of models are complementary, we have conducted parallel studies of 1/2 and 3/4 with the goal of illuminating the role of 3His coordination in promoting CDO/ADO catalysis. Therefore, this manuscript describes and compares the geometric structures, electronic properties, and O2 reactivites of complexes 1–4, as revealed through a combination of crystallographic, spectroscopic, kinetic, and computational methods. The similitude of these models to the enzyme active site is evaluated by juxtaposing their structural and spectroscopic features to those reported in previous studies of CDO.
Scheme 2.
Formation of an iron-superoxo intermediate is the putative first step of O2 activation in both TDOs and related complexes; however, the spin state and degree of S-radical character in the Fe/O2 adduct remain matters of debate. In this report, the nature of O2-bound 2 and 4 in various spin states has been probed via density functional theory (DFT) calculations, with a particular focus on the effect of ligand charge (TIP vs. Tp−) on energetics and Fe-S covalency. We have also explored the surprisingly complicated reactivity of 1 and 2 with NO – a well-established surrogate for O2. Electron paramagnetic resonance (EPR) and IR experiments revealed that multiple Fe/NO species (both high- and low-spin) are formed, including a cationic dinitrosyl iron complex (DNIC). These results highlight the hemilability of scorpionate chelates, as well as the critical role of the active-site pocket in controlling the relative positioning of first-sphere ligands in CDO and ADO.
2. Experimental and Computational Methods
General Information
Unless otherwise noted, all reagents and solvents were purchased from commercial sources and used as received. Dichloromethane (CH2Cl2) was purified and dried using a Vacuum Atmospheres solvent purification system and stored under N2. The synthesis and handling of air-sensitive materials were performed under inert atmosphere using a Vacuum Atmospheres Omni-Lab glovebox equipped with a freezer set to −30 °C. Nitric oxide (NO) from a cylinder (Airgas, Inc.) was purified by passage through an ascarite II column, followed by a cold trap at −78 °C to remove higher NxOy impurities. 15N-labeled NO was prepared via reaction of Na15NO2 with ascorbic acid and aqueous Cu(II) chloride under an argon (Ar) atmosphere.47 The LN3 supporting ligands K(Ph,MeTp) and Ph2TIP, as well as complexes 3 and 4, were prepared according to literature procedures.36–37,44,48
Physical Methods
Elemental analyses were performed at Midwest Microlab, LLC in Indianapolis, IN. UV-vis absorption spectra were collected with an Agilent 8453 diode array spectrometer equipped with a cryostat from Unisoku Scientific Instruments (Osaka, Japan) for experiments at reduced temperatures. Infrared (IR) spectra were measured with a Nicolet Magna-IR 560 spectrometer. X-band EPR experiments were performed using a Bruker EleXsys E650 instrument equipped with an ER4415DM cavity, an Oxford Instruments ITC503 temperature controller, and an ESR-900 He flow cryostat. The program EasySpin (version 5) was used to simulate the experimental spectra.49 1H NMR spectra were recorded in deuterated solvents using a Varian 400 MHz spectrometer. Mass spectrometric data were measured using either an Agilent 6850 gas chromatography-mass spectrometer (GC-MS) with a HP-5 column, or a Brucker matrix-assisted laser desorption/ionization (MALDI) instrument. Magnetic circular dichroism (MCD) data were collected using a Jasco Model J-715 spectropolarimeter, in conjunction with an Oxford Instrument SM-4000 8T magnetocryostat. The samples for these studies were prepared in CH2Cl2 and then diluted with butyronitrile to a final ratio of 3:7 (v/v), or 1:1 (v/v) for the NO-treated samples, thereby yielding glassy solutions upon freezing in liquid nitrogen. Potential artifacts due to glass strain were eliminated by taking the difference between spectra collected with the magnetic field aligned parallel and antiparallel to the direction of light propagation.
X-ray diffraction (XRD) data were collected at 100 K with an Oxford Diffraction SuperNova kappa-diffractometer (Rigaku Corp.) equipped with dual microfocus Cu/Mo X-ray sources, X-ray mirror optics, an Atlas CCD detector, and a low-temperature Cryojet device. The data were processed with the CrysAlis Pro program package, followed by numerical absorption correction based on Gaussian integration over a multifaceted crystal model and then empirical absorption correction using spherical harmonics, as implemented in SCALE3 ABSPACK scaling algorithm. Structures were solved using the SHELXS program and refined with the SHELXL program50 within the Olex2 crystallographic package.51 X-ray crystallographic parameters are provided in Table S1 and further experimental details are available in the CIFs supplied in the Supporting Information.
[Fe(Ph2TIP)(CysOEt)]BPh4 (1)
L-cysteine ethyl ester hydrochloride (38 mg, 0.20 mmol) and triethylamine (62 μL, 0.44 mmol) were combined in CH2Cl2 and stirred until the solid completely dissolved. A solution of the [Fe(Ph2TIP)(MeCN)3](OTf)2 precursor44 (0.242 g, 0.20 mmol) in CH2Cl2 was added dropwise, giving rise to a bright yellow solution that was stirred for 30 minutes. The solvent was removed under vacuum and the resulting yellow residue was taken up in MeOH, and filtered. Addition of NaBPh4 (68 mg, 0.20 mmol) caused immediate formation of a yellow precipitate. After solvent removal under vacuum, the resulting yellow powder was dissolved in a minimal amount of 1,2-dichloroethane and layered with MeOH. The yellow needles that formed after one day were collected, washed with MeOH, and dried under vacuum. Yield = 0.138 g (55 %). X-ray quality crystals were obtained by allowing the mother liquor to stand for one week at room temperature and under inert atmosphere. Crystallographic analysis revealed the presence of a MeOH solvate in the unit cell. Elemental analysis calculated (%) for C77H69BFeN7O2PS•CH3OH: C, 72.84; H, 5.72; N, 7.62. Found: C, 72.56; H, 5.61; N, 7.70. IR (solution in CH2Cl2, cm−1): ν = 3345 (w, ν(N-H)), 1728 (s, ν(C=O)). 1H NMR (400 MHz, CDCl3): δ = 15.54 (s, 1H, -NH), 15.14 (s, 1H, -NH), 14.86 (s, 9H, N-CH3), 8.08 (s, 3H), 7.63 (s, 12H, BPh4), 6.90 (s, 6H), 6.37 (s, 8H, BPh4), 6.04 (s, 6H), 4.22 (s, 6H), 2.96 (q, 2H, -CO2CH2CH3), 2.41 (broad s, 2H, -SCH2C-), 2.01 (s, 1H, -H2NCH-), 1.31 (t, 3H, -COOCH2CH3), −17.46 (broad s, 6H) ppm.
[Fe(Ph2TIP)(CysAm)]BPh4 (2)
Cysteamine hydrochloride (23 mg, 0.20 mmol) and triethylamine (62 μL, 0.44 mmol) were combined in CH2Cl2 and stirred until all the solid dissolved. A solution of [Fe(Ph2TIP)(MeCN)3](OTf)2 (0.242 g, 0.20 mmol) in CH2Cl2 was added dropwise, giving an orange solution that was stirred for 30 minutes. The solvent was removed under vacuum and the resulting yellow residue taken up in MeOH. Addition of NaBPh4 (68 mg, 0.20 mmol) caused immediate formation of a yellow precipitate. After evaporation of the solvent under vacuum, the resulting yellow powder was dissolved in a minimal amount of 1,2-dichloroethane and layered with MeOH. X-ray quality yellow crystals formed after one day at room temperature. The crystals were collected, washed with MeOH, and dried under vacuum. Yield = 0.015 g (18 %). Elemental analysis calculated (%) for C74H65BFeN7PS: C, 75.19; H, 5.54; N, 8.29. Found: C, 76.02; H, 6.02; N, 7.89. IR (solution in CH2Cl2, cm−1): ν = 3353 (w, ν(N-H)). 1H NMR (400 MHz, CDCl3): δ = 14.86 (s, 9H, N-1-CH3), 8.19 (s, 8H, BPh4), 8.05 (s, 4H, BPh4), 7.34 (s, 8H, BPh4), 6.85 (s, 6H), 6.03 (s, 3H), 5.93 (s, 3H), 4.08 (s, 6H), 3.89 (s, 2H, -NH2), 3.21 (s, 6H), 2.63 (t, 1H, -CH2CH2-), 1.35 (t, 1H, -CH2CH2-), 1.23 (t, 1H, -CH2CH2-), 1.13 (t, 1H, -CH2CH2-), −19.26 (broad s, 6H, o-4-Ph) ppm.
Reactivity with Dioxygen
Rate measurements were generally performed under conditions of O2 saturation by bubbling O2 (Airgas, Inc.) through CH2Cl2 solutions of 1 or 3 for several minutes. For both complexes, rates of reaction were measured four times at 20 °C, and changes in absorption intensity at two wavelengths were fit to exponential curves using the program IGOR. The reported k1-values and uncertainties correspond to the average and standard deviation, respectively, of the resulting pseudo first-order rate constants. The concentration of O2 in solution at various temperatures (T) was estimated using the formula: S = (LPO2)/(TR), where L is the Ostwald coefficient (0.257 for CH2Cl2), PO2 is the partial pressure of O2, and R is the gas constant.52–53 The determination of PO2 accounted for the vapor pressure of CH2Cl2 (Psolv) as a function of T: PO2 = 1 atm − Psolv. Lower concentrations of O2 were obtained by injecting anaerobic solutions of the Fe(II) complex into O2-saturated solutions of CH2Cl2 in varying amounts. Following established procedures,36–37 the products of the oxygenation reactions were isolated by addition of 3M HCl to the reaction mixture and stirred for 3 hours. The aqueous layer was collected and the solvent removed under vacuum. The remaining residue was dissolved in MeOH and stirred with chelex for 12 hours. The solvent was removed by vacuum after filtration, and the resulting white solid was washed with toluene. 1H NMR data of the reaction products were interpreted with the aid of published spectra and/or by comparison to spectra measured with commercially available material. Product of 1 + O2 reaction: Yield = XX %; 1H NMR (400 MHz, CD3OD): δ = 1.28 (t, 3H, −OCH2CH3), 2.49 (dd, 1H, -CH2S), 2.61 (dd, 1H, -CH2S), 4.02 (m, 1 H, -CHNH2), 4.20 (q, 2H, −OCH2CH3) ppm. MALDI-MS: m/z = 169.20, (calc. ). Product of 2 + O2 reaction: Yield = XX %; 1H NMR (400 MHz, CD3OD): δ = 2.45 (t, 2H, -CH2S), 3.15 (t, 2H, -CH2NH2) ppm. MALDI-MS: m/z = 169.20, (calc. ).
Density Functional Theory (DFT) Computations
Electronic-structure calculations were carried out using the ORCA 3.0 software package developed by Dr. F. Neese (MPI for Chemical Energy Conversion).54 Computational models of 2 and 4 were obtained via unrestrained DFT geometry optimizations, using the X-ray crystal structures as starting points. For calculations involving 2, the Ph2TIP ligand was truncated by replacing the 4,5-diphenyl-1-methylimidazole donors with 4-methylimidazole rings; similarly, in calculations for 4 employed the Ph,MeTp− ligand was modeled as Me,HTp−. Numerical frequency calculations verified that all structures corresponded to a local minimum with only real vibrational frequencies. The zero-point energies, thermal corrections, and entropy terms (vibrational, rotational, translational) were obtained from these frequency calculations. All calculations were carried out spin-unrestricted and utilized Ahlrichs’ valence triple-ζ basis set (TZV) and TZV/J auxiliary basis set, in conjunction with polarization functions on main-group and transition-metal elements (default-basis 3 in ORCA).55–57 Solvent effects were accounted for using the conductor-like screening model (COSMO)58 with a dielectric constant (ε) of 9.08 for CH2Cl2.
The DFT calculations employed different functionals depending on the nature of the species under examination and the property being computed. Geometric structures for the Fe(II) complexes and Fe/O2 adducts were optimized using the Perdew-Burke-Ernzerhof (PBE) functional59 with 10% Hartree-Fock exchange. The “spin-flip” feature of ORCA was employed to generate broken-symmetric wavefunctions for the Stot = 2 and 1 states of Fe/O2 adducts containing high-spin Fe centers. The transition state for the S-Od bond forming reaction (where Od is the distal oxygen atom of the iron-superoxo unit) was located by performing a relaxed surface scan along the S···Od distance. The existence of the transition state was confirmed by the presence of one imaginary frequency, corresponding to the ν(S–Od) mode.
Calculations of the Fe/NO adducts employed either the Becke-Perdew (BP86)60–61 or TPSSh62–63 functionals. In calculations of EPR parameters, the “core properties” with extended polarization [CP(PPP)] basis set64 was used for the Fe atom and Kutzelnigg’s NMR/EPR (IGLO-III) basis set65 for the NO ligand. The contribution of spin-orbit coupling to the g- and A-tensors was evaluated by solving the coupled-perturbed self-consistent field (CP-SCF) equations.66–69 To compute the hyperfine coupling constants, a high resolution grid with an integration accuracy of 7.0 was used for the Fe and N atoms. Time-dependent DFT (TD-DFT) calculations employed the cam-B3LYP range-separated hybrid functional,70 previously shown to yield good agreement between experimental and TD-DFT computed absorption spectra for CDO.71 Absorption energies and intensities were computed for 40 excited states with the Tamm-Dancoff approximation.72–73 Isosurface plots of molecular orbitals were prepared using the ChemCraft program.
3. Results and Discussion
3.A. Synthesis and Solid State Structures
Complexes 1 and 2 were synthesized by first treating the previously-reported [Fe(Ph2TIP)(MeCN)3](OTf)2 precursor44 with either CysOEt•HCl or CysAm•HCl and two equivalents of triethylamine in CH2Cl2. After solvent removal, the resulting triflate salts were dissolved in MeOH and addition of NaBPh4 caused immediate precipitation of 1 and 2 as bright yellow solids. X-ray quality crystals of 2 were grown by layering concentrated 1,2-dichloroethane (DCE) solutions with MeOH. In the case of 1, this procedure yielded analytically pure but poorly-diffracting needles; however, crystals suitable for X-ray analysis were eventually obtained by allowing the decanted mother liquor to sit for one week. The resulting crystal structure revealed that, during this time, the CysOEt ligand had undergone trans-esterification to give the complex [Fe(Ph2TIP)(CysOMe)]BPh4 (1Me) instead. Despite this, 1H NMR spectra of the first crop of crystals, which were employed in all spectroscopic and reactivity studies, indicated retention of the ethyl substituent (vide infra).
The X-ray structures of 1Me and 2 reveal the presence of 5C monoiron(II) complexes with a bidentate S,N-cysteinato anion and κ3-Ph2TIP chelate (Figure 1). In each case, the thiolate donor forms a hydrogen bond with a MeOH solvate molecule. The Fe(II) coordination geometries lie half-way between the square-pyramidal and trigonal bipyramidal limits, as quantified by the τ-values of 0.54 (1Me) and 0.55 (2).74 Significantly, the facial coordination mode of the Ph2TIP ligand ensures that a vacant site for O2-binding is available cis to the thiolate ligand – a known requirement for dioxygenation of the sulfur atom.31 Selected metric parameters for the solid-state structures of 1Me and 2 are provided in Table 1, in addition to previously-reported values for 3 and 4. The Fe-NTIP bond distances are typical for high-spin Fe(II) complexes and fall within a narrow range of 2.15–2.20 Å. The Fe-NTp distances in 3 and 4 are slightly shorter on average, while also displaying greater variation with two short bonds of 2.12 Å (ave) and one long bond near 2.275 Å. The Fe-S1 and Fe-NCys bond distances change little across the 1–4 series with average values of 2.31 and 2.26 Å, respectively.
Figure 1.

(a) Thermal ellipsoid plot (50% probability) obtained from the X-ray crystal structure of 2. The BPh4− counteranion, non-coordinating solvent molecules, and all hydrogen atoms have been omitted for clarity. (b) Overlays of the crystallographically-derived structures of 1Me (light gray) and the Cys-bound active-site of CDO (pink) derived from PDB 4TJO. The phenyl substituents of the TIPPh2 ligand are not shown.
Table 1.
Selected Bond Distances (Å) and Bond Angles (°) for Complexes 1–4 and the Cys-Bound CDO Active Site as Determined by X-ray Crystallography.
| Bond Distances | 1 | 2 | 3 a | 4 b | CDO c |
|---|---|---|---|---|---|
| Fe1-N1 | 2.170(2) | 2.169(2) | 2.112(2) | 2.153(2) | 1.893 |
| Fe1-N3 | 2.153(2) | 2.178(2) | 2.106(2) | 2.110(2) | 2.199 |
| Fe1-N5 | 2.195(2) | 2.183(2) | 2.275(2) | 2.249(2) | 2.110 |
| Fe1-S1 | 2.3107(6) | 2.3051(5) | 2.3122(9) | 2.3175(6) | 2.291 |
| Fe1-N7 | 2.245(2) | 2.248(2) | 2.290(3) | 2.252(2) | 2.262 |
|
| |||||
| Bond Angles | |||||
| N1-Fe1-N3 | 96.73(6) | 98.67(6) | 95.36(9) | 95.97(6) | 99.9 |
| N1-Fe1-N5 | 87.90(6) | 86.32(6) | 82.29(9) | 86.08(6) | 98.6 |
| N1-Fe1-S1 | 125.08(5) | 120.73(4) | 134.21(7) | 129.17(5) | 112.9 |
| N1-Fe1-N7 | 89.01(6) | 88.43(6) | 89.30(9) | 90.71(6) | 85.7 |
| N3-Fe1-N5 | 83.02(6) | 82.88(6) | 85.29(9) | 82.16(6) | 86.0 |
| N3-Fe1-S1 | 137.22(5) | 139.15(4) | 130.07(7) | 134.18(5) | 146.8 |
| N3-Fe1-N7 | 87.80(6) | 86.20(6) | 92.04(9) | 87.38(7) | 96.9 |
| N5-Fe1-S1 | 104.99(4) | 108.23(4) | 104.07(6) | 105.47(5) | 94.1 |
| N5-Fe1-N7 | 169.90(6) | 167.03(6) | 170.88(9) | 168.67(6) | 174.3 |
| S1-Fe1-N7 | 84.63(5) | 84.64(4) | 84.32(7) | 84.96(5) | 80.8 |
The superposition of complex 1Me upon the active-site structure of Cys-bound Fe(II)-CDO75 in Figure 1(b) highlights the structural similarities between the synthetic and enzymatic coordination environments. The most noticeable discrepancy concerns the relative orientations of the imidazole rings. The His donors in the enzyme active site possess greater rotational freedom than their synthetic counterparts, as the latter are tethered to a central P-atom at the 2-position. The CDO crystal structure exhibits Fe-NHis distances of 2.20, 2.11, and 1.89 Å (Table 1); the smallest value (corresponding to His86) is much shorter than expected based on synthetic CDO models and typical Fe-NHis bond distances in proteins.76 The Fe-NCys and Fe-SCys bond distances of CDO nicely match those measured for 1Me (Table 1) and the orientations of the cysteinato ligands with respect to the facial N3 triad are quite similar, although CDO lies somewhat closer to the square-pyramidal limit (τ-value of 0.46). The high degree of structural resemblance between CDO and 1Me suggests that these five-coordinate sites are similarly primed for O2 activation.
3.B. Solution-State Spectroscopic Properties
Examination of CDCl3 solutions of 1 and 2 using the Evans NMR method provided effective magnetic moments of 5.16 and 5.02 μB, respectively, consistent with mononuclear, high-spin (S = 2) ferrous complexes. The 1H NMR spectra of 1 and 2 exhibit paramagnetically-shifted peaks in the 15 to −20 ppm range(Figures S1 and S2). The three imidazole donors are spectroscopically equivalent due to dynamic averaging of the CysOEt/CysAm positions on the NMR time scale. Based on our prior analysis of Ph2TIP-based Fe(II) complexes,44 the intense downfield peak at 15 ppm is assigned to the N-1-CH3 groups, while the broad upfield peak near −18 ppm is attributed to ortho protons of the 4-Ph substituents. Resonances arising from the CysOEt and CysAm ligands experience only modest shifts and broadening upon ligand binding to Fe. The splitting pattern due to the ethyl ester moiety is clearly discernable in the spectrum of 1, indicating that the CysOEt ligand remains intact. In short, the 1H NMR data confirm that the solution- and solid-states structures are essentially the same.
Solutions of 1 and 2 in CH2Cl2 are pale yellow due to the presence of overlapping absorption bands (ε ~ 1000 M−1cm−1) in the near-UV region that are attributed to S→Fe(II) charge transfer (CT) transitions (vide infra). To obtain further information regarding the electronic excited states of these complexes, magnetic circular dichroism (MCD) spectra of 1 and 2 were measured in frozen 3:7 solutions of CH2Cl2:butyronitrile. The absorption and MCD spectra of the two complexes are nearly identical, suggesting that the cysteinato substituent (–CO2Et or –H) has little effect on overall electronic structure. As shown in Figures 2 and S3, the MCD spectra exhibit two intense, positive features near 23,800 and 26,800 cm−1. The broadness of the lower-energy band suggests that it is composed of at least two electronic transitions. In all cases, the intensities of the MCD bands increase with decreasing temperature, displaying the C-term behavior expected for paramagnetic S = 2 systems. Significantly, the MCD spectra of 1 and 2 also exhibit a temperature-dependent band at 11,270 cm−1 (887 nm) that is barely perceptible in the absorption spectra. Based on its large C0/D0 ratio of ~2.0 (where C0 and D0 are related to the MCD and absorption intensities, respectively77), this peak likely arises from an Fe(II) d-d transition. This assignment is supported by previous spectroscopic studies, which revealed that the highest-energy d-d transition of 5C Fe(II) complexes lies between 8,400 and 13,500 cm−1.78
Figure 2.

Electronic absorption and MCD spectra collected for complexes 2 (top) and 4 (bottom). The absorption spectra were measured at room temperature in CH2Cl2. The MCD spectra were obtained for frozen, glassy solutions in a 3:7 mixture (v/v) of CH2Cl2:butyronitrile at 4 K with a magnetic field of 7 T.
For the sake of comparison, Figure 2 also displays the absorption and MCD spectra of complex 4 measured under identical conditions. Only two S→Fe(II) CT bands are apparent, and these are blue-shifted by 2700 cm−1 relative to those of complex 2. The d-d band experiences a smaller energy increase of 300 cm−1. Similar shifts in band energies are observed when spectra of CysOEt-containing 1 and 3 are compared (Figure S3).
3.C. Computational Analysis of Fe(II) Complexes
The spectroscopic results were interpreted with the aid of time-dependent DFT (TD-DFT) studies that employed the cam-B3LYP range-separated hybrid functional.70 These calculations were performed with geometry-optimized structures of 2 and 4 that are in good agreement with the crystallographic structures (Table S2). Consistent with the experimental data, the TD-DFT computed absorption spectrum for 2 is dominated by three S→Fe(II) CT transitions in the region below 30,000 cm−1 (Figure S4). The energy-level diagram provided in Figure 3 depicts the ligand- and Fe(II)-based molecular orbitals (MOs) involved in the relevant transitions. The donor MOs are localized on the CysAm ligand and contain primarily S(3p) character: the Sσ MO lies along the Fe-S bond, while the Sπ MO is perpendicular to the bond. The two lowest-energy CT bands in the TD-DFT spectrum arise from Sπ→Fe(dxz)/Fe(dyz) transitions centered around 25,000 cm−1, which nicely matches the experimental band at 23,800 cm−1. To higher energy, TD-DFT predicts a Sσ→Fe(II) (σ→σ*) transition at 29,100 cm−1 that likely corresponds to the experimental feature at 26800 cm−1. The highest-energy Fe d-d transition, attributed to the Fe(dxy)→Fe(dx2–y2) excitation, is predicted to appear as a weak feature at 13,750 cm−1. Thus, the calculated energies agree resonably well with the experimental values, with a root-mean-square deviation of only 2100 cm−1 for the bands observed by MCD.
Figure 3.

Energy-level diagram for the spin-down (β) molecular orbitals (MOs) obtained from a spin-unrestricted DFT calculation for complex 2. MOs are labeled according to their principal contributor. DFT-generated isosurface plots of the MOs are also provided.
Comparison of the DFT results obtained for 2 and 4 indicate that substitution of neutral Ph2TIP with anionic Ph,MeTp− causes an nearly uniform 4450 cm−1 increase in the energies of the Fe 3d set of orbitals. Hence, the S→Fe(II) CT bands experience a substantial blue-shift upon conversion of 2→4, while the highest-energy d-d transition is largely unaffected – consistent with the spectroscopic data described above. The Sσ→Fe(II) (σ→σ*) transition of complex 4, predicted to occur at 33,270 cm−1, is likely obscured by the onset of π–π* transitions in the near-UV region, which explains the presence of only two CT bands in the absorption/MCD spectra of 3 and 4. Moreover, an analysis of the relative compositions of the half-occupied Fe 3d-based MOs indicates that the Fe–S bond in 2 is more covalent than its counterpart in complex 4, which is also reflected by minor differences in the Mulliken spin populations of the S atoms (0.19 spins in 2 and 0.15 spins in 4). This finding is consistent with the fact that the TIP-based complexes exhibit more intense S→Fe(II) CT transitions than the Tp-based complexes (Figures 2 and S3), given that CT intensity is proportional to the amount of ligand character in the metal-based MOs.
Substrate-bound Fe(II)-CDO likewise exhibits two S→Fe(II) CT bands in its MCD spectrum; however, both of these features appear above 30,000 cm−1, much higher in energy than the corresponding bands of complexes 1–4.79 This large discrepancy in CT energies is rather surprising given the high-degree of structural similarity between the first coordination spheres of 1 and Cys-bound Fe(II)-CDO, as discussed above (Figure 1). Noncovalent interactions within the active site of CDO presumably modulate the relative energies of the thiolate- and Fe 3d-based MOs, thereby increasing the CT energies.
3.D. O2 Reactivity: Experimental Results
Not surprisingly, the O2 reactivity of the Ph2TIP-based complexes closely mirrors that reported previously by Limberg and coworkers for 3 and 4. As shown in Figure 4, treatment of 1 with O2 results in the slow decay of the S→Fe(II) CT bands to eventually yield a featureless spectrum, indicating loss of the iron-thiolate bond. Similar spectral changes were observed upon O2 exposure of complexes 3 and 4,36–37 as well as Cys-bound Fe(II)-CDO.79 The reaction products were isolated after stirring solutions of 1 and 2 in O2-saturated CH2Cl2 for six hours, followed by acidic work-up. 1H NMR spectra revealed that the 1 + O2 reaction yields the ethyl ester of L-cysteine sulfinic acid as the only observable CysOEt-derived product (Figure S5). Similarly, exposure of 2 to O2 yields hypotaurine (HTau) as the dominant product, although an unidentified CysAm-derived compound is also present in smaller amounts (HTau:unknown ratio of 5:1; Figure S6). The identities of the sulfinic acid products were confirmed by comparison to literature spectra (cysteine sulfinic acid ethyl ester) or commercial samples (HTau), in addition to mass spectrometric analysis. Thus, each complex in the 1–4 series behaves as a structural and functional TDO mimic.
Figure 4.
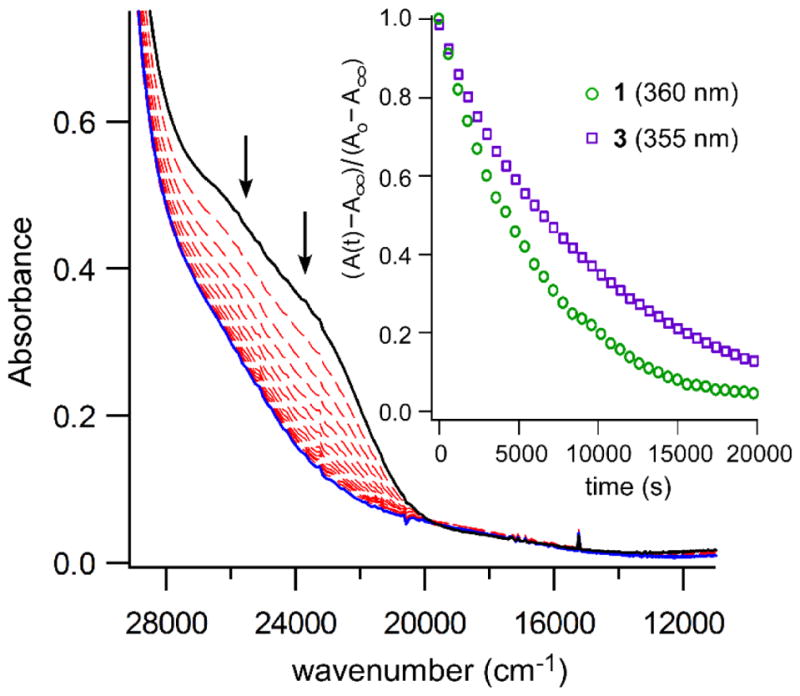
Time-resolved absorption spectra for the reaction of complex 1 (conc. = 0.53 mM) with O2; the spectra shown here were collected at intervals of 30 min. The reaction was performed at 20 °C in O2-saturated CH2Cl2. The path length of the cuvette was 1.0 cm. Inset: Plots of absorption intensity as a function of time for reaction of 1 and 3 with O2 at 20 °C in O2-saturated CH2Cl2. The wavelengths monitored were 360 and 355 nm for 1 and 3, respectively. Absorption intensities were normalized using the equation provided in the plot.
Rate measurements for the reaction of 1 and 3 with O2 were generally performed in O2-saturated CH2Cl2 solutions ([O2] = 5.8 mM at 20 °C) with Fe(II) concentrations of less than 0.60 mM. Under these conditions, the decay in absorption intensity versus time followed first-order behavior past three half-lives (Figures 4 and S7) for both complexes, and initial rates increased linearly with O2 concentration (Figure S8). Thus, the reactions are first-order with respect to both Fe and O2, permitting a detailed kinetic analysis. At 20 °C, the 1 + O2 reaction proceeds with a pseudo-first-order rate constant (k1) of 1.9(4) × 10−4 s−1. The 3 + O2 reaction exhibits a smaller k1-value of 1.2(3) × 10−4 s−1.80 Thus, the identity of the LN3 supporting ligand has only a modest impact on the O2 reaction rate, with the neutral Ph2TIP ligand offering a slight rate advantage over the anionic Ph,MeTp− ligand.
Activation parameters for the 1 + O2 reaction were determined by monitoring rates at temperatures between 35 and −30 °C. Analysis of the linear Eyring plot (Figure 5) provided an activation enthalpy (ΔH‡) of 12(1) kcal/mol and activation entropy (ΔS‡) of −25(4) cal/(mol·K), resulting in an activation free energy (ΔG‡) of 19(2) kcal/mol at 293 K. These values are similar to previously-reported parameters for O2 activation by mononuclear Fe complexes,52 including closely-related 5C complexes with Ph2TIP and Tp− ligands.81–82 The large negative value for ΔS‡ indicates that the rate-determining step has an associative nature, which is common for reactions involving O2. However, as discussed in the next section, the activation parameters likely reflect multiple elementary reactions, and thus the values must be interpreted with caution.
Figure 5.
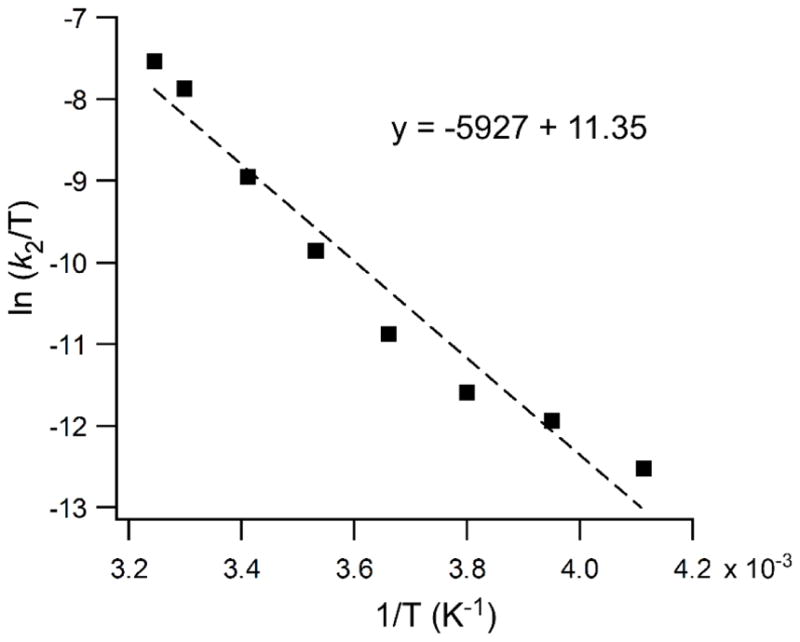
Eyring plot for the reaction of 1 with O2 in O2-saturated CH2Cl2 over a temperature range of 35 to −30 °C. Second-order rate constants (k2) were obtained by dividing the pseudo-first-order rate constant by [O2] at the specified temperature.
3.E. O2 Reactivity: Computational Results
The first step in the proposed O2 activation mechanisms of CDO and related models complexes is the generation of an iron-superoxo species, followed by nucleophilic attack of the distal O atom on the thiolate ligand to give a four-membered Fe-O-O-S ring. Computational studies suggested that this second step is rate-limiting in both the enzyme and complex 3.39,83 To better understand the kinetic data reported above, we have examined the thermodynamics of O2 binding and O–S bond formation via DFT calculations for complexes 2 and 4.84 These calculations employed the PBE functional with 10% HF exchange, which has been shown provide reliable geometries and relative energies for Fe/O2 species.85
The adduct that forms upon O2 binding to a high-spin Fe(II) complex has three possible spin states: triplet (S = 1), quintet (S = 2), or septet (S = 3). The septet and quintet states are best described as high-spin Fe(III) centers coupled to a superoxide radical (O2•−) in either a ferromagnetic or antiferromagnetic manner, respectively.86 The triplet state arises from antiferro-magnetic coupling between a high-spin Fe(II) center and a neutral O2 ligand. However, we also considered the possibility that the S = 1 Fe/O2 adduct consists of a low-spin Fe(III) ion ferromagnetically coupled to O2•−, as this configuration yielded the lowest-energy Fe/O2 model in de Visser’s study of complex 3.39 The resulting computational models are labeled spin[2-O2](HS,LS) and spin[4-O2](HS,LS), where HS and LS indicate whether the Fe center is high-spin or low-spin, respectively. Metric parameters of the geometry-optimized structures are provided in the Supporting Information (Table S3). Whereas de Visser found that O2 binding to 3 triggers dissociation of a pyrazole donor regardless of spin state,39 nearly allo of our optimized Fe/O2 models are 6C with Fe-NTIP/Tp bonds lengths ≤ 2.31 Å; the only exceptions being the S=3[2-O2]HS and S=2[2-O2]HS models, in which one of the Fe-NTIP bonds elongates to ~2.4 Å during geometry optimization to yield quasi-6C structures (Table S3).
Table 2 summarizes the computed thermodynamic parameters for the eight Fe(II) + O2 → Fe/O2 reactions examined here. For both [2-O2] and [4-O2], the four spin states lie with 3 kcal/mol of each other. This result is typical of ferric-superoxo species, which commonly possess a large number of close-lying electronic states.86 The enthalpic (ΔHrxn) contributions to O2 binding are generally insignificant or slightly favorable, yet the overall process is endergonic (ΔGrxn = 8–13 kcal/mol) due to the unfavorable entropic effects characteristic of bimolecular reactions. Although the differences are within the uncertainty of the methodology, the computed ΔGrxn-values are uniformly smaller for the [4-O2] models relative to their [2-O2] counterparts, suggesting that the Tp-based complexes have a higher affinity for O2 than the TIP-based complexes. This conclusion is supported by the fact that the computed ν(O-O) frequencies for the [4-O2] models are 15–25 cm−1 lower in energy compared to their counterparts in the analogous [2-O2] models (Table 2), implying that the Ph,MeTp− ligand promotes greater charge transfer from Fe(II) to O2. Analysis of the Mulliken populations revealed that the S-donors in the Fe/O2 adducts possess a significant amount of spin-density, with alpha-spin-values of 0.33 and 0.27 in the S = 2 structures of [2-O2] amd [4-O2], respectively. This partial radical character, which is consistently larger in the TIP-containing models, likely promotes formation of the S-O bond in the next step of the mechanism.
Table 2.
Energetic Parameters Computed for O2 Binding to Complexes 2 and 4, and Comparison of Superoxo Stretching Frequencies in the Resulting [FeO2] Adducts.
| Stot (Fe spin) a | reactants | ΔHrxn | ΔSrxn b | ΔGrxn b | ν(O-O) (cm−1) |
|---|---|---|---|---|---|
| S = 3 (HS) | 2 + O2 | 0.5 | 10.3 | 10.8 | 1222 |
| 4 + O2 | −1.3 | 10.6 | 9.3 | 1197 | |
|
| |||||
| S = 2 (HS) | 2 + O2 | 2.3 | 10.8 | 13.1 | 1213 |
| 4 + O2 | 0.7 | 11.2 | 11.9 | 1198 | |
|
| |||||
| S = 1 (HS) | 2 + O2 | 1.7 | 11.2 | 12.9 | 1269 |
| 4 + O2 | −0.3 | 12.3 | 12.0 | 1243 | |
|
| |||||
| S = 1 (LS) | 2 + O2 | −3.3 | 14.6 | 11.3 | 1183 |
| 4 + O2 | −7.1 | 15.1 | 8.0 | 1160 | |
Stot is the overall spin of the [FeO2] adduct, whereas HS and LS indicate the spin-state of the Fe center itself.
Thermodyanmic parameters were computed assuming a temperature of 298.15 K.
The majority of computational studies of CDO and synthetic mimics suggest that formation of the critical Fe(II)-O-O-S cyclic intermediate occurs along the S = 2 potential energy surface (PES). 39–40,83 Our DFT calculations revealed that conversion of S=2[2-O2]HS and S=2[4-O2]HS to the corresponding peroxo species occurs with overall transition-state barriers of 25 and 23 kcal/mol, respectively, relative to the Fe(II) and O2 starting materials (Scheme 3). The barrier of ~12 kcal/mol for Od-S bond formation is in line with previous DFT studies of WT CDO and related models, which reported values around 10 kcal/mol for the same mechanistic step.40,87 The difference in transition-state energies between S=2[2-O2]HS and S=2[4-O2]HS lies within the estimated error of the computations (± 2 kcal/mol), consistent with our experimental finding that the identity of LN3 does not have a major impact on dioxygenation rates. Significantly, the computed activation parameters for S=2[2-O2]HS (ΔH‡ = 10.5 kcal/mol, ΔS‡ = −48 cal/(mol•K)) are in reasonably good agreement with the experimentally-determined values of ΔH‡ = 12(1) kcal/mol, ΔS‡ = −25(4) cal/(mol•K). In the DFT-optimized transition-state geometry derived from S=2[2-O2]HS (Scheme 3), the O-O bond is positioned directly over the Fe-S bond, giving rise to a S···Od distance of 2.18 Å. The O–O and Fe–S bonds are elongated by 0.040 and 0.212 Å, respectively, compared to the S=2[2-O2]HS adduct. The degree of S-radical character in the transition state, as indicated by Mulliken spin populations, is significantly greater for TIP-based 2 (0.18 spins) than Tp-based 4 (0.11 spins). The distribution of unpaired spin density in the transition-state structure (Figure S9) indicates that the S–Od bond arises from overlap between the S-based pz-orbital and the O2-based π* orbital pointed along the S–Od axis.
Scheme 3.

Relative energies (in kcal/mol) for the initial steps in the reaction of 2 (red lines) and 4 (blue lines) with O2 at 298.15 K. Geometry-optimized structures of S=2[2-O2]HS (left) and S=2[2-O2]TS (right) are also provided and select bond lengths are indicated in angstroms (Å).
For the sake of completeness, we also examined the cyclization reaction along the S = 1 PES originating from the S=1[2-O2]LS and S=1[4-O2]LS species, as this process yielded the lowest-energy transition state in de Visser’s study of complex 3.39 However, the barrier for the S =1 route is nearly 10 kcal/mol higher in energy than the S = 2 pathway, suggesting that the former is not a viable option for the dioxygenation mechanism if the Fe center remains six coordinate.
As shown in Figure S10, the energies of the resulting Fe(II)-O-O-S cyclic intermediates on the S = 2 PES are roughly equal to those of the preceding Fe/O2 adducts. The next step in the mechanism requires O–O bond cleavage to yield an high-spin oxoiron(IV) unit bound to a sulfoxide ligand. Our DFT calculations indicate that the transition-state barrier for O-O bond cleavage is quite small (~2 kcal/mol; Figure S10), providing further evidence that formation of the Fe-O-O-S species is the rate-limiting step in the dioxygenation mechanism.
3.F. Nitric Oxide Reactivity: Experimental Results
Using nitric oxide (NO) as a surrogate for O2 is a common strategy for gaining insights into the electronic structures of ferrous centers in MNIDs and related complexes, since the reaction gives rise to an EPR-active {FeNO}7 species (using the Enemark-Feltham notation, where the superscript indicates the sum of Fe 3d and NO π* electrons). A notable feature of CDO is that the enzyme-substrate complex binds NO to yield a low-spin (S = 1/2) Fe/NO adduct, as opposed to a high-spin (S = 3/2) adduct as is generally observed for MNID enzymes with the 2H1C triad.88
Our studies found that exposure of 1 and 2 to NO yields different species depending on the temperature of the reaction. For both complexes, the Fe/NO adducts generated at −70 °C (1NO and 2NO) feature two absorption bands near 440 and 660 nm. As shown in Figure 6, these spectra closely resemble those reported previously for high-spin, six-coordinate (6C) {FeNO}7 complexes with similar ligand sets, such as [Fe(NO)(4-TIPPh)(acac)]+ and [Fe(NO)(Ph2Tp)(acacPhF3)] (where 4-TIPPh = tris(2-phenylimidazol-4-yl)phosphine and acacPhF3 = anion of 4,4,4-trifluoro-1-phenyl-1,3-butanedione; the structure of the latter complex was verified crystallographically).45 The corresponding X-band EPR spectra (Figure 6; inset) exhibit an axial S = 3/2 signal (g ~ 4.0, 2.0), also characteristic of 6C {FeNO}7 species.89 The intensity of the S = 3/2 signal was measured between 10 and 50 K for 1NO, and simulation of the data provided a D-value of 6.7(3) cm−1 and E/D-ratio of 0.005. We also generated Fe/NO adducts of 3 and 4 (3NO and 4NO, respectively), as Limberg and coworkers did not report NO-binding studies for these complexes. The electronic absorption and EPR data of 3NO and 4NO (not shown) are nearly identical to those obtained for 1NO and 2NO, indicating that the LN3 ligand has little effect on NO reactivity or the spectroscopic features of the resulting species. Therefore, only the iron-nitrosyl adducts derived from 1 and 2 will be discussed in detail.
Figure 6.
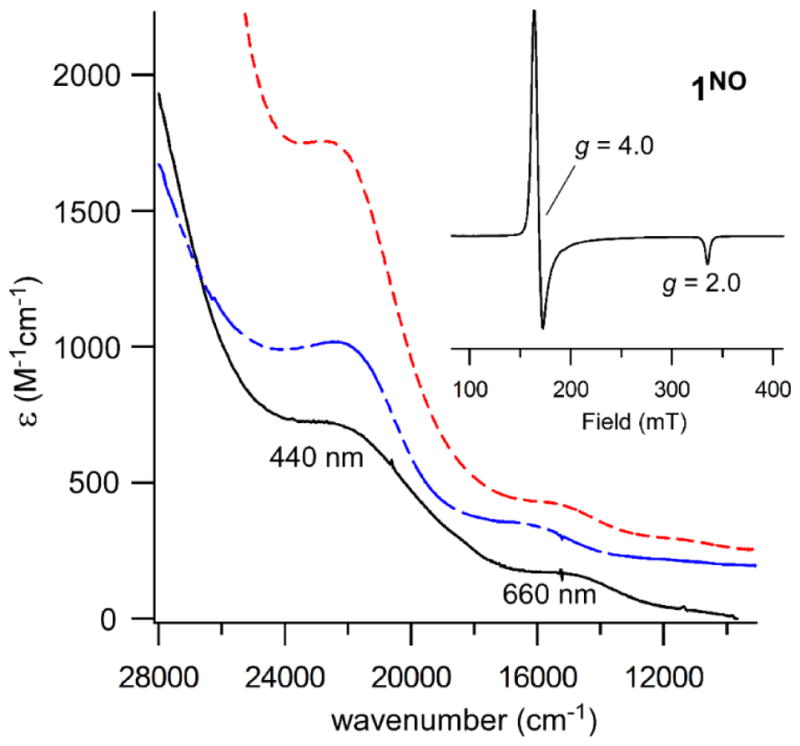
Absorption spectrum of 1NO (black solid line) generated via reaction of 1 with NO at −70 °C in CH2Cl2. Previously-reported45 UV-vis spectra of [Fe(NO)(4-TIPPh)(acac)]+ (blue dashed line) and [Fe(NO)(Ph2Tp)(acacPh,F3)] in MeCN and CH2Cl2, respectively, are shown for comparison (these spectra have been shifted upwards for clarity). Inset: X-band EPR spectrum of 1NO in frozen CH2Cl2. Parameters: frequency = 9.380 GHz; power = 2.0 mW; modulation amplitude = 2.0 G; T = 10 K.
In addition to the dominant S = 3/2 resonances arising from 1NO and 2NO, EPR spectra of samples exposed to NO at low temperatures contained variable amounts of a minor S = 1/2 signal (Figures 6 and S11). The concentrations of the low-spin species, labeled 1NO-LS and 2NO-LS, were generally less than 10% of total Fe as determined by double integration of the signal at 77 K.90 Notably, these rhombic spectra exhibit 1:1:1 triplet superhyperfine splitting in each of the three g-values, corresponding to a 14N nucleus with Ax,y,z = 36, 36, and 45 MHz in the case of 2NO-LS (the complete set of spin-Hamiltonian parameters is provided in Table 3). This pattern coverts to a 1:1 doublet when the samples are prepared with 15NO (Figures 6 and S11), confirming that the splitting is due to the N-atom of a single NO ligand. Therefore, we can rule out the possibility that the 1NO-LS and 2NO-LS signals are due to formation of a DNIC. Possible structures for these S = 1/2 species derived from DFT calculations will be discussed in the next section.
Table 3.
Experimental and Computed EPR Parameters for Low-Spin {FeNO}7 Species
| 14N A-values (MHz) a | |||||||
|---|---|---|---|---|---|---|---|
| Species | Method | gx | gy | gz | Ax | Ay | Az |
| 1NO-LS | exper | 2.009 | 2.037 | 2.072 | 46 | 41 | 44 |
| 2NO-LS | exper | 2.007 | 2.035 | 2.070 | 45 | 37 | 36 |
| S=1/2[2-NO]6C | DFT/BP86 | 1.985 | 2.014 | 2.042 | 43 | 68 | 39 |
| DFT/TPSSh | 1.986 | 2.004 | 2.022 | 31 | 81 | 36 | |
| S=1/2[2-NO]5C | DFT/BP86 | 2.005 | 2.021 | 2.047 | 50 | 46 | 41 |
| DFT/TPSSh | 2.007 | 2.031 | 2.054 | 53 | 34 | 28 | |
| [Fe(NO)(N3PyS)]+ | exper b | 1.962 | 2.007 | 2.047 | 40 | 59 | 40 |
| 6C complex | DFT/BP86 | 1.976 | 2.006 | 2.025 | 29 | 62 | 32 |
| DFT/TPSSh | 1.975 | 2.001 | 2.019 | 19 | 82 | 23 | |
| Fe(OEP)(NO) | exper c | 2.015 | 2.057 | 2.106 | 41 | 50 | 43 |
| Fe(II)CDO-Cys/NO | exper d | 1.979 | 2.028 | 2.071 | <30 | 100 | <30 |
The reaction of 1 (or 2) with NO at room temperature (RT) initially generates a chromophore with absorption features matching those observed at −70 °C. However, 1NO and 2NO are not stable at elevated temperatures, quickly converting to the same new species (RTNO) characterized by a weak absorption band at 910 nm (Figure 8). Time-dependent absorption spectra for this reaction display an isosbestic point at 820 nm, indicating clean conversion of 1NO or 2NO to RTNO without the build-up of intermediates (Figure S12). EPR spectra collected for frozen solutions of RTNO exhibit a sharp, rhombic S = 1/2 signal that lacks hyperfine splitting (Figure 8; inset), along with a very weak peak at g = 4.0 arising from residual 1NO or 2NO. Unfortunately, despite repeated attempts, we were unable to grow crystals of RTNO suitable for X-ray crystallography.
Figure 8.

Electronic absorption (top, black) and MCD (bottom, red) spectra of RTNO prepared via reaction of NO with complex 1 at ambient temperature. The absorption spectrum was measured in CH2Cl2. The MCD spectrum was obtained for a frozen, glassy solution in 1:1 (v/v) CH2Cl2:butyronitrile at a temperature of 4 K and a magnetic field of 7 T. Inset: X-band EPR spectrum (blue line) of RTNO in frozen CH2Cl2. Parameters: frequency = 9.497 GHz; power = 0.5 mW; modulation amplitude = 1.0 G; T = 77 K.
Notably, the absorption and EPR features of RTNO are quite similar to those reported for cationic, dinitrosyl iron complexes (DNICs) with the general formula [Fe(NO)2(LN2)]+ (where LN2 is a neutral N,N-donor).91–94 Like RTNO, these S = 1/2 {Fe(NO)2}9 species commonly exhibit a rhombic g-tensor centered at 2.03 and a broad absorption band in the near-IR region with an extinction coefficient of ~100 M−1 cm−1. To determine whether RTNO corresponds to a DNIC, IR spectra of NO-treated 1 and 2 were measured at RT. The resulting spectra display two intense peaks at 1745 and 1817 cm−1, both of which downshift by ~34 cm−1 upon 15NO substitution (Figure S13). These frequencies and isotope shifts are consistent with prior studies of [Fe(NO)2(LN2)]+ complexes, which revealed that the out-of-phase and in-phase combinations of ν(NO) modes occur within energy ranges of 1720–1770 and 1790–1840 cm−1, respectively. Furthermore, the separation between NO stretching frequencies (ΔνNO) is diagnostic of coordination number and nuclearity,95 and the ΔνNO-value of 72 cm−1 measured for RTNO is typical of four-coordinate (4C) {Fe(NO)2}9 species.91–94
The formation of {Fe(NO)2}9 DNICs from Fe(II)-thiolate precursors requires formation of 0.5 equivalent of disulfide to provide the additional electron. Analysis with GC-MS found that L-cystine ethyl ester was the only CysOEt-derived product generated during the 1 + NO reaction at RT. Thus, our collective experimental results suggest that DNIC formation occurs via the reaction:
where R = -CH2CH(NH2)CO2Et
This conclusion is supported by the fact that the spectroscopic parameters of RTNO are identical regardless of whether 1 or 2 serves as the precursor, indicating that the thiolate ligand is no longer attached to the iron-containing product.
To further probe the electronic properties of RTNO, we collected MCD spectra for frozen solutions in 1:1 (v/v) CH2Cl2:butyronitrile. Whereas the absorption spectrum of RTNO exhibits few well-defined features, the MCD spectrum measured at 4 K and 7 T shows four distinct bands between 10,000 and 25,000 cm−1 (Figure 8). Variable-temperature variable-field (VTVH) MCD data collected at 907 nm (Figure S14) confirm that the MCD bands arise from a S = 1/2 species, consistent with the EPR results. To the best of our knowledge, these data represent the first MCD analysis of a DNIC. The origins of the observed absorption/MCD features were elucidated with the help of TD-DFT calculations, as discussed in the following section.
3.G. Nitric Oxide Reactivity: Computational Results
DFT calculations for Fe/NO species derived from complexes 1 and 2 utilized either the nonhybrid BP86 or hybrid meta-GGA TPSSh functionals, which are known to provide accurate geometries and spectroscopic parameters for both mono- and dinitrosyl iron complexes.94 Geometry optimizations of the high-spin {FeNO}7 adduct obtained upon NO binding to 2 yielded quasi-6C structures, labeled S=3/2[2-NO]6C, with an elongated distance of 2.5 Å for the Fe–NTIP bond trans to the nitrosyl (Table S4) – indicative of the strong trans influence of NO ligands.96 Interestingly, during the BP86 geometry optimizations of the low-spin {FeNO}7 adduct, the trans imidazole fully dissociated and formed an intramolecular H-bond with the amine group of CysAm, yielding the 5C structure S=1/2[2-NO]5C. In order to generate a low-spin 6C model (i.e., S=1/2[2-NO]6C), it was necessary to constrain the trans Fe–NTIP bond distance to 2.3 Å. The structures and relative energies of the three BP86-optimized models are provided in Figure 9. The S=1/2[2-NO]5C structure is more stable than both S=3/2[2-NO]6C and S=1/2[2-NO]6C by several kcal/mol. When the TPSSh functional is employed instead of BP86, a low-spin {FeNO}7 species with a quasi-6C structure (trans Fe-NTIP distance of 2.41 Å) was found to exist in a local minimum. While the three TPSSh-generated structures are fairly close in energy, the S=1/2[2-NO]5C structure remains the most stable (Figure 9).
Figure 9.

DFT optimized structures and relative energies (in kcal/mol) of the S=3/2[2-NO]6C, S=1/2[2-NO]6C, and S=1/2[2-NO]5C models computed using the BP86 and TPSSh functionals. The structures were derived from the BP86 calculations.
To determine whether the S=1/2[2-NO]5C structures corresponds to the mononitrosyl S = 1/2 species observed via EPR spectroscopy (Figure 7), we calculated g-values and 14N A-tensors using both functionals. We also performed parallel calculations for [Fe(NO)(N3PyS)]+ – a structurally characterized 6C, low-spin {FeNO}7 complex prepared by Goldberg and coworkers.34 As shown in Table 3, our DFT methodology accurately reproduces the EPR features of this complex, namely, its rhombic g-tensor centered near ge = 2.00 and large Ay-value. Importantly, the calculations are consistent with prior studies of low-spin {FeNO}7 complexes, both heme and nonheme, which have revealed that certain spin-Hamiltonian parameters are diagnostic of coordination number.97–100 Specifically, the gx-values of 6C species are always less than the free-electron value (ge = 2.002), whereas gx > ge for 5C {FeNO}7 species. Additionally, the A-tensors of 6C complexes exhibit relatively large anisotropy and a dominant Ay-value, whereas 5C complexes possess relatively isotropic 14N A-tensors in which the Ax-value is the largest component (Table 3). The EPR parameters of 1NO-LS and 2NO-LS closely align with those computed for S=1/2[2-NO]5C, as well as those observed for 5C NO adducts of ferrous-porphyrin complexes.97–100 Thus, we conclude that the observed low-spin signal is due to dissociation of the trans imidazole donor in a fraction of the {FeNO}7 adducts prepared at low temperature.
Figure 7.
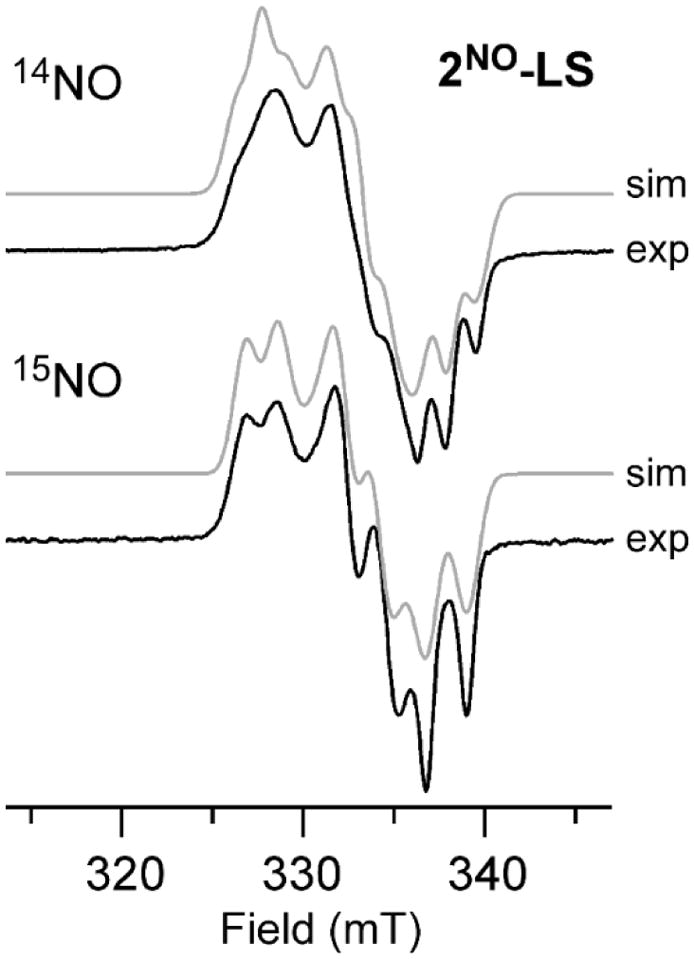
EPR spectra of 2NO-LS (black solid line) generated via reaction of 1 with 14NO (top) or 15NO (bottom) at −70 °C in CH2Cl2. Parameters: frequency = 9.492 GHz; power = 2.0 mW; modulation amplitude = 1.0 G; T = 77 K. Simulations of the experimental data (gray lines) were generated using the spin-Hamiltonian parameters listed in Table 3.
We also generated computational models with the formula [Fe(NO)2(MeTIP)]+ to further confirm that RTNO corresponds to a cationic DNIC. These calculations employed the broken-symmetry approach to generate the proper S = 1/2 wavefunction, best described as a high-spin Fe(III) center coupled antiferromagnetically to two NO− (S = 1) ligands.101 Regardless of the functional used, geometry optimizations of the DNICDFT models converged to four-coordinate (4C) structures with a κ2-TIP ligand, further demonstrating the hemilability of the TIP framework. The computed spectroscopic properties of the DNICDFT models are summarized in Table 4. DFT is known to overestimate ν(NO) frequencies, with hybrid functionals generally performing worse than nonhybrid functionals.94,101 While our results follow this pattern, the BP86 functional proved quite effective in estimating the energy splitting between the in-phase and out-of-phase combinations of ν(NO) modes of RTNO. Both functionals yielded rhombic g-tensors centered near g = 2.03, consistent with our EPR data and prior studies of DNICs.91–94 The computed A-tensors are highly anisotropic with two small A-values (< 10 MHz) and one moderate value of 25 or 39 MHz, in accordance with the absence of any detectable 14N hyperfine splitting in the experimental RTNO EPR spectrum.
Table 4.
Comparison of Computed Spectroscopic Parameters for DNICDFT Models with Experimental Values Measured for RTNO.
| DNICDFT (BP86) | DNICDFT (TPSSh) | RTNO (exper) | |
|---|---|---|---|
|
| |||
| ν(NO)sym, ν(NO)asym | 1843, 1776 cm−1 | 1897, 1816 cm−1 | 1817, 1745 cm−1 |
| gx, gy, gz | 2.013, 2.028, 2.058 | 2.027, 2.033, 2.061 | 2.01, 2.04, 2.07 |
| Ax, Ay, Az (14N7) | 3, 22, 3 MHz | 7, 36, 13 MHz | not observed |
| Ax, Ay, Az (14N8) | 4, 5, 25 MHz | 1, 8, 39 MHz | |
Consistent with the experimental absorption spectrum of RTNO, TD-DFT calculations of DNICDFT predict a series of weak bands (ε-values < 400 M−1cm−1) in the visible region (Figure S15). The donor MOs for these transitions contain approximately equal amounts of Fe 3d and NO π* character due to the highly-covalent nature of the iron-nitrosyl bonds. In contrast, the acceptor orbital is the Fe(dx2-y2)-based MO that lies in the NTIP-Fe-NTIP plane and lacks contributions from the NO ligands (Figure S15). Thus, the transitions have both ligand-field and NO−→Fe(III) CT character, but their intensities are limited by the poor spatial overlap between donor and acceptor MOs.
4. Conclusions
The syntheses, X-ray crystallographic structures, electronic properties, and O2/NO reactivities of two novel TDO active-site models (1 and 2) have been reported. These 5C Fe(II) complexes consist of a bidentate “substrate” ligand (CysOEt or CysAm) and the facially-coordinating Ph2TIP supporting ligand (Scheme 2). In our estimation, 1 and 2 are the most structurally accurate models of the CDO and ADO active sites prepared to date, as the Ph2TIP supporting ligand reproduces the neutral charge and all-imidazole coordination of the 3His triad. The electronic structures of 1 and 2, along with those of the previously-reported Ph,MeTp-based TDO models (3 and 4), were elucidated with spectroscopic (absorption, MCD) and computational (DFT) techniques. These studies yielded specific assignments for the three overlapping S→Fe(II) CT transitions in the near-UV region, and MCD analysis permitted observation of the highest-energy d-d transition at ~11,300 cm−1. In addition, they revealed that substitution of neutral Ph2TIP with anionic Ph,MeTp− destabilizes the Fe 3d manifold by ~0.13 eV and significantly reduces the covalency of the Fe–S bond.
Like similar complexes generated by the Limberg and Goldberg groups, 1 and 2 are functional TDO mimics that react with O2 to yield the corresponding sulfinic acid product. Our kinetic studies determined that the identity of the LN3 ligand (Ph2TIP versus Ph,MeTp−) has little effect on O2 reaction rates. DFT methods were then employed to evaluate the energetics of formation of the Fe/O2 adduct and its conversion to the cyclic Fe-O-O-S intermediate; the latter reaction is the putative rate-determining step in the dioxygenation mechanism. These calculations indicated that O2 binding to the high-spin Fe(II) centers of 2 and 4 is endergonic by 9–13 kcal/mol due to an unfavorable entropic contribution. The transition state leading to the putative Fe-O-O-S intermediate along the S = 2 surface is further uphill by ~12 kcal/mol for 2 and 4, resulting in an overall barrier of ~24 kcal/mol relative to the starting complexes and O2 (Scheme 3). These computed values are roughly consistent with the experimental activation parameters of ΔH‡ = 12(1) kcal/mol and ΔS‡ = −25(4) cal/(mol•K) measured experimentally for the reaction of 1 with O2. In both the Fe/O2 adducts and transition-state structures, the charge of the supporting ligand modulates the amount of unpaired spin density on the S atom of the thiolate ligand, with the TIP-based models exhibiting greater S-radical character than the Tp-based models. This correlation offers a possible rationale for the preference of TDOs for the 3His triad instead of the 2H1C triad, since the development of (partial) thiyl radical character is thought to facilitate formation of the critical Fe-O-O-S species in the enzymatic mechanism.102
Since our efforts to trap intermediates in the dioxygenation reactions were unsuccessful, we explored the reactivity of 1 and 2 with NO – an established proxy for O2. As summarized in Scheme 4, initial NO binding generates a 6C, high-spin {FeNO}7 adduct (1NO and 2NO) that is metastable at reduced temperatures, but converts at RT to a more stable 4C DNIC (RTNO) with concomitant formation of disulfide. Samples prepared at low-temperatures also revealed a minor S = 1/2 signal (1NO-LS and 2NO-LS) that was shown, through a combination of spectroscopic and computational analysis, to correspond to a 5C species featuring a κ2-TIP ligand. The dissociation of the trans imidazole donor may be required for the conversion of the 6C {FeNO}7 adducts into the 4C {Fe(NO)2}9 species.
Scheme 4.

Interestingly, even though the first-sphere coordination geometries of 1 and 2 are nearly identical to those of CDO and ADO, there are dramatic differences in electronic structure, spectroscopic properties, and reactivity between the synthetic models and enzyme active sites. For example, the S→Fe(II) CT transitions observed for CDO are much higher in energy than those observed for complexes 1–4. In addition, whereas 1NO and 2NO are high-spin and decompose at RT to form a DNIC, the 6C {FeNO}7 adduct of CDO is low-spin and relatively stable.88 These differences highlight the importance of the protein environment in fine-tuning the geometric and electronic structures of the nonheme iron active-site. The rotational freedom of the imidazolyl and pyrazolyl donors allows the Ph2TIP and Ph,MeTp ligands to adopt κ2-N,N binding modes, thereby encouraging the formation of 5C and 4C Fe/NO adducts. In contrast, QM/MM studies have determined that the binding of NO to substrate-bound CDO causes little change in Fe-NHis bond distances,102 suggesting that the positions of the His ligands are fixed within the active-site. Thus, despite the considerable advantages of scorpionate ligands, our future TDO modeling studies will employ more constrained ligands that prevent elongation or breaking of the Fe-N bond trans to the O2/NO binding site. Moreover, the incorporation of second-sphere donors capable of hydrogen-bonding interactions would serve to modulate spectroscopic features and stabilize reactive intermediates in a manner similar to the protein environment.
Supplementary Material
Acknowledgments
A.T.F thanks the U.S. National Science Foundation (CHE-1056845) for supporting this research. The laboratories of Profs. Judith Burstyn and Shannon Stahl are thanked for their assistance. T.C.B. acknowledges support by the National Institutes of Health under award number R01GM117120.
Footnotes
1H NMR spectra of complexes 1–2 and products of the O2 reactions, metric parameters for DFT-computed structures, absorption and MCD spectra of 1 and 3, results from TD-DFT calculations, kinetic data and Eyring plot for reaction of 1 with O2, EPR spectra of 1NO-LS, UV-vis absorption and IR spectra of RTNO, VTVH-MCD data collected for RTNO, and crystallographic data in CIF format. This material is available free of charge at http://pubs.acs.org.
References
- 1.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Geometric and electronic structure/function correlations in non-heme iron enzymes. Chem Rev. 2000;100:235–349. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 2.Costas M, Mehn MP, Jensen MP, Que L. Dioxygen activation at mononuclear nonheme iron active sites: Enzymes, models, and intermediates. Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 3.Vaillancourt FH, Bolin JT, Eltis LD. The ins and outs of ring-cleaving dioxygenases. Crit Rev Biochem Mol Biol. 2006;41:241–267. doi: 10.1080/10409230600817422. [DOI] [PubMed] [Google Scholar]
- 4.Parales RE, Haddock JD. Biocatalytic degradation of pollutants. Curr Opin Biotechnol. 2004;15:374–379. doi: 10.1016/j.copbio.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 5.Lipscomb JD. Mechanism of extradiol aromatic ring-cleaving dioxygenases. Curr Opin Struct Biol. 2008;18:644–649. doi: 10.1016/j.sbi.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stipanuk MH, Simmons CR, Andrew Karplus P, Dominy JE., Jr Thiol dioxygenases: unique families of cupin proteins. Amino Acids. 2011;41:91–102. doi: 10.1007/s00726-010-0518-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joseph CA, Maroney MJ. Cysteine dioxygenase: structure and mechanism. Chem Commun. 2007:3338–3349. doi: 10.1039/b702158e. [DOI] [PubMed] [Google Scholar]
- 8.Yamaguchi K, Hosokawa Y. Cysteine dioxygenase. Methods Enzymol. 1987;143:395–403. doi: 10.1016/0076-6879(87)43069-3. [DOI] [PubMed] [Google Scholar]
- 9.Stipanuk MH, Ueki I. Dealing with methionine/homocysteine sulfur: cysteine metabolism to taurine and inorganic sulfur. J Inherited Metab Dis. 2011;34:17–32. doi: 10.1007/s10545-009-9006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ueki I, Roman HB, Valli A, Fieselmann K, Lam J, Peters R, Hirschberger LL, Stipanuk MH. Knockout of the murine cysteine dioxygenase gene results in severe impairment in ability to synthesize taurine and an increased catabolism of cysteine to hydrogen sulfide. Am J Physiol. 2011;301:E668–E684. doi: 10.1152/ajpendo.00151.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Slivka A, Cohen G. Brain ischemia markedly elevates levels of the neurotoxic amino acid, cysteine. Brain Research. 1993;608:33–7. doi: 10.1016/0006-8993(93)90770-n. [DOI] [PubMed] [Google Scholar]
- 12.Pean AR, Parsons RB, Waring RH, Williams AC, Ramsden DB. Toxicity of sulfur-containing compounds to neuronal cell lines. J Neurol Sci. 1995;129:107–8. doi: 10.1016/0022-510x(95)00078-g. [DOI] [PubMed] [Google Scholar]
- 13.Heafield MT, Fearn S, Steventon GB, Waring RH, Williams AC, Sturman SG. Plasma cysteine and sulphate levels in patients with motor neurone, Parkinson’s and Alzheimer’s disease. Neurosci Lett. 1990;110:216–20. doi: 10.1016/0304-3940(90)90814-p. [DOI] [PubMed] [Google Scholar]
- 14.Perry TL, Norman MG, Yong VW, Whiting S, Crichton JU, Hansen S, Kish SJ. Hallervorden-Spatz disease: cysteine accumulation and cysteine dioxygenase deficiency in the globus pallidus. Annals of Neurology. 1985;18:482–9. doi: 10.1002/ana.410180411. [DOI] [PubMed] [Google Scholar]
- 15.Bradley H, Gough A, Sokhi RS, Hassell A, Waring R, Emery P. Sulfate metabolism is abnormal in patients with rheumatoid arthritis. Confirmation by in vivo biochemical findings. J Rheumatol. 1994;21:1192–6. [PubMed] [Google Scholar]
- 16.Emery P, Bradley H, Gough A, Arthur V, Jubb R, Waring R. Increased prevalence of poor sulphoxidation in patients with rheumatoid arthritis: effect of changes in the acute phase response and second line drug treatment. Ann Rheum Dis. 1992;51:318–20. doi: 10.1136/ard.51.3.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dominy JE, Jr, Simmons CR, Hirschberger LL, Hwang J, Coloso RM, Stipanuk MH. Discovery and Characterization of a Second Mammalian Thiol Dioxygenase, Cysteamine Dioxygenase. J Biol Chem. 2007;282:25189–25198. doi: 10.1074/jbc.M703089200. [DOI] [PubMed] [Google Scholar]
- 18.Bruijnincx PCA, van Koten G, Gebbink RJMK. Mononuclear non-heme iron enzymes with the 2-His-1-carboxylate facial triad: recent developments in enzymology and modeling studies. Chem Soc Rev. 2008;37:2716–2744. doi: 10.1039/b707179p. [DOI] [PubMed] [Google Scholar]
- 19.Koehntop KD, Emerson JP, Que L. The 2-His-1-carboxylate facial triad: a versatile platform for dioxygen activation by mononuclear non-heme iron(II) enzymes. J Biol Inorg Chem. 2005;10:87–93. doi: 10.1007/s00775-005-0624-x. [DOI] [PubMed] [Google Scholar]
- 20.McCoy JG, Bailey LJ, Bitto E, Bingman CA, Aceti DJ, Fox BG, Phillips GN., Jr Structure and mechanism of mouse cysteine dioxygenase. Proc Natl Acad Sci USA. 2006;103:3084–3089. doi: 10.1073/pnas.0509262103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simmons CR, Liu Q, Huang QQ, Hao Q, Begley TP, Karplus PA, Stipanuk MH. Crystal structure of mammalian cysteine dioxygenase - A novel mononuclear iron center for cysteine thiol oxidation. J Biol Chem. 2006;281:18723–18733. doi: 10.1074/jbc.M601555200. [DOI] [PubMed] [Google Scholar]
- 22.Ye S, Wu Xa, Wei L, Tang D, Sun P, Bartlam M, Rao Z. An Insight into the Mechanism of Human Cysteine Dioxygenase. Key Roles of the Thioether-Bonded Tyrosine-Cysteine Cofactor. J Biol Chem. 2007;282:3391–3402. doi: 10.1074/jbc.M609337200. [DOI] [PubMed] [Google Scholar]
- 23.Diebold AR, Neidig ML, Moran GR, Straganz GD, Solomon EI. The Three-His Triad in Dke1: Comparisons to the Classical Facial Triad. Biochemistry. 2010;49:6945–6952. doi: 10.1021/bi100892w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leitgeb S, Straganz GD, Nidetzky B. Biochemical characterization and mutational analysis of the mononuclear non-haem Fe2+ site in Dke1, a cupin-type dioxygenase from Acinetobacter johnsonii. Biochem J. 2009;418:403–411. doi: 10.1042/BJ20081161. [DOI] [PubMed] [Google Scholar]
- 25.Leitgeb S, Nidetzky B. Structural and functional comparison of 2-His-1-carboxylate and 3-His metallocentres in non-haem iron(II)-dependent enzymes. Biochem Soc Trans. 2008;36:1180–1186. doi: 10.1042/BST0361180. [DOI] [PubMed] [Google Scholar]
- 26.Chen J, Li W, Wang M, Zhu G, Liu D, Sun F, Hao N, Li X, Rao Z, Zhang XC. Crystal structure and mutagenic analysis of GDOsp, a gentisate 1,2-dioxygenase from Silicibacter Pomeroyi. Protein Sci. 2008;17:1362–1373. doi: 10.1110/ps.035881.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matera I, Ferraroni M, Buerger S, Scozzafava A, Stolz A, Briganti F. Salicylate 1,2-dioxygenase from Pseudaminobacter salicylatoxidans: Crystal structure of a peculiar ring-cleaving dioxygenase. J Mol Biol. 2008;380:856–868. doi: 10.1016/j.jmb.2008.05.041. [DOI] [PubMed] [Google Scholar]
- 28.Buongiorno D, Straganz GD. Structure and function of atypically coordinated enzymatic mononuclear non-heme-Fe(II) centers. Coord Chem Rev. 2013;257:541–563. doi: 10.1016/j.ccr.2012.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Badiei YM, Siegler MA, Goldberg DP. O2 Activation by Bis(imino)pyridine Iron(II)-Thiolate Complexes. J Am Chem Soc. 2011;133:1274–1277. doi: 10.1021/ja109923a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang Y, Widger LR, Kasper GD, Siegler MA, Goldberg DP. Iron(II)-Thiolate S-Oxygenation by O2: Synthetic Models of Cysteine Dioxygenase. J Am Chem Soc. 2010;132:12214–12215. doi: 10.1021/ja105591q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McQuilken AC, Goldberg DP. Sulfur oxygenation in biomimetic non-heme iron-thiolate complexes. Dalton Trans. 2012;41:10883–10899. doi: 10.1039/c2dt30806a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Widger LR, Jiang Y, Siegler MA, Kumar D, Latifi R, de Visser SP, Jameson GNL, Goldberg DP. Synthesis and Ligand Non-Innocence of Thiolate-Ligated (N4S) Iron(II) and Nickel(II) Bis(imino)pyridine Complexes. Inorg Chem. 2013;52:10467–10480. doi: 10.1021/ic4013558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McQuilken AC, Jiang Y, Siegler MA, Goldberg DP. Addition of Dioxygen to an N4S(thiolate) Iron(II) Cysteine Dioxygenase Model Gives a Structurally Characterized Sulfinato-Iron(II) Complex. J Am Chem Soc. 2012;134:8758–8761. doi: 10.1021/ja302112y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McQuilken AC, Ha Y, Sutherlin KD, Siegler MA, Hodgson KO, Hedman B, Solomon EI, Jameson GNL, Goldberg DP. Preparation of Non-heme {FeNO}7 Models of Cysteine Dioxygenase: Sulfur versus Nitrogen Ligation and Photorelease of Nitric Oxide. J Am Chem Soc. 2013;135:14024–14027. doi: 10.1021/ja4064487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McQuilken AC, Matsumura H, Durr M, Confer AM, Sheckelton JP, Siegler MA, McQueen TM, Ivanovic-Burmazovic I, Moenne-Loccoz P, Goldberg DP. Photoinitiated Reactivity of a Thiolate-Ligated, Spin-Crossover Nonheme {FeNO}7 Complex with Dioxygen. J Am Chem Soc. 2016;138:3107–3117. doi: 10.1021/jacs.5b12741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sallmann M, Siewert I, Fohlmeister L, Limberg C, Knispel C. A Trispyrazolylborato Iron Cysteinato Complex as a Functional Model for the Cysteine Dioxygenase. Angew Chem Int Ed. 2012;51:2234–2237. doi: 10.1002/anie.201107345. [DOI] [PubMed] [Google Scholar]
- 37.Sallmann M, Braun B, Limberg C. Dioxygenation of cysteamine to hypotaurine at a tris(pyrazolyl)borate iron(II) unit - cysteamine dioxygenase mimicking? Chem Commun. 2015;51:6785–6787. doi: 10.1039/c5cc01083g. [DOI] [PubMed] [Google Scholar]
- 38.Sallmann M, Limberg C. Utilizing the trispyrazolyl borate ligand for the mimicking of O2-activating mononuclear nonheme iron enzymes. Acc Chem Res. 2015;48:2734–2743. doi: 10.1021/acs.accounts.5b00148. [DOI] [PubMed] [Google Scholar]
- 39.Sallmann M, Kumar S, Chernev P, Nehrkorn J, Schnegg A, Kumar D, Dau H, Limberg C, de Visser SP. Structure and mechanism leading to formation of the cysteine sulfinate product complex of a biomimetic cysteine dioxygenase model. Chemistry - A European Journal. 2015;21:7470–7479. doi: 10.1002/chem.201500644. [DOI] [PubMed] [Google Scholar]
- 40.Kumar D, Sastry GN, Goldberg DP, de Visser SP. Mechanism of S-Oxygenation by a Cysteine Dioxygenase Model Complex. J Phys Chem A. 2012;116:582–591. doi: 10.1021/jp208230g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gonzalez-Ovalle LE, Quesne MG, Kumar D, Goldberg DP, de Visser SP. Axial and equatorial ligand effects on biomimetic cysteine dioxygenase model complexes. Org Biomol Chem. 2012;10:5401–5409. doi: 10.1039/c2ob25406a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alberto ME. A trispyrazolylborato iron cysteinato complex efficiently mimics the cysteine dioxygenation process: mechanistic insights. Chem Commun. 2015;51:8369–8372. doi: 10.1039/c5cc00813a. [DOI] [PubMed] [Google Scholar]
- 43.Park H, Baus JS, Lindeman SV, Fiedler AT. Synthesis and Characterization of Fe(II) beta-Diketonato Complexes with Relevance to Acetylacetone Dioxygenase: Insights into the Electronic Properties of the 3-Histidine Facial Triad. Inorg Chem. 2011;50:11978–11989. doi: 10.1021/ic201115s. [DOI] [PubMed] [Google Scholar]
- 44.Bittner MM, Baus JS, Lindeman SV, Fiedler AT. Synthesis and Structural Characterization of Iron(II) Complexes with Tris(imidazolyl)phosphane Ligands: A Platform for Modeling the 3-Histidine Facial Triad of Nonheme Iron Dioxygenases. Eur J Inorg Chem. 2012:1848–1856. [Google Scholar]
- 45.Park H, Bittner MM, Baus JS, Lindeman SV, Fiedler AT. Fe(II) Complexes That Mimic the Active Site Structure of Acetylacetone Dioxygenase: O-2 and NO Reactivity. Inorg Chem. 2012;51:10279–10289. doi: 10.1021/ic3012712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baum AE, Lindeman SV, Fiedler AT. Mononuclear Iron-(hydro/semi)-quinonate Complexes Featuring Neutral and Charged Scorpionates: Synthetic Models of Intermediates in the Hydroquinone Dioxygenase Mechanism. Eur J Inorg Chem. 2016;2016:2455–2464. [Google Scholar]
- 47.Smith AT, Majtan T, Freeman KM, Su Y, Kraus JP, Burstyn JN. Cobalt Cystathionine β-Synthase: A Cobalt-Substituted Heme Protein with a Unique Thiolate Ligation Motif. Inorg Chem. 2011;50:4417–4427. doi: 10.1021/ic102586b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tietz T, Limberg C, Stoesser R, Ziemer B. Four-Coordinate Trispyrazolyl-boratomanganese and -iron Complexes with a Pyrazolato Co-ligand: Syntheses and Properties as Oxidation Catalysts. Chem - Eur J. 2011;17:10010–10020. doi: 10.1002/chem.201100343. [DOI] [PubMed] [Google Scholar]
- 49.Stoll S, Schweiger A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J Magn Reson. 2006;178:42–55. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 50.Sheldrick GM. A short history of SHELX. Acta Crystallogr Section A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 51.Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J Appl Crystallogr. 2009;42:339–341. [Google Scholar]
- 52.Kryatov SV, Rybak-Akimova EV, Schindler S. Kinetics and Mechanisms of Formation and Reactivity of Non-heme Iron Oxygen Intermediates. Chem Rev. 2005;105:2175–2226. doi: 10.1021/cr030709z. [DOI] [PubMed] [Google Scholar]
- 53.Battino R, Cleve HL. Solubility of gases in liquids. Chem Rev. 1966;66:395–463. [Google Scholar]
- 54.Neese F. ORCA - An Ab Initio, DFT and Semiempirical Electronic Structure Package, version 3.0. Max Planck Institute for Chemical Energy Conversion; Muelheim (Germany): 2013. [Google Scholar]
- 55.Schafer A, Huber C, Ahlrichs R. FULLY OPTIMIZED CONTRACTED GAUSSIAN-BASIS SETS OF TRIPLE ZETA VALENCE QUALITY FOR ATOMS LI TO KR. J Chem Phys. 1994;100:5829–5835. [Google Scholar]
- 56.Schafer A, Horn H, Ahlrichs R. FULLY OPTIMIZED CONTRACTED GAUSSIAN-BASIS SETS FOR ATOMS LI TO KR. J Chem Phys. 1992;97:2571–2577. [Google Scholar]
- 57.Weigend F, Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys Chem Chem Phys. 2005;7:3297–3305. doi: 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- 58.Klamt A, Schueuermann G. COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J Chem Soc, Perkin Trans 2. 1993:799–805. [Google Scholar]
- 59.Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett. 1996;77:3865–3868. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 60.Becke AD. DENSITY FUNCTIONAL CALCULATIONS OF MOLECULAR-BOND ENERGIES. J Chem Phys. 1986;84:4524–4529. [Google Scholar]
- 61.Perdew JP. DENSITY-FUNCTIONAL APPROXIMATION FOR THE CORRELATION-ENERGY OF THE INHOMOGENEOUS ELECTRON-GAS. Phys Rev B. 1986;33:8822–8824. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- 62.Perdew JP, Tao J, Staroverov VN, Scuseria GE. Meta-generalized gradient approximation: Explanation of a realistic nonempirical density functional. J Chem Phys. 2004;120:6898–6911. doi: 10.1063/1.1665298. [DOI] [PubMed] [Google Scholar]
- 63.Staroverov VN, Scuseria GE, Tao J, Perdew JP. Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes. J Chem Phys. 2003;119:12129–12137. [Google Scholar]
- 64.The “core properties” basis set is derived from the TurboMole DZ basis set developed by Ahlrichs and coworkers. It was obtained from the basis set library under ftp.chemie.uni-karlsruhe.de/pub/basen.
- 65.Kutzelnigg W, Fleischer U, Schindler M. The IGLO Method: Ab Initio Calculation and Interpretation of NMR Chemical Shifts and Magnetic Susceptibilities. Vol. 23 Springer-Verlang; Heidelberg: 1990. [Google Scholar]
- 66.Neese F. Quantum chemical calculations of spectroscopic properties of metalloproteins and model compounds: EPR and Moessbauer properties. Curr Opin Chem Biol. 2003;7:125–135. doi: 10.1016/s1367-5931(02)00006-6. [DOI] [PubMed] [Google Scholar]
- 67.Neese F. Prediction of electron paramagnetic resonance g values using coupled perturbed Hartree-Fock and Kohn-Sham theory. J Chem Phys. 2001;115:11080–11096. [Google Scholar]
- 68.Sinnecker S, Neese F, Noodleman L, Lubitz W. Calculating the electron paramagnetic resonance parameters of exchange coupled transition metal complexes using broken symmetry density functional theory: Application to a Mn(III)/Mn(IV) model compound. J Am Chem Soc. 2004;126:2613–2622. doi: 10.1021/ja0390202. [DOI] [PubMed] [Google Scholar]
- 69.Neese F. Metal and ligand hyperfine couplings in transition metal complexes: the effect of spin-orbit coupling as studied by coupled perturbed Kohn-Sham theory. J Chem Phys. 2003;118:3939–3948. [Google Scholar]
- 70.Yanai T, Tew DP, Handy NC. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP) Chem Phys Lett. 2004;393:51–57. [Google Scholar]
- 71.Blaesi EJ, Fox BG, Brunold TC. Spectroscopic and computational Investigation of the H155A variant of cysteine dioxygenase: Geometric and electronic consequences of a third-sphere amino acid substitution. Biochemistry. 2015;54:2874–2884. doi: 10.1021/acs.biochem.5b00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hirata S, Head-Gordon M. Time-dependent density functional theory within the Tamm-Dancoff approximation. Chem Phys Lett. 1999;314:291–299. [Google Scholar]
- 73.Hirata S, Head-Gordon M. Time-dependent density functional theory for radicals - An improved description of excited states with substantial double excitation character. Chem Phys Lett. 1999;302:375–382. [Google Scholar]
- 74.The geometric parameter τ is defined as τ = |(α – β)|/60, where α and β are the two basal angles in psuedo-square pyramidal geometry. The τ-value is 0.0 in idealized square-planar geometries and 1.0 in idealized but trigonal bipyramidal geometries. For more details, see: Addison AW, Rao TN, Reedijk J, Vanrijn J, Verschoor GC. J Chem Soc, Dalton Trans. 1984:1349–1356.
- 75.Driggers CM, Cooley RB, Sankaran B, Hirschberger LL, Stipanuk MH, Karplus PA. Cysteine Dioxygenase Structures from pH 4 to 9: Consistent Cys-Persulfenate Formation at Intermediate pH and a Cys-Bound Enzyme at Higher pH. J Mol Biol. 2013;425:3121–3136. doi: 10.1016/j.jmb.2013.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zheng H, Chruszcz M, Lasota P, Lebioda L, Minor W. Data mining of metal ion environments present in protein structures. J Inorg Biochem. 2008;102:1765–1776. doi: 10.1016/j.jinorgbio.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.The C0/D0 ratio was calculated using the following expression: C0/D0 = (kT/βH)[ΔεMCD/εabs]. The CT bands of complex 2 exhibit C0/D0 ratios between 0.05 and 0.10
- 78.Pavel EG, Kitajima N, Solomon EI. Magnetic circular dichroism spectroscopic studies of mononuclear non-heme ferrous model complexes. Correlation of excited- and ground-state electronic structure with geometry. J Am Chem Soc. 1998;120:3949–3962. [Google Scholar]
- 79.Gardner JD, Pierce BS, Fox BG, Brunold TC. Spectroscopic and Computational Characterization of Substrate-Bound Mouse Cysteine Dioxygenase: Nature of the Ferrous and Ferric Cysteine Adducts and Mechanistic Implications. Biochemistry. 2010;49:6033–6041. doi: 10.1021/bi100189h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.The concentration of O2 in CH2Cl2 at 20 °C is 5.8 mM. Therefore, the second-order rate constants (k2) for the O2 reaction are 3.3(7) × 10−2 M−1s−1 for 1 and 2.1(5) × 10−2 M−1s−1 for 3.
- 81.Bittner MM, Lindeman SV, Popescu CV, Fiedler AT. Dioxygen Reactivity of Biomimetic Fe(II) Complexes with Noninnocent Catecholate, o-Aminophenolate, and o-Phenylenediamine Ligands. Inorg Chem. 2014;53:4047–4061. doi: 10.1021/ic403126p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mehn MP, Fujisawa K, Hegg EL, Que L. Oxygen activation by nonheme iron(II) complexes: alpha-keto carboxylate versus carboxylate. J Am Chem Soc. 2003;125:7828–7842. doi: 10.1021/ja028867f. [DOI] [PubMed] [Google Scholar]
- 83.Kumar D, Thiel W, de Visser SP. Theoretical Study on the Mechanism of the Oxygen Activation Process in Cysteine Dioxygenase Enzymes. J Am Chem Soc. 2011;133:3869–3882. doi: 10.1021/ja107514f. [DOI] [PubMed] [Google Scholar]
- 84.For the sake of computational efficiency, DFT studies of the O2 reaction were performed using truncated models of 2 and 4 that lack the ethyl ester moiety of 1 and 3. Calculations the Fe(II) precursors indicate that identity of the LS,N ligand (CysOEt or CysAm) has little impact on coordination geometry and electronic structure.
- 85.Schenk G, Pau MYM, Solomon EI. Comparison between the Geometric and Electronic Structures and Reactivities of {FeNO}7 and {FeO2}8 Complexes: A Density Functional Theory Study. J Am Chem Soc. 2004;126:505–515. doi: 10.1021/ja036715u. [DOI] [PubMed] [Google Scholar]
- 86.Ye S, Riplinger C, Hansen A, Krebs C, Bollinger JM, Neese F. Electronic Structure Analysis of the Oxygen-Activation Mechanism by FeII- and α-Ketoglutarate (αKG)-Dependent Dioxygenases. Chem Eur J. 2012;18:6555–6567. doi: 10.1002/chem.201102829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.de Visser SP, Straganz GD. Why Do Cysteine Dioxygenase Enzymes Contain a 3-His Ligand Motif Rather than a 2His/1Asp Motif Like Most Nonheme Dioxygenases? J Phys Chem A. 2009;113:1835–1846. doi: 10.1021/jp809700f. [DOI] [PubMed] [Google Scholar]
- 88.Pierce BS, Gardner JD, Bailey LJ, Brunold TC, Fox BG. Characterization of the nitrosyl adduct of substrate-bound mouse cysteine dioxygenase by electron paramagnetic resonance: Electronic structure of the active site and mechanistic implications. Biochemistry. 2007;46:8569–8578. doi: 10.1021/bi700662d. [DOI] [PubMed] [Google Scholar]
- 89.Berto TC, Speelman AL, Zheng S, Lehnert N. Mono- and dinuclear non-heme iron-nitrosyl complexes: Models for key intermediates in bacterial nitric oxide reductases. Coord Chem Rev. 2013;257:244–259. [Google Scholar]
- 90.At 77 K the S = 3/2 signal is absent due to rapid relaxation
- 91.Tran CT, Skodje KM, Kim E. Monomeric dinitrosyl iron complexes: synthesis and reactivity. Prog Inorg Chem. 2014;59:339–379. [Google Scholar]
- 92.D’Autreaux B, Horner O, Oddou JL, Jeandey C, Gambarelli S, Berthomieu C, Latour JM, Michaud-Soret I. Spectroscopic Description of the Two Nitrosyl-Iron Complexes Responsible for Fur Inhibition by Nitric Oxide. J Am Chem Soc. 2004;126:6005–6016. doi: 10.1021/ja031671a. [DOI] [PubMed] [Google Scholar]
- 93.Tonzetich ZJ, Do LH, Lippard SJ. Dinitrosyl Iron Complexes Relevant to Rieske Cluster Nitrosylation. J Am Chem Soc. 2009;131:7964–7965. doi: 10.1021/ja9030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Speelman AL, Zhang B, Silakov A, Skodje KM, Alp EE, Zhao J, Hu MY, Kim E, Krebs C, Lehnert N. Unusual Synthetic Pathway for an {Fe(NO)2}9 Dinitrosyl Iron Complex (DNIC) and Insight into DNIC Electronic Structure via Nuclear Resonance Vibrational Spectroscopy. Inorg Chem. 2016;55:5485–5501. doi: 10.1021/acs.inorgchem.6b00510. [DOI] [PubMed] [Google Scholar]
- 95.Tsai ML, Tsou CC, Liaw WF. Dinitrosyl Iron Complexes (DNICs): From Biomimetic Synthesis and Spectroscopic Characterization toward Unveiling the Biological and Catalytic Roles of DNICs. Acc Chem Res. 2015;48:1184–1193. doi: 10.1021/ar500459j. [DOI] [PubMed] [Google Scholar]
- 96.Hunt AP, Lehnert N. Heme-Nitrosyls: Electronic Structure Implications for Function in Biology. Acc Chem Res. 2015;48:2117–2125. doi: 10.1021/acs.accounts.5b00167. [DOI] [PubMed] [Google Scholar]
- 97.Hayes RG, Ellison MK, Scheidt WR. Definitive assignment of the g tensor of [Fe(OEP)(NO)] by single-crystal EPR. Inorg Chem. 2000;39:3665–3668. doi: 10.1021/ic000159c. [DOI] [PubMed] [Google Scholar]
- 98.Praneeth VKK, Neese F, Lehnert N. Spin Density Distribution in Five- and Six-Coordinate Iron(II)-Porphyrin NO Complexes Evidenced by Magnetic Circular Dichroism Spectroscopy. Inorg Chem. 2005;44:2570–2572. doi: 10.1021/ic050144k. [DOI] [PubMed] [Google Scholar]
- 99.Praneeth VKK, Naether C, Peters G, Lehnert N. Spectroscopic Properties and Electronic Structure of Five- and Six-Coordinate Iron(II) Porphyrin NO Complexes: Effect of the Axial N-Donor Ligand. Inorg Chem. 2006;45:2795–2811. doi: 10.1021/ic050865j. [DOI] [PubMed] [Google Scholar]
- 100.Lehnert N, Scheidt WR, Wolf MW. Structure and Bonding in Heme-Nitrosyl Complexes and Implications for Biology. Struct Bonding. 2014;154:155–223. [Google Scholar]
- 101.Ye S, Neese F. The Unusual Electronic Structure of Dinitrosyl Iron Complexes. J Am Chem Soc. 2010;132:3646–3647. doi: 10.1021/ja9091616. [DOI] [PubMed] [Google Scholar]
- 102.Blaesi EJ, Gardner JD, Fox BG, Brunold TC. Spectroscopic and computational characterization of the NO adduct of substrate-bound Fe(II) cysteine dioxygenase: Insights into the mechanism of O2 activation. Biochemistry. 2013;52:6040–6051. doi: 10.1021/bi400825c. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.