

Abstract
This study shows that fibroblasts migrating into a collagen matrix release numerous microvesicles into the surrounding medium. By spreading in regions of the matrix far distant from cells of origin, microvesicles carry metalloproteinase 9 (MMP-9) to act upon the collagen fibrils. As a result, the collagen matrix is gradually transformed from a laminar to a fibrillar type of architecture. As shown by western blots and gelatin zymography, MMP-9 is secreted as a 92 kDa precursor and activated upon release of 82 kDa product into the culture medium. Activation is more efficient under three-dimensional than in two-dimensional culturing conditions. While MMP-9 labeling is associated with intraluminal vesicles clustered inside the microvesicles, the microvesicle’s integrin β1 marker is bound to the outer membrane. The intraluminal vesicles are recruited from the cortical cytoplasm and eventually released following uploading inside the microvesicle. Here, we propose that fusion of the intraluminal vesicles with the outer microvesicle’s membrane could work as a mechanism controlling the extent to which MMP-9 is first activated and then released extracellularly.
Keywords: Microvesicles, exosome, ectosome, metalloproteinase, collagen matrix
Introduction
Cell communication plays a key role in multicellular organisms.1 Cells communicate by either ligand–receptor interaction, surface recognition of specific cell adhesion molecules, or transfer of cytoplasmic components through junctional coupling.2 In recent years, it has become apparent that cells may also interact through the release of extracellular vesicles. Originally regarded as cellular debris with no apparent function,3 microvesicles are now recognized as specific structures released by cells under a variety of physiological and pathological conditions.4–8 They may either originate as exosomes upon fusion of the endosomal compartment with the plasma membrane or be directly released from the cell membrane in the form of shedding vesicles.9,10 The role played by microvesicles in cell communication is largely dependent upon the molecular cargo they carry as well as on the mechanism(s) that target them to other cells. While cell destination is defined by the type of ligands or receptor(s) expressed on their limiting membrane, the variety of molecules—enzymes, messenger RNA, microRNA, or RNA interference—carried by microvesicles will ultimately affect the developmental fate of the target cells.11–13 Because of this capacity, microvesicles are involved in many pathologies, including cancer metastasis, besides having a beneficial role in a variety of cell responses under physiological conditions.14–17 Microvesicles are also involved in every step of the wound healing process, from blood clotting to neogenesis of the scar tissue. Interestingly, in each of these circumstances, the proteolytic activity of the extracellular matrix has been shown to be associated with the release of microvesicles. Through this mechanism, various cell types may contribute to generate an appropriate microenvironment for tissue repair.5,18
The primary objective of wound healing is to reestablish the integrity of the injured tissues.19 To this end, a variety of skin cells, including keratinocytes and dermal fibroblasts, is expected to migrate from the wound edges and to invade the underlying connective tissue.20 The mechanism by which migrating cells interact with the extracellular matrix and eventually regulate collagen metabolism during tissue formation has been the subject of extensive investigations in recent years.21–23 It is generally recognized that wound healing entails degradation of the damaged matrix through recruitment of inflammatory cells, followed by deposition of new collagen fibers by migrating fibroblasts.20,24 A proper balance between degradation and synthesis of collagen is thus essential for a new granulation tissue to be formed on the wound bed and to be later transformed into a mature scar.25–27 During this transition a number of specific growth factors are known to stimulate fibroblasts to synthesize and release a number of matrix metalloproteinases (MMPs).28–30 By acting upon the damaged extracellular matrix, these enzymes are known to degrade collagen and to allow nearby cells to take up the resulting peptides.31,32
MMPs constitute a class of collagen-degrading enzymes known to play key roles in many physiological and pathological tissue remodeling processes, including embryogenesis and cancer, besides wound repair.33 Expression of MMPs can be regulated by either gene transcription,34 pro-enzyme activation of the catalytic domain,35 or inhibition through specific tissue matrix proteinases.36 However, metalloproteinase activity may also be controlled through selective anchoring on specific membrane domains37 or by confinement into membrane-enclosed compartments.38 In each case, enzyme activation is delayed until release or detachment from the membrane compartment.
In this study, we show that fibroblasts migrating in vitro into an accessible collagen matrix release a number of microvesicles into the culture medium. These vesicles act as carriers conveying MMP-9 to nearby collagen fibers. The primary objective of this study is thus to verify whether the release of microvesicles can actually account for (i) the diffusion of pro-metalloproteinases through the collagen matrix; (ii) the activation of metalloproteinase upon interaction with collagen fibers and eventually; and (iii) the extracellular processing of prosthetic collagen prior to cell uptake and intracellular digestion by fibroblasts.
Materials and methods
Cell culturing
Stabilized murine fibroblasts (NIH 3T3) were used throughout this study. To collect enough cells for plating on collagen matrices, NIH 3T3 fibroblasts were first cultured in 75 cm2 flasks containing Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum, 1% glutamine, and 1% penicillin/streptomycin, and maintained in a humidified incubator at 37°C supplied with a 5% carbon dioxide (CO2) atmosphere. Upon reaching confluence, cells were passed and the medium changed every 3 days. Three-dimensional (3-D) collagen scaffolds were cut into blocks of approximately 1 cm2 of surface area with a sterile razor blade and each block inserted into 24-well plates. Aliquots of 2 × 105 cells were seeded on the upper surface of every collagen block and incubated at 37°C in 1.5 mL of culture medium under a continuous flow of 5% CO2 for time intervals ranging from 4 h to 14 days. Medium was then renewed every 3 days.
Scanning electron microscopy
Collagen scaffolds inclusive of their cellular loads were cut into a number of small blocks with a sterile razor blade and fixed overnight at 4°C with 3% glutaraldehyde in 0.05 M phosphate buffer (PB) at pH 7.2. Following extensive rinsing with the same buffer at 4°C, blocks were soaked for 1 h in 0.5% tannic acid in PB at 4°C. Then they were rinsed four times in the same buffer for 15 min at 4°C, and postfixed with 1% osmium tetroxide in PB for 1 h at 4°C. Samples were washed in distilled water, dehydrated in a graded series of ethanol, and freeze-dried in a Balzers Union CPD 020 (Balzers, Liechtenstein) using the procedure of critical point drying. Samples were eventually sputter coated with gold in a Balzers MED 010 unit and observed in a JEOL JSM 6010LA electron microscope (Japan).
Transmission electron microscopy
Samples were cut into a number of small blocks with a sterile razor blade and fixed as described above. Following postfixation in 1% osmium tetroxide, samples were washed in distilled water, block stained with 1% uranyl acetate in distilled water, and then dehydrated in a graded series of ethanol. Subsequently, they were infiltrated for 2 days in graded mixtures of LR white resin/ethanol (London Resin Company, Berkshire, England). By the end of this procedure, samples were embedded in fresh LR white resin and cut with a Reichert Ultracut ultramicrotome (Depew, New York, USA) equipped with a diamond knife. Ultrathin sections (60–80 nm thick) were collected on copper grids, stained with uranyl acetate and lead citrate, and observed in a JEOL 1200 EX II electron microscope. Micrographs were acquired using an Olympus SIS VELETA CCD camera (Japan) equipped with an The TEM imaging platform (iTEM) software version, iTEM 5.1 (Build 1700).
Immunohistochemistry
Immunohistochemistry was performed using the ABC-peroxidase reaction followed by nickel enhancement.39 Collagen and 3T3 samples were first fixed in 4% paraformaldehyde in 0.05 M phosphate buffer saline (PBS) at 4°C and then embedded in paraffin wax. Serial sections of 5–7 μm were incubated for 18 h at 4°C with a rabbit polyclonal antiserum anti-MMP-9 (Ab 38898, Abcam, UK) diluted 1:500. Negative controls were run by omitting the primary antibody. Subsequently, sections were incubated for 60 min with a biotinylated goat anti-rabbit immunoglobulin G (IgG) serum (Vector Labs, Burlingame, California, USA), diluted 1:1000 with PBS containing 0.1% sodium azide and 1% bovine serum albumin (BSA), followed by 60 min incubation in the avidin–biotin–peroxidase complex (ABC Elite Vectastain, Vector laboratories). Following extensive rinsing in distilled water, sections were dehydrated, mounted, and examined with a Zeiss light microscope (Germany) under a bright-field illumination. Images were acquired with a color camera (Axio Cam MRC, Arese, Milan, Italy) equipped with an AxioVision-KS 300 software version, AxioVision 40V 4.1.1.0.
Immune gold labeling
Samples were fixed for 2 h at room temperature with a mixture of 4% paraformaldehyde and 0.25% glutaraldehyde in 0.05 M piperazine-N, N′-bis-2-ethanesulfonic acid at pH 7.3. After rinsing for 10 min in the same buffer, samples were dehydrated in a graded series of ethanol and embedded in medium grade LR white resin, following addition of the LR white accelerator. The resin was then polymerized for 20 min at room temperature in tightly capped gelatin capsules. Ultrathin sections were obtained with a Reichert Ultracut ultramicrotome equipped with a diamond knife and collected on nickel grids. For immune gold labeling, 60–80 nm-thick sections were processed following the incubation protocol originally developed by Aurion (Vageningen, the Netherlands). Unspecific binding of antibody was prevented by treating sections for 20 min with 0.05 M glycine in PBS, at pH 7.4, followed by a 30 min blocking step in 5% BSA, 5% goat serum, and 0.1% fish gelatin in cold distilled water. Sections were subsequently washed in an incubation buffer containing 0.1% Aurion BSA-C™ in PBS and incubated overnight in a moist chamber in the presence of one of the following antibodies: anti-MMP-9 (ab38898 Abcam) polyclonal rabbit antibody, anti-Integrin β1 (TS2/16; ab119333 Abcam) mouse monoclonal antibody or anti-type I collagen (Ab34710, Abcam) rabbit polyclonal antibody. Antibodies were diluted 1:100 (if polyclonal) or 1:10 (if monoclonal) in the incubation buffer. Following this labeling step, sections were thoroughly washed in incubation buffer (6 × 5 min) and incubated for an additional hour in a secondary antibody. MMP-9 and type I collagen were revealed with a goat anti-rabbit antibody conjugated to 10 nm gold particles (British BioCell International, UK), whereas integrin β1 was revealed with a goat anti-mouse antibody conjugated to 20 nm gold particles (British BioCell International), diluted 1:10 in incubation buffer. After rinsing in incubation buffer (6 × 5 min), grids were washed in PBS (6 × 5 min). Sections were hence stained with uranyl acetate and observed in a JEOL JEM EXII transmission electron microscope (TEM) set at 100 kV. Micrographs were acquired by means of an Olympus SIS VELETA CCD camera equipped with the iTEM software. Control samples were obtained by omitting the primary antibody from the labeling mixture. Double immune labeling was carried out by first exposing sections to the primary antibody—either rabbit polyclonal or mouse monoclonal antibody—and subsequently to their respective goat secondary antibodies, sequentially applied for each of the antigen to be revealed.
Negative staining
Droplets of sample suspensions (10 µL) from either 2-D or 3-D culture media were placed on formvar–carbon-coated grids and allowed to be adsorbed for 60 s. Excess liquid was gently removed from each droplet with a strip of filter paper. Processing for negative staining was carried out by placing a drop of negative stain (2% uranyl acetate in distilled water) on the specimen grids, followed by quick blotting. This step was repeated a second time on a new drop of negative staining solution and prolonged for 60 s. Samples were eventually observed at a JEOL 1200 EX II electron microscope and micrographs acquired by the Olympus SIS VELETA CCD camera equipped with the iTEM software.
Western blot
By the end of every incubation period (1, 3, or 7 days from the time of seeding) samples consisting of either fibroblasts (2-D: 3T3) or collagen blocks containing migrating fibroblasts (3-D: Coll3T3) were washed twice in sterile PBS. Both samples were then frozen in liquid nitrogen for 2 min, pulverized, and incubated for 30 min at room temperature in RIPA buffer (Sigma-Aldrich, St Louis, Missouri, USA) with protease inhibitors added. The protein concentration was determined in each extract using the Bradford microassay method (Sigma-Aldrich) with BSA as a standard. Samples were loaded on a 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel in the concentration of 50 µg of proteins per well and resolved by electrophoresis at 20 mA constant current until the dye front reaches the bottom line. To detect antibody-binding proteins, samples were transferred onto nitrocellulose membranes (Millipore, Burlington, Massachusetts, USA) using a wet transfer system (Bio-Rad, Hercules, California, USA). These membranes were then blocked in Tris-buffer saline with 0.01% Tween-20, pH 7.4 with 5% BSA (TBST) for 1 h at room temperature, washed, and incubated overnight at 4°C with a rabbit polyclonal antiserum anti-MMP-9 (Ab 38898, Abcam) diluted 1:1000. Nitrocellulose membranes were washed and incubated for 4 h with a horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody diluted 1:1000 with TBST. Western blots (WB) were developed using the ECL chemiluminescence detection system (ChemiDoc, Bio-Rad).
Gelatin zymography
To detect the MMP activity in 3-D (Coll+3T3) or 2-D (3T3) culture samples, the protocol of Toth and Fridman40 for gelatin zymography was routinely used. Sample solutions were processed under native conditions in the absence of reducing agents and without boiling. They were then electrophoresed in a 10% SDS-polyacrylamide gel containing 0.1 mg/mL of porcine gelatin (Sigma-Aldrich) and subsequently incubated for 30 min at room temperature in a 2.5% (v/v) Triton X-100 solution to remove SDS from the resolved polypeptides. Following a thorough rinse in double distilled water for enzyme renaturation, gels were incubated at 37°C for 20 h in developing buffer (50 mM Tris/hydrochloric acid buffer, pH 7.8 containing 20 mM sodium chloride, 5 mM calcium chloride, and 0.02% Brij-35), stained with 0.1% Coomassie Brilliant Blue R-250 and eventually destained in 10% acetic acid and 30% methanol. Gelatinolytic activity was detected as unstained bands on a blue background and revealed with the ChemiDoc instrument (Bio-Rad). Acquired pictures were quantitatively evaluated using the ImageJ program version, ImageJ 1.48v – Wayne Rasband, National Institute of Health, USA.
Results
Fibroblasts cocultured under 3-D conditions until 7 days, that is, in the presence of a prosthetic collagen scaffold, can be shown to migrate rapidly inside the matrix and take close contact with the collagen fibers.41 Figure 1(a) to (c) depicts an initial condition (1 day) of this in vitro culture. At this stage, roundish fibroblasts appear firmly attached onto a collagen matrix, still characterized by a prevailing laminar structure. By the end of this culture period (7 days), cells had already become highly intermingled with collagen fibers, the matrix itself having lost most its laminar structure and acquired, in the meantime, a rather fibrillar appearance (Figure 1(d)). The collagen fibers themselves are no longer aligned in continuous laminar sheets, but are rather scattered in the form of a fragmented network with no recognizable orientational order (Figure 1(e)).
Figure 1.
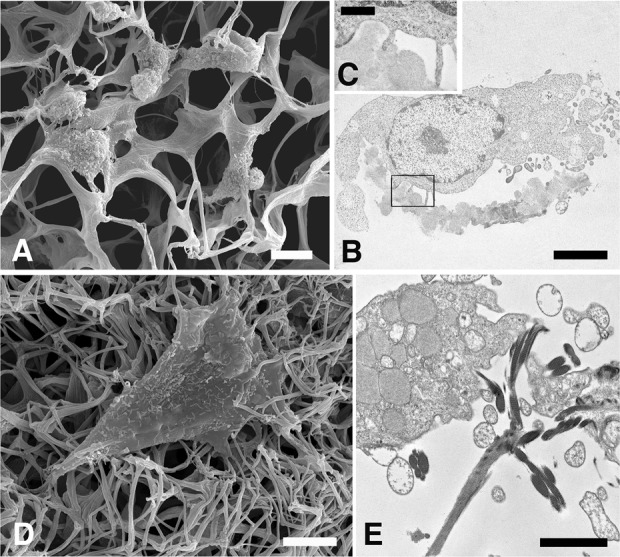
SEM and TEM micrographs showing how the collagen matrix is structurally modified in the presence of migrating fibroblasts. Figures (a), (b), and (c) show the collagen matrix at day 1 following cell seeding. Notice the laminar structural appearance of the collagen matrix (a) and the highly intertwined connections with fibroblasts’ filopodia (b, c). Bars: (a) 10 μm; (b) 5 μm; (c) 1 μm. Figures (d) and (e) depict fibroblasts after 7 days of in vitro culture. Notice that the collagen matrix has acquired a prevailing fibrillar organization. Bars: (d) 5 μm; (e) 2 μm. SEM: scanning electron microscopic; TEM: transmission electron microscopy.
To account for the laminar-to-fibrillar transition of the prosthetic collagen, two aspects of the in vitro culturing setup were further examined: (i) migrating fibroblasts were analyzed for their capacity to release metalloproteinases in the surrounding medium and, (ii) the enzymes released by fibroblasts were visualized in their structural relationship with collagen processing. Of the numerous metalloproteinases identified so far, we chose to study the MMP-9 because it is already known to be constitutively expressed in many cell types, including fibroblasts, besides being endowed with the capacity to remodel the extracellular matrix and degrading a broad variety of collagen types.35 Figure 2 is a gelatin zymography of fibroblasts cocultured in a 3-D collagen matrix showing the presence of two major protein fractions. These are identifiable as bona fide pro-MMP-9 and active MMP-9 on the basis of their respective molecular weights and antibody reactivity, both data being highly indicative of a precursor–product relationship.42 When detected by WB analysis (see Figure 2, Western Blot (WB) line), the anti-MMP-9 antibody appears to bind preferentially to the 92 kD fraction, suggesting that the antibody may be more specific for the MMP-9 precursor epitope(s). However, only the lower molecular weight fraction should be expected to be enzymatically active by zymography. Indeed, activation of the pro-MMP-9 is thought to result from an artificial enzyme exposure to the denaturing conditions of the gel electrophoresis.43
Figure 2.

Immune detection of pro-MMP-9 and relative enzyme activity in cell lysates following in vitro culture in the presence of a collagen matrix 3-D (Coll+3T3) or in 2-D culture medium alone (3T3) for 1, 3, or 7 days. GZ; WB with anti-MMP-9 antibody. Zymograms were scanned with densitometer equipped with ImageJ program. MMP: matrix metalloproteinase; GZ: gelatin zymography; WB: Western blot.
Results from gelatin zymography show that the relative concentration of the active MMP-9 form increases from 1 to 7 days of in vitro culture, while the pro-MMP-9 is gradually declining during the same time period. This temporal correlation is interpreted as indicating that coculture with the collagen matrix is a condition necessary, though not sufficient, for the MMP-9 to be released from the fibroblasts and subsequently activated in the extracellular milieu. By contrast, fibroblasts cultured under 2-D conditions, that is, in the absence of any collagen interference, appear to have a much lower MMP-9 activity and a higher enzyme turn over, both MMP fractions having already disappeared following 3 days of in vitro culture (see right panel in Figure 2). MMP-9 expression, as detected by gelatin zymography, was also extended to the analysis of culture media under 2-D and 3-D conditions. Interestingly, MMP-9 expression in 3-D culture media follows a kinetics similar to that already recorded for cell lysates with the pro- and active enzymes being temporally correlated in a precursor–product relationship. By comparison, MMP-9 expression in 2-D culture media was restricted to the sole 82 kDa active form (Figure 3). These observations are highly suggestive of the possibility that in vitro cultured fibroblasts may actually release “something” in the culture medium that is potentially capable of acting as a carrier for conveying the MMP-9 activity onto the collagen matrix.
Figure 3.

Immune detection of MMP-9 enzyme activity in the culture medium extracted from the 3-D collagen matrix (Coll+3T3)) or following in vitro 2-D cell culturing (3T3) for 1, 3, or 7 days. GZ; WB with anti-MMP-9 antibody reacting exclusively with the pro-active enzyme form. Zymograms were scanned with densitometer equipped with ImageJ program. MMP: matrix metalloproteinase; GZ: gelatin zymography; WB: Western blot.
To verify whether that something spreading the enzyme activity in the culture medium could be accounted for by the release of microvesicles from fibroblasts, extracts from all culture media were routinely checked by negative staining. Results show that vesicles recovered from these extracts fall within the size range of 30–300 nm (see Figure 4), suggesting that these samples may be comprised of both intact microvesicles and their relative intraluminal vesicles that are likely to be released upon shedding from the fibroblasts or during extraction from the culture medium.
Figure 4.
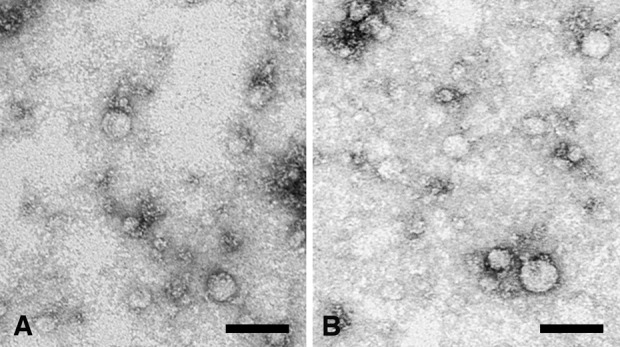
Negative staining of representative samples taken from culture media under 2-D (a) or 3-D conditions; (b) showing the vesicular composition of the relative extracts. The size range of the vesicles detected through this technique is 30–300 nm. Bars: 500 nm.
An extensive ultrastructural analysis of 2-D or 3-D cultured fibroblasts has shown that microvesicles are composite organelles containing numerous smaller intraluminal vesicles ranging in size from 30 nm to 200 nm (Figure 5(a)). To verify whether MMP-9 activity and the release of microvesicles are indeed causally related events, fibroblasts from both in vitro culturing conditions were examined by immune anti-MMP-9 labeling. The low magnification micrograph depicted in Figure 5(b) shows that under these experimental conditions many of the microvesicles dispersed along the fibroblast cell periphery become heavily labeled with gold-tagged anti-MMP-9 antibodies. Interestingly, anti-MMP-9 labeling is not associated with the entire microvesicle, but is instead restricted to some, if not all, of the intraluminal vesicles present inside it. The micrographs shown in Figure 5(c) to (e) were selected to depict a hypothetical sequence of MMP-9 release through microvesicle shedding from the fibroblast cell surface. Intraluminal vesicles are likely to be uploaded in the microvesicles following transfer from the cortical cytoplasm and docking to the cell plasma membrane (Figure 5(c) and (d)). MMP-9 activity may be eventually released in the extracellular space through fusion of the intraluminal vesicles with the outer microvesicle membrane (Figure 5(e)).
Figure 5.

Immune gold labeling of anti-MMP-9. (a) high-resolution TEM micrograph showing a microvesicle continuous with the cell membrane loaded with numerous intraluminal vesicles, Bar 500 nm; (b), low magnification micrograph of a 3-D cultured fibroblast showing numerous microvesicles heavily labeled with anti-MMP-9 gold-tagged antibody along the entire cell periphery, Bar 1 µm; (c) to (e), a hypothetical sequence of an MMP-9 activity uploaded via intraluminal vesicles from the cortical cytoplasm and eventually released from a shed microvesicle. Bars 500 nm. MMP: matrix metalloproteinase; TEM: transmission electron microscopic.
To identify the fibroblast microvesicles described in this study as real intercellular organelles transferring MMP-9 in membrane-bound intraluminal vesicles, some specific cell markers had to be used. Integrin β1 is a cell adhesion molecule recognized by many investigators as a reliable microvesicle marker for a variety of cell types.44 Accordingly, 10 nm gold-tagged anti-MMP-9 antibodies were used to detect the MMP-9 activity as the intraluminal microvesicle content, while 20 nm gold-tagged anti-integrin β1 antibodies were used to identify the enclosing microvesicle as the respective enzyme container. As shown in Figure 6(a), anti-integrin antibodies bind the microvesicle’s outer membrane, while anti-MMP-9 antibodies are restricted to the vesicle’s inner space. Since the prolonged treatment required for double immune labeling is known to affect the binding efficiency of gold-tagged antibodies,45 MMP-9 and Integrin β1 were also colocalized on alternate thin sections. Very briefly, this consisted in identifying the same microvesicle in subsequent grids and exposing it to different gold-tagged antibodies, followed by merging of the resulting images (Figures 6(b) to (d)).
Figure 6.

In vitro cultured fibroblasts subjected to double immune labeling with 10 nm gold-tagged anti-MMP-9 antibodies and 20 nm gold-tagged anti-integrin β1 antibodies. (a) microvesicles shed along the fibroblast cell periphery are internally labeled with 10 nm gold particles (arrows) and on the outer membrane with 20 nm gold particles (double arrows). Bar 1 µm; (b) microvesicle labeled with anti-integrin β1 antibodies along the outer membrane; (c) the same vesicle as in (b) labeled with anti-MMP-9 antibodies; and (d), merging of (b) and (c) micrographs obtained by overlapping 50% transparent images. Bars: 250 nm. MMP: matrix metalloproteinase.
Vesicular shedding was also shown to occur in 2-D cultures; fibroblasts proved capable of synthesizing and releasing MMP-9 even in the absence of the collagen matrix. However, the extent to which microvesicles are released into the surrounding milieu under these conditions is by far much less pronounced than in 3-D cultures. A detailed analysis of 3-D cell cultures from 1 to 7 days has clearly shown that microvesicles can gain access to regions of the collagen matrix far away from their cells of origin. It is not clear whether microvesicles actually move away from their budding site or simply remain anchored to the collagen fibrils, while cells migrate along the collagen fibers. Whatever mechanism(s) sustains their migration, microvesicles occur either as isolated clusters or in association with numerous frayed collagen fibrils (Figure 7(a) and (b)). Under both circumstances, microvesicles appear still loaded with a MMP-9 cargo, as shown by their binding capacity to MMP-9 antibody (Figure 7(c)). However, when observed in close contact with collagen, gold label appears to spread onto frayed fibrils (Figure 7(d)), indicating that the MMP-9 cargo has been released from microvesicles and dispersed in the surrounding extracellular environment. That the amorphous or fuzzy material contacting microvesicles should actually be identified as collagen, in spite of the difficulty of recognizing it in the absence of a native banding pattern, it is clearly demonstrated by its reactivity to the anti-collagen antibody (see Figure 7(e)).
Figure 7.

(a) High-resolution TEM micrograph showing microvesicles dispersed in the collagen matrix, Bar 1 µm; (b) SEM micrograph showing a cluster of microvesicles associated with collagen fibrils, Bar 2,5 µm; (c) immune gold localization of anti-MMP-9 in association with a cluster of isolated microvesicles. Note that gold particles are both inside the intraluminal vesicles as well as along the enclosing membrane of the microvesicles, Bar 4 µm; (d) immune gold localization of anti-MMP-9 in a microvesicle associate with a bundle of frayed collagen fibrils; the arrowhead points to gold labeling released from the microvesicle onto the collagen fibrils, Bar 200 nm; and (e) immune gold localization of anti-collagen antibody on a bundle of frayed collagen fibrils, Bar 500 nm. TEM: transmission electron microscopic; SEM: scanning electron microscopy; MMP: matrix metalloproteinase.
The ultimate result of MMP-9 release from microvesicles is the spread of the enzyme throughout the entire collagen matrix. As clearly indicated by the sequence of images depicted in Figure 8(a) to (c), labeling with anti-MMP-9 antibody becomes progressively more pronounced on the collagen matrix as culturing times prolong from 1 to 7 days. This observation can also be verified at the ultrastructural level where collagen fibers exposed to the anti-MMP-9 antibody appear progressively more labeled with gold particles (Figure 8(d) to (f)).
Figure 8.

Immunohistochemical localization of MMP-9 on collagen matrix following 1, 3, or 7 days of in vitro 3-D culture in the presence of fibroblasts. (a to c) thick sections of formaldehyde-fixed and paraffin-embedded collagen matrix exposed to anti-MMP-9 antibody and stained with DAB and nickel enhancement as described in material and methods. Bars 10 µm. (d to f) thin sections of formaldehyde-fixed and glycol-methacrylate embedded collagen matrix exposed to anti-MMP-9 antibody and treated with a gold labeled secondary antibody. Note how gold particles remain associated with the collagen fibrils even in case where the collagen banding pattern is lost. Bars 500 nm. MMP: matrix metalloproteinase.
Discussion
The main objective of this study was to find out what causal relationship holds between collagen degradation, microvesicle release, and metalloproteinase activities. When cocultured with fibroblasts in vitro, the collagen matrix is gradually transformed from a prevailing laminar to an exclusive fibrillar type of architecture, indicating that collagen fibers are gradually frayed, disassembled, and, possibly, degraded into smaller peptide fragments. Given their temporal coincidence over the time period tested in this study, both microvesicle release and metalloproteinase activity could be potential candidates for causing this collagen transformation. Indeed, the use of gelatin zymography on fibroblast cell lysates demonstrates that the 82 kDa MMP-9 form increases gradually in activity as the relative 92 kDa pro-MMP-9 progressively declines. During the same time period, the extent to which microvesicles are released from the fibroblast plasma membrane is also significantly enhanced, as clearly documented by the numerous micrographs depicting fibroblast cell peripheries and various regions of the collagen matrix. However, temporally coincident occurrences can be meaningful only if causally related, for they could be independent of each other and act on collagen degradation through different mechanisms. The evidence that microvesicles and MMP secretion are causally related has come from the finding that microvesicles are labeled with anti-MMP-9 antibody, indicating that they may indeed act as vesicular carriers for this enzyme activity. The causal role played by microvesicles is also demonstrated by the progressive enhancement of MMP-9 expression as they spread in regions of the matrix far distant from their cells of origin. During this time period, MMP-9 expression becomes progressively more pronounced as the collagen matrix is gradually transformed from a laminar to a fibrillar type of fibril assembly. This finding is in line with the observation that the amount of collagen deposited in damaged tissues is directly correlated with the level of MMP-9 expression, suggesting that wound healing is somehow conditioned by the tissue capacity to remove any impaired collagen matrix.46
The present finding that metalloproteinase and microvesicle shedding are causally related events is consonant with earlier studies demonstrating how microvesicles from various cell types affect persistence and stability of the extracellular matrix under a variety of normal and pathological conditions. A growing body of evidence provides additional support to the notion that many tissue-specific stem cells can manipulate their environment during wound healing and cancer progression through the release of microvesicles47 and that, in all of these circumstances, microvesicles act as vehicles transferring metalloproteinases to modify the extracellular matrix and increase tumor cell mobility.48 However, to the best of our knowledge, the evidence reported in this study is the first demonstrating such a high resolution localization of a specific metalloproteinase by immune labeling on an in vitro cultured collagen matrix. Interestingly, MMP-9 is heterogeneously distributed inside the microvesicle, its localization being exclusively restricted to the intraluminal vesicles contained therein (see Figure 5(c) to (e)). The ultrastructural evidence gathered in this study indicates that these intraluminal vesicles are recruited from the cortical cytoplasm, conveyed to the shedding site of the fibroblast cell membrane and eventually uploaded inside the microvesicle.
We are not aware of whether similar structures have been reported in other cell types, except for the so-called multivesicular cargo that Fertig et al.49 have recently described in cardiac telocytes, a new cell type of mesenchymal origin, supposedly playing key roles in tissue regeneration and repair.50,51 However, the presence of intraluminal vesicles inside microvesicles is at variance from what is currently known about their origin and role(s) in cell communication and information transfer. Usually, intraluminal vesicles are sorted out in the endosomal compartment through specific tetraspanin interaction before they are actually clustered in multivesicular bodies and released as extracellular exosomes.52,53 Whether the same or similar mechanism(s) is responsible for the vesicular recruitment in both types of organelles is yet to be established, although their functional similarity suggests that they may be largely analogous and likely to make use of similar molecular interactions. In spite of their morphological differences, they could in fact avail of the same endosomal sorting complexes required for transport (ESCRT) to segregate endocytic membrane domains destined to become intraluminal vesicles. For this very reason, they have recently been termed ectosomes, rather than being generically referred to as microvesicles.54 Their identity as ectosomes was further confirmed in this study through the use of specific vesicular markers. Integrin β1 is cell adhesion molecule frequently used as a reliable marker for distinguishing ectosomes from exosomes.6,54 The observation that anti-integrin β1 antibody binds the microvesicle’s outer membrane, while anti-MMP-9 antibody localization remains confined to the intraluminar vesicles is a clear-cut evidence that these microvesicles are indeed ectosomes acting as carriers delivering their metalloproteinase cargo onto the extracellular matrix.
In wound healing, ectosomes are primarily addressed to sustain cell-to-matrix relationship, rather than cell-to-cell communication. In a cell-to-matrix relationship they may break directly in the extracellular environment and release their content upon shedding from the cell of origin or at some distance from it.8 Under these conditions, cells may activate their metalloproteinase cargo by controlling the extent to which intraluminal vesicles are either allowed or impeded to fuse with the outer membrane. Since metalloproteinases remain inactive as long as they are confined within these multivesicular compartments, the possibility to be released in the surrounding milieu, and consequently to act on the nearby collagen substrate, can only occur when the two membranes—the outer membrane and the intraluminal membrane—are transiently fused by exocytosis. How this could occur in vivo is as yet to be established, although a number of plausible mechanisms may be at work in different tissues.
As MMP-9 is released extracellularly in the form of a stable, inactive pro-MMP-9 zymogen, a number of factors could potentially convert it to an active form. On the whole, many of the factors triggering this conversion could act in combination at different levels, from gene transcription, to zymogen activation or endogenous inhibition.34 Binding to specific cell surface antigens, such as CD44 or integrin receptors, could also facilitate the proenzyme activation by allosteric interference with its catalytic site.38 It is well known that adhesive interactions with the extracellular matrix play important roles in tissue remodeling, especially if mediated by MMP activation.55 It is also likely that activation of metalloproteinase may eventually result in the release of a number of collagen breakdown products that, by themselves, may have some chemotactic effect on cells migrating along the wound bed during formation of the granulation tissue.56
In conclusion, the experimental evidence reported in this study demonstrates that collagen breakdown in vitro, and perhaps wound healing in vivo, are causally dependent on the release of metalloproteinases via microvesicle shedding. Given this relationship, what remains to be verified is how pro-MMP-9 come to be uploaded inside the shedding microvesicles, and eventually how they are targeted to specific matrix substrates following release in the extracellular milieu.
Footnotes
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
- 1. Gilbert SF. Cell-cell comunication in development In: Sinauer Associates. (ed) Developmental biology. 7th ed Sunderland: Elsevier Science, 2000, pp. 143–179. [Google Scholar]
- 2. Goodenough DA, Paul DL. Gap junctions. Cold Spring Harb Perspect Biol 2009; 1: a002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. De Broe ME, Wieme RJ, Logghe GN, et al. Spontaneous shedding of plasma membrane fragments by human cells in vivo and in vitro. Clin Chim Acta 1977; 81: 237–245. [DOI] [PubMed] [Google Scholar]
- 4. Vittorelli ML. Shed membrane vesicles and clustering of membrane-bound proteolytic enzymes. Curr Top Dev Biol 2003; 54: 411–432. [DOI] [PubMed] [Google Scholar]
- 5. Ratajczak J, Wysoczynski M, Hayek F, et al. Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia 2006; 20(9): 1487–1495. [DOI] [PubMed] [Google Scholar]
- 6. Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol 2009; 19(2): 43–51. [DOI] [PubMed] [Google Scholar]
- 7. Pap E, Pállinger É, Falus A. The role of membrane vesicles in tumorigenesis. Crit Rev Oncol Hematol 2011; 79(3): 213–223. [DOI] [PubMed] [Google Scholar]
- 8. Turturici G, Tinnirello R, Sconzo G, et al. Extracellular membrane vesicles as a mechanism of cell-to-cell communication: advantages and disadvantages. Am J Physiol Cell Physiol 2014; 306(7): C621–C633. [DOI] [PubMed] [Google Scholar]
- 9. Heijnen HF, Schiel AE, Fijnheer R, et al. Activated platelets release two types of membrane vesicles: microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood 1999; 94: 3791–3799. [PubMed] [Google Scholar]
- 10. Théry C. Exosomes: secreted vesicles and intercellular communications. Biol Rep 2011; 3: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cocucci E, Racchetti G, Podini P, et al. Enlargeosome traffic: exocytosis triggered by various signals is followed by endocytosis, membrane shedding or both. Traffic 2007; 8: 742–757. [DOI] [PubMed] [Google Scholar]
- 12. Deregibus MC, Cantaluppi V, Calogero R, et al. Endothelial progenitor cell derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA. Blood 2007; 110: 2440–2448. [DOI] [PubMed] [Google Scholar]
- 13. Mathivanan S, Ji H, Simpson RJ. Exosomes: extracellular organelles important in intercellular communication. J Proteomics 2010; 73(10): 1907–1920. [DOI] [PubMed] [Google Scholar]
- 14. Ginestra A, La Placa MD, Saladino F, et al. The amount and proteolytic content of vesicles shed by human cancer cell lines correlates with their in vitro invasiveness. Anticancer Res 1998; 18: 3433–3437. [PubMed] [Google Scholar]
- 15. Ginestra A, Miceli D, Dolo V, et al. Membrane vesicles in ovarian cancer fluids: a new potential marker. Anticancer Res 1999; 19: 3439–3445. [PubMed] [Google Scholar]
- 16. Sidhu SS, Mengistab AT, Tauscher AN, et al. The microvesicle as a vehicle for EMMPRIN in tumor-stromal interactions. Oncogene 2004; 23: 956–963. [DOI] [PubMed] [Google Scholar]
- 17. Collino F, Deregibus MC, Bruno S, et al. Microvesicles derived from adult human bone marrow and tissue specific mesenchymal stem cells shuttle selected pattern of miRNAs. PLoS One 2010; 5(7): e11803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Camussi G, Deregibus MC, Bruno S, et al. Exosome/microvesicle-mediated epigenetic reprogramming of cells. Am J Cancer Res 2011; 1(1): 98–110. [PMC free article] [PubMed] [Google Scholar]
- 19. Gurtner GC, Werner S, Barrandon Y, et al. Wound repair and regeneration. Nature 2008; 453(7193): 314–321. [DOI] [PubMed] [Google Scholar]
- 20. Diegelmann RF, Evans MC. Wound healing: an overview of acute, fibrotic and delayed healing. Front Biosci 2004; 9: 283–289. [DOI] [PubMed] [Google Scholar]
- 21. Carlson MA, Longaker MT. The fibroblast-populated collagen matrix as a model of wound healing: a review of the evidence. Wound Repair Regen 2004; 12(2): 134–147. [DOI] [PubMed] [Google Scholar]
- 22. Yun YR, Won JE, Jeon E, et al. Fibroblast growth factors: biology, function, and application for tissue regeneration. Tissue Eng 2010; 2010: 218142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guo S, DiPietro LA. Factor affecting wound healing. J Dent Res 2010; 89(3): 219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McDougall S, Dallon J, Sherratt J, et al. Fibroblast migration and collagen deposition during dermal wound healing: mathematical modelling and clinical implications. Philos Trans A Math Phys Eng Sci 2006; 364(1843): 1385–1405. [DOI] [PubMed] [Google Scholar]
- 25. Nedelec B, Ghahary A, Scott PG, et al. Control of wound contraction. Basic and clinical features. Hand Clin 2000; 16(2): 289–302. [PubMed] [Google Scholar]
- 26. Madsen DH, Engelholm LH, Ingvarsen S, et al. Extracellular collagenases and the endocytic receptor, urokinase plasminogen activator receptor-associated protein/Endo180, cooperate in fibroblast-mediated collagen degradation. J Biol Chem 2007; 282(37): 27037–27045. [DOI] [PubMed] [Google Scholar]
- 27. Rohani MG, Chow YH, Razumova MV, et al. uPARAP function in cutaneous wound repair. PLoS One 2014; 9(3): e92660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Werner S, Grose R. Regulation of wound healing by growth factors and cytokines. Physiol Rev 2003; 83: 835–870. [DOI] [PubMed] [Google Scholar]
- 29. Armstrong DG, Jude EB. The role of matrix metalloproteinases in wound healing. J Am Podiatr Med Assoc 2002; 92(1): 12–18. [DOI] [PubMed] [Google Scholar]
- 30. Grotendorst GR, Rahmanie H, Duncan MR. Combinatorial signaling pathways determine fibroblast proliferation and myofibroblast differentiation. FASEB J 2004; 18(3): 469–479. [DOI] [PubMed] [Google Scholar]
- 31. Everts V, van der Zee E, Creeemers L, et al. Phagocytosis and intracellular digestion of collagen, its role in turnover and remodelling. Histochem J 1996; 28(4): 229–245. [DOI] [PubMed] [Google Scholar]
- 32. Abraham LC, Dice JF, Lee K, et al. Phagocytosis and remodeling of collagen matrices. Exp Cell Res 2007; 313(5): 1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Löffek S, Schilling O, Franzke CW. Series “matrix metalloproteinases in lung health and disease”: biological role of matrix metalloproteinases: a critical balance. Eur Respir J 2011; 38(1): 191–208. [DOI] [PubMed] [Google Scholar]
- 34. Gaffney J, Solomonov I, Zehorai E, et al. Multilevel regulation of matrix metalloproteinases in tissue homeostasis indicates their molecular specificity in vivo. Matrix Biol 2015; 44–46C: 191–199. [DOI] [PubMed] [Google Scholar]
- 35. Fanjul-Fernández M, Folgueras AR, Cabrera S, et al. Matrix metalloproteinases: evolution, gene regulation and functional analysis in mouse models. Biochim Biophys Acta 2010; 1803(1): 3–19. [DOI] [PubMed] [Google Scholar]
- 36. Chakraborti S, Mandal M, Das S, et al. Regulation of matrix metalloproteinases: an overview. Mol Cell Biochem 2003; 253: 269–285. [DOI] [PubMed] [Google Scholar]
- 37. Maretzky T, Yang G, Ouerfelli O, et al. Characterization of the catalytic activity of the membrane-anchored metalloproteinase ADAM15 in cell-based assays. Biochem J 2009; 420(1): 105–113. [DOI] [PubMed] [Google Scholar]
- 38. Tocchi A, Parks WC. Functional interactions between matrix metalloproteinases and glycosaminoglycans. FEBS J 2013; 280(10): 2332–2341. [DOI] [PubMed] [Google Scholar]
- 39. Adam JC. Heavy metal intensification of DAB-based HRP reaction product HRP reaction product. J Histochem Cytochem 1981; 29(6): 775. [DOI] [PubMed] [Google Scholar]
- 40. Toth M, Fridman R. Assessment of gelatinases (MMP-2 and MMP-9) by gelatin zymography. Methods Mol Med 2001; 57: 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Laghezza Masci V, Taddei AR, Gambellini G, et al. Ultrastructural investigation on fibroblast interaction with collagen scaffold. J Biomed Mater Res A 2016; 104(1): 272–282. [DOI] [PubMed] [Google Scholar]
- 42. Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res 2006; 69: 562–573. [DOI] [PubMed] [Google Scholar]
- 43. Frankowski H, Gu YH, Heo JH, et al. Use of gel zymography to examine matrix metalloproteinase (gelatinase) expression in brain tissue or in primary glial cultures. Methods Mol Biol 2012; 814: 221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee TH, D’Asti E, Magnus N, et al. Microvesicles as mediators of intercellular communication in cancer – the emerging science of cellular ‘debris’. Semin Immunopathol 2011; 33(5): 455–467. [DOI] [PubMed] [Google Scholar]
- 45. Furuhata S, Nemoto N, Miyazawa S, et al. Pitfalls in the double labelling immunoelectron microscopy using one face of the grid. J Electron Microsc (Tokyo) 1992; 41(2): 120–122. [PubMed] [Google Scholar]
- 46. Agren M, Jorgensen LN, Andersen M, et al. Matrix metalloproteinase 9 level predicts optimal collagen deposition during early wound repair in humans. Br J Surg 1998; 85(1): 68–71. [DOI] [PubMed] [Google Scholar]
- 47. Dittmer J, Leyh B. Paracrine effects of stem cells in wound healing and cancer progression (Review). Int J Oncol 2014; 44: 1789–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Muralidharan-Chari V, Clancy JW, Sedgwick A, et al. Microvesicles: mediators of extracellular communication during cancer progression. J Cell Sci 2010; 123: 1603–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fertig ET, Gherghiceanu M, Popescu LM. Extracellular vesicles release by cardiac telocytes: electron microscopy and electron tomography. J Cell Mol Med 2015; 18: 1938–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Faussone Pellegrini M-S, Popescu LM. Telocytes. BioMol Concepts 2011; 2(6): 481–489. [DOI] [PubMed] [Google Scholar]
- 51. Ceafalan L, Gherghiceanu M, Popescu LM, et al. Telocytes in human skin – are they involved in skin regeneration? J Cell Mol Med 2012; 16: 1405–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Akers JC, Gonda D, Kim R, et al. Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J Neurooncol 2013; 113: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Andreu Z, Yáñez-Mó M. Tetraspanins in extracellular vesicle formation and function. Front Immunol 2014; 5: 442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cocucci E, Meldolesi J. Ectosomes and exosomes: shedding the confusion between extracellular vesicles. Trends Cell Biol 2015; 25(6): 364–372. [DOI] [PubMed] [Google Scholar]
- 55. Abel M, Vliagoftis H. Mast cell-fibroblast interactions induce MMP-9 release from fibroblasts: role for IgE-mediated mast cell activation. J Immunol 2008; 180: 3543–3550. [DOI] [PubMed] [Google Scholar]
- 56. Brett D. A review of collagen and collagen-based wound dressings. Wounds 2008; 20(12): 347–356. [PubMed] [Google Scholar]