

SUMMARY
Polarized vesicle transport plays an important role in cell polarization, but the mechanisms underlying this process and its role in innate immune responses are not well understood. Here, we describe a phosphorylation-regulated polarization mechanism that is important for neutrophil adhesion to endothelial cells during inflammatory responses. We show that the protein kinase PKN1 phosphorylates RPH3A, which enhances binding of RPH3A to GTP-bound RAB21. These interactions are important for polarized localization of RAB21 and RPH3A in neutrophils, which leads to PIP5K1C90 polarization. Consistent with the roles of PIP5K1C90 polarization, the lack of PKN1 or RPH3A impairs neutrophil integrin activation, adhesion to endothelial cells, and infiltration in inflammatory models. Furthermore, myeloid-specific loss of PKN1 decreases tissue injury in a renal ischemia-reperfusion model. Thus, this study characterizes a mechanism for protein polarization in neutrophils and identifies a potential protein kinase target for therapeutic intervention in reperfusion-related tissue injury.
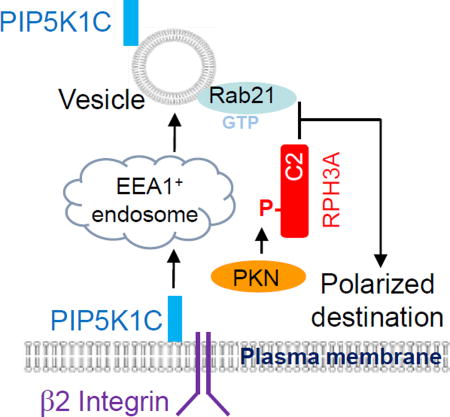
Graphical Abstract
INTRODUCTION
The neutrophil is a key player in inflammatory responses. It not only plays an important role in host defense, but also contributes to inflammation-related tissue damages in various chronic and acute inflammatory diseases, including ischemia reperfusion (Kolaczkowska and Kubes, 2013; Nauseef and Borregaard, 2014). During the inflammation, neutrophils are recruited to the sites of injury and infection. They extravasate across the endothelium that lines the blood vessel wall through a multi-step process, which includes rolling, firm adhesion, intravascular crawling, diapedesis, and extravascular chemotaxis (Abram and Lowell, 2007; Gerhardt and Ley, 2015; Imhof and Aurrand-Lions, 2004; Luo et al., 2007; Nourshargh and Alon, 2014; Rose et al., 2007; Williams et al., 2011). This infiltration process, which is also shared by other types of leukocytes, requires neutrophil polarization, namely the transformation of the apolar, circulating cells to being polarized through a spatial reorganization of signaling and cytoskeletal molecules. Chemoattractant stimulation can polarize the neutrophils anteroposteriorly to form the lamellar F-actin-rich leading edge and actomyosin-containing uropod, and this polarization is crucial for neutrophil migration (Afonso and Parent, 2011; Bear and Haugh, 2014; Graziano and Weiner, 2014; Kolsch et al., 2008; Ridley, 2011; Shelef et al., 2013; Swaney et al., 2010; Weninger et al., 2014). However, in vivo before neutrophils undergo chemoattractant-stimulated migration, they have to first adhere to the endothelium via integrin engagement, which can induce a different form of neutrophil polarization from that induced by chemoattractants. Integrins induce the polarization of PIP5K1C90 independently of chemoattractant signaling initially at the site of attachment, which becomes the uropod when the cell polarizes anteroposteriorly upon chemoattractant stimulation (Xu et al., 2010). Polarization of PIP5K1C90, which is a lipid kinase involved in PtdIns4,5P2 synthesis, leads to polarized RHOA activation and increases in integrin affinity and neutrophil adhesion to the endothelium (Nourshargh and Alon, 2014; Williams et al., 2011; Xu et al., 2010).
One mechanism for establishment and maintenance of cell polarity is polarized vesicle trafficking. Secretory vesicles generated from the trans-Golgi network carrying newly synthesized cargos or from endosomal compartments often carrying recycled cargos are transported to specific areas of the cell, leading to cell polarization. The evolutionarily conserved exocyst, an octameric protein complex, plays an important role in polarized exocytosis during many cellular activities including cell migration and epithelial polarization (He and Guo, 2009; Mukherjee et al., 2014; Orlando and Guo, 2009), including the control of directional vesicle trafficking to leading edges in migrating cells (Fletcher and Rappoport, 2010; He and Guo, 2009; Ulrich and Heisenberg, 2009). One of the components of the exocyst is the RAB small GTPases. In addition to their involvement in the exocyst, the superfamily of the RAB small GTPases is the master regulator of vesicle trafficking in almost every step of the process. However, the role of RAB GTPases or directional vesicle trafficking in neutrophil protein polarization and infiltration has not yet been well investigated.
Protein Kinase N (PKN)1 together with its close homologs PKN2 and 3 are also known as protein kinase c-related kinases (PRKs) (Mukai, 2003). They belong to the AGC superfamily of protein kinases. PKN1 was reported to be involved in several functions including smooth muscle migration and division (Singh et al., 2012), cardiacmyocyte survival (Kajimoto et al., 2011), neurofilament organization and axonal transport in neurons and enhancement of germinal center formation (Yasui et al., 2012). Known substrates for PKN1 are the intermediate filament proteins, neurofilaments, vimentin, and the microfilament protein α-actinin (Mukai, 2003). The role of PKN1 in myeloid functions has not been investigated.
PIP5K1C90 is a plasma membrane-associated protein and interacts with talin and endocytic structural protein adaptin-2 (AP2) (Di Paolo et al., 2002; Lee et al., 2005; Ling et al., 2002; Thieman et al., 2009; Xu et al., 2010). Upon β2 integrin engagement, PIP5K1C90 is endocytosed with integrins and shows polarized localization in mouse neutrophils (Xu et al., 2010). Polarization of PIP5K1C90 leads to polarized RHOA activation and is important for neutrophil firm adhesion to the endothelium (Nourshargh and Alon, 2014; Williams et al., 2011; Xu et al., 2010). Although vesicle trafficking is implicated for its involvement in PIP5K1C90 polarization (Xu et al., 2010), the mechanism for this polarization is unknown. In this study, we elucidate the mechanism by which PKN1 regulates directional trafficking of RAB21 vesicles via its phosphorylation of a newly characterized RAB21 effector RPH3A, which underlies PIP5K1C90 polarization.
RESULTS
PKN1-deficiency impedes neutrophil adhesion to endothelial cells
The Pkn1 gene is expressed at high levels in mouse leukocytes and the highest in neutrophils among the leukocytes based on gene expression data of the Immunological Genome Project (www.immgen.org). The PKN1 protein was also readily detected in mouse neutrophils by Western blot analysis (Fig. 1A). To investigate the importance of Pkn1 in mouse myeloid cells, we generated the myeloid-specific knockout mouse line (Pkn1m/m) by crossing the Pkn1-floxed mouse line (Fig. S1A) with the Lyzstm1(cre)Ifo/ J mice. The PKN1 protein was not detectable in the Pkn1m/m neutrophils by Western blot analysis (Fig. 1A). Myeloid-specific PKN1 deficiency did not affect the numbers of neutrophils or monocytes in circulation (Table S1).
Figure 1. PKN1-deficiency attenuates neutrophil adhesion to endothelial cells.

A. Validation of the lack of PKN1 proteins in neutrophils isolated from Pkn1m/m mice by Western blot analysis. PKN2 and tubulin were detected as internal controls.
B–D. PKN1-deficiency impairs neutrophil infiltration into inflamed mouse peritonea (B), adhesion to endothelial cells under shear flow (C), and binding to ICAM1 (D). Data are presented as means ± SEM (*, p<0.05, Student’s t-Test, n>5)
E–G. Effect of PKN1-deficiency on neutrophil-endothelial cell interaction in inflamed cremaster muscle venues. Adhesion (E), transmigration of neutrophils (F), and rolling flux (G), were determined after stimulation of the cremaster muscle with TNFα (0.5 µg) for 4h. All values are means of n = 5 animals (with 5 to 7 vessels per animal) ± SEM. (*, p<0.05; **, p<0.01, Student’s t-Test).
Next, we investigated the effect of PKN1 deficiency on neutrophil recruitment in vivo by using a peritonitis model. When isolated WT and PKN1-null neutrophils were labelled with different fluorescent dyes, mixed, and injected intravenously to WT mice in which peritonitis was elicited by thioglycollate, significantly fewer PKN1-null neutrophils were detected than the WT cells in the peritonea (Fig. 1B). To understand how PKN1-deficiency affects neutrophil infiltration in vivo, we first examined if PKN1 deficiency affects neutrophil chemotaxis in vitro using a Dunn chamber. PKN1-deficiency did not significantly affect the motility even though it increased the directional errors (Fig. S1B). The defect in chemotactic directionality reflected by the increase in the directional errors does not appear to be substantial enough to explain the strong in vivo infiltration defects. We thus examined if PKN1-deficiency affected the neutrophil adhesion to endothelial cells, another key step in neutrophil infiltration. We first assessed the adhesion in vitro using a flow chamber coated with mouse endothelial cells, a condition resembling what occurs in vivo. We found that PKN1-deficiency significantly decreased the number of neutrophils adherent to endothelial cells under shear flow (Fig. 1C). Consistent with this observation, we also found that PKN1-null neutrophils showed significantly reduced binding to the integrin ligand ICAM-1 (Fig. 1D), suggesting that PKN1 may regulate the adhesion through the regulation of integrin activity.
To assess the role of PKN1 in neutrophil adhesion to endothelial cells in vivo, we performed intravital microscopic examination of the cremaster muscle venules. PKN1-deficiency markedly reduced the number of cells adherent to the endothelium of the venules treated with TNFα (Figure 1E) and the number of transmigrated cells (Fig. 1F), while increasing the number of rolling cells on the endothelium (Fig. 1G). The decrease in the adhesion would result in a decrease in transmigration and an increase in rolling. Therefore, these intravital observations are consistent with those made in the flow chambers, and they together demonstrate that PKN1 is important for firm neutrophil adhesion to endothelial cells.
PKN1 is required for PIP5K1C90 polarization
The adhesion-related phenotypes described in Fig. 1B–G for PKN1-deficiency are very similar to what we have previously reported for PIP5K1C-deficiency (Xu et al., 2010). We hence wondered if PKN1 was involved in PIP5K1C90 polarization. PIP5K1C90 polarization is induced by β2 integrin engagement and has an important role in firm adhesion of neutrophils to endothelial cells (Xu et al., 2010). Indeed, PKN1-deficiency significantly inhibited PIP5K1C90 polarization in response to fibrinogen (Fb), an integrin ligand (Fig. 2A–C). Instead of being localized at one side of the WT cells, PIP5K1C90 was primarily localized at the vicinity of EEA1-postive early endosomes with a partial colocalization with EEA1-staining in the majority of PKN1-null neutrophils (Fig. 2A–B). This result, together with the knowledge of the association of PKN1 with endosomes (Mellor et al., 1998) and involvement of vesicle transport in PIP5K1C90 polarization (Xu et al., 2010), prompted us to postulate that PKN1 may regulate PIP5K1C90 polarization via its regulation of vesicle trafficking.
Figure 2. PKN1-deficiency impairs PIP5K1C90 polarization in neutrophils.

A–B. Mouse neutrophils transfected with HA-PIP5K1C90 were placed on Fb-coated coverslips for 30 min. They were then fixed and stained by anti-HA and anti-EEA1 antibodies followed by secondary antibodies conjugated with Alexa633 (colored in red) and Alexa488 (colored in green). Cell contours are outlined and scale bars are 2.5 µm for all of the figures.
C. Quantification of the effect of PKN1-deficiency on PIP5K1C90 polarization. The quantification was done from three independent observations, and more than 20 cells were examined in each observation. Data are presented as means ± SD (**, p<0.01, Student’s t-Test).
RAB21 is polarized and regulates PIP5K1C90 polarization
Given the central role of the RAB GTPases in directing vesicle trafficking, we reasoned a likely involvement of at least one RAB GTPase in PIP5K1C90 polarization. Identification of the RAB GTPase may help us to understand how PKN1 regulates PIP5K1C90 polarization. We tested 8 RAB GTPases, including RAB3D, RAB21, RAB11A, RAB9, RAB14, RAB32, RAB35 and RAB22A that are highly expressed in neutrophils (Carroll et al., 2001; Fletcher and Rappoport, 2010; He and Guo, 2009; Kauppi et al., 2002; Klinkert and Echard, 2016; Lu and Wilson, 2016; Millar et al., 2002; Ortiz Sandoval and Simmen, 2012; Simpson et al., 2004; Stenmark, 2009; Ulrich and Heisenberg, 2009). Each of the dominant negative (DN) mutants of these small GTPase was expressed in neutrophils, and PIP5K1C90 polarization on Fb was quantified (Fig. S1C). We found that the DN mutant of RAB21 (T31N) inhibited the PIP5K1C90 polarization induced by Fb (Fig. S1C, 3A–C, & S2A). RAB21 belongs to the RAB5 subfamily of small GTPases that includes RAB5, RAB21 and RAB22. These RABs are localized to early endosomes and plasma membrane and play an important role in vesicle trafficking (Caswell et al., 2009; Pellinen et al., 2006; Stenmark, 2009). However, RAB5 (Xu et al., 2010) and RAB22 (Fig. S1C) are not involved in PIP5K1C90 polarization in mouse neutrophils. Of note, neither RAB21DN expression nor PKN1-deficiency affected static adhesion of neutrophils to Fb or PK-coated surfaces; hence, their effects on PIP5K1C90 polarization cannot be due to a secondary effect of reduced neutrophil adhesion (Fig. S2B). Importantly, the localization of RAB21 was also polarized in the majority of cells placed on Fb-coated surfaces compared to cells placed on poly-lysine (PK)-coated surfaces that do not elicit strong integrin signaling (Fig. 3D–F, S2F–G, Movie 1). In addition, RAB21 and PIP5K1C90 were polarized towards the same locations in Fb stimulated neutrophils (Fig. 3G). Concordantly, there was increased colocalization of RAB21 and PIP5K1C90 in Fb-stimulated cells compared to cells on a PK-coated surface (Fig. 3G & S2C). Unlike Fb, which does not stimulate overtly polarized F-actin formation (Xu et al., 2010) (Fig. 3E), chemoattractants such as fMLP stimulate strong F-actin formation at the leading edge (Fig. S2E). Chemoattractant-stimulated leading edge F-actin serves as a useful reference marker for neutrophil polarity. In fMLP-treated neutrophils, PIP5K1C90 polarized to the uropods opposite to the leading-edge F-actin (Xu et al., 2010), and so did RAB21 (Fig. S2E). ICAM-1 is a more specific β2-integrin ligand expressed on the endothelial cell surfaces. We previously showed that it also could promote PIP5K1C90 polarization as Fb does (Xu et al., 2010). It stimulated polarization of RAB21 as well (data not shown). Moreover, the addition of chemoattractant CXCL2 to neutrophils seeded on an ICAM-1-coated surface, which is more closely relevant to the in vivo settings, resulted in RAB21 polarization to the uropods (Fig. S2D) as occurred under the Fb and fMLP stimulation. These results together demonstrate that PIP5K1C90 and RAB21 share the same polarity in polarized neutrophils. In contrast to RAB21, RAB5 showed a different localization pattern (Fig. S2E). Thus, we conclude that RAB21 is involved in PIP5K1C90 polarization in mouse neutrophils.
Figure 3. RAB21 is polarized and involved in PIP5K1C90 polarization in neutrophils.

A–C. RAB21DN mutant inhibits PIP5K1C90 polarization in mouse neutrophils. Mouse neutrophils were cotransfected with HA-PIP5K1C90 and GFP (A) or RAB21DN-GFP (B) and sorted by FACS. GFP-positive cells were stimulated by Fb for 30 min and stained by anti-HA and anti-EEA1 antibodies, followed by secondary antibodies conjugated with Alexa633 (colored in red) and Alexa488 (colored in green). Two representative cells are shown in A and B. C shows the quantification of the percentage of neutrophils showing PIP5K1C90 polarization from three independent experiments. More than 20 cells were examined in each experiment. Data are presented as means ± SD (*, p<0.01, Student’s t-Test). Fig S2A shows localization of PIP5K1C90 and EEA1 staining in representative cells placed on PK-coated surfaces as a control.
D–F. Polarization of RAB21 in mouse neutrophils upon Fb stimulation. Mouse neutrophils were placed on coverslips coated with PK (D) or Fb (E) for 30 min and stained by anti-RAB21 and Alexa-488-conjugated secondary antibody (green) in presence of Alexa647-phalloidin (red). F shows the quantification of the percentage of neutrophils showing RAB21 polarization from three independent experiments. More than 20 cells were examined in each experiment. Data are presented as means ± SD (*, p<0.01, Student’s t-Test).
G. Polarization of RAB21 and PIP5K1C90 in neutrophils. Mouse neutrophils transfected with HA-PIP5K1C90 were placed on Fb-coated coverslips for 30 min and stained by anti-HA and anti-RAB21 antibody followed by secondary antibodies conjugated with Alexa633 (colored in red) and Alexa488 (colored in green). Fig S2C shows localization of PIP5K1C90 and RAB21 staining in cells placed on PK-coated surfaces as a control.
H. PKN1-deficiency impairs RAB21 polarization. Mouse neutrophils were placed on Fb-coated coverslips for 30 min and fixed and stained by anti-RAB21 and anti-EEA1 antibodies, followed by secondary antibodies conjugated with Alexa633 (colored in red) and Alexa488 (colored in green). The quantification for percentage of neutrophils showing RAB21 polarization was done from three independent observations, and more than 20 cells were examined in each observation. Data are presented as means ± SD (*, p<0.01, Student’s t-Test).
Like PIP5K1C90 localization in PKN1-null neutrophils, PIP5K1C90 was also found in the vicinity of EEA1-positive endosomes with partial colocalization in neutrophils expressing RAB21DN (Fig. 3B). This result suggests that functional RAB21, like PKN1, is required for trafficking of PIP5K1C90-associated vesicles to their polarized destination from the endosomes. However, when a constitutively active form of RAB21 (Q76L) was expressed in neutrophils, PIP5K1C90 polarization was impaired as well, in contrast to normal PIP5K1C90 polarization in neutrophils expressing wildtype (WT) RAB21 (Fig. 3C). This result suggests that the GTP/GDP cycling may be important for RAB21 to regulate PIP5K1C90-associaed vesicles trafficking.
We next examined if PKN1 regulated RAB21 polarization and found that PKN1-deficiency inhibited RAB21 polarization (Fig. 3H). The localization of RAB21 in the vicinity of EEA1 endosomes in PKN1-null neutrophils (Fig. 3H) implies that PKN1 may regulate RAB21-associated vesicles trafficking away from the early endosomes for PIP5K1C90 polarization.
RPH3A interacts with RAB21 and regulates the polarization
To understand how RAB21 regulates PIP5K1C90 polarization, a search for RAB21 effectors was conducted. The recombinant protein of a constitutively active form (Q76L) of RAB21 fused to GST was used in a pull-down experiment with the cell extract of HEK293 cells, followed by mass spectrometry (MS) analysis. Among the proteins identified by MS (Table S2), Rabphilin 3A (RPH3A) (Fig. S3A) caught our attention for its role in vesicle trafficking in neurons (Gonzalez and Scheller, 1999; Sasaki et al., 1997) and importantly for being one of numerous potential substrates of PKN1 identified in a previous proteomic study (Amano et al., 2010). Given that PKN1-deficiency and RAB21DN acted similarly in impairment of PIP5K1C90 polarization, we suspected that RPH3A might be the link between RAB21 and PKN1. We went on to validate the direct interaction between RAB21 or RAB21-QL and RPH3A using purified recombinant proteins (Fig. 4A & S3B). RPH3A showed a preference for GTP-bound RAB21 or RAB21-QL in a Ca2+-dependent manner (Fig. 4A & S3B). Deletional mutagenesis revealed that RAB21 directly binds to the region that contains the C2 domains (Fig. 4B & S3C). The finding of the interaction of the C-terminal portion of RPH3A with RAB21 is rather unexpectedly, as the Zn-binding domain at RPH3A N-terminus was shown to interact with RAB3A and RAB27 previously (Fukuda et al., 2004; Gonzalez and Scheller, 1999).
Figure 4. RPH3A binds to RAB21.

A,B. RAB21 binds to RPH3A via RPH3A’s C2 domain in a Ca2+ and GTP-dependent manner. Purified recombinant proteins were used in a GST-pull down assay under the conditions described in the figure. Proteins were then detected by Western blot analysis. Zn, the Zinc binding domain; C2, the C2 domain.
The role of RPH3A polarization in neutrophils was then examined using a RPH3A-null mouse line (Schluter et al., 1999). RPH3A-deficiency significantly impeded polarization of PIP5K1C90 and RAB21 in mouse neutrophils (Fig. 5A, B). The lack of RPH3A also caused the localization of PIP5K1C90 and RAB21 in the vicinity of the EEA1-positve endosomes (Fig. 5A, B). These results indicate that RPH3A regulates the polarization of both PIP5K1C90 and RAB21 in mouse neutrophils. Supporting this conclusion, RPH3A-deficiency neutrophils showed phenotypes similar to those of PIP5K1C-null and PKN1-null neutrophils: RPH3A-null neutrophils showed significant reductions in binding to ICAM and adhesion to endothelial cells under flow in vitro, and infiltration to inflamed peritonea in mice (Fig. 5C–E). Moreover, RPH3A-deficient neutrophils showed significantly reduced adhesion to the WT endothelium of inflamed cremaster muscle venules in WT mice receiving transfer from Rph3a−/− bone marrow, compared to WT neutrophils in mice receiving transfer from WT bone marrow (Fig. 5F). In addition, RPH3A-deficient neutrophils showed reduced transmigration and increased rolling in the same intravital microscopic observations (Fig. 5G–H). These phenotypes are similar to those of PKN1-deficiency described in Fig. 1B–G and PIP5K1C-deficiency described previously (Xu et al., 2010), further confirming that these proteins function in the same pathway.
Figure 5. RPH3A is involved in PIP5K1C90 and RAB21 polarization.

A,B. RPH3A-deficiency impairs polarization of PIP5K1C90 and RAB21 in mouse neutrophils. Mouse neutrophils transfected with HA-PIP5K1C90 were placed on fibrinogen (Fb)-coated coverslips for 30 min and stained by anti-EEA1 and anti-HA (A) or anti-RAB21 (B) antibodies, followed by secondary antibodies conjugated with Alexa488 (colored in green) and Alexa633 (colored in red). The percentage of neutrophils showing polarization was quantified from three independent observations, and more than 15 cells were examined in each observation. Data are presented as means ± SD (**, p<0.01, Student’s t-Test).
C–E. RPH3A-deficiency impairs neutrophil binding to ICAM1 (C), adhesion to endothelial cells under shear flow (D), and infiltration into inflamed mouse peritonea (E). Data are presented as means ± SEM (*, p<0.05, **, p<0.01, Student’s t-Test, n>4)
F–H. Effect of RPH3A-deficiency on neutrophil-endothelial cell interaction in inflamed cremaster muscle venues. Adhesion (F), transmigration of neutrophils (G), and rolling flux (H), were determined after stimulation of the cremaster muscle with TNFα (0.5 µg) for 4h. All values are means of n = 3 animals (with 5 to 7 vessels per animal) ± SEM. (**, p<0.01, Student’s t-Test).
In addition to regulating integrin mediated neutrophil firm adhesion during infiltration, PIP5K1C90 was reported to regulate myosin II-mediated uropod retraction during chemotaxis in differentiated HL-60 cells (Lokuta et al., 2007). This might be another mechanism accounting for our neutrophil recruitment defect in PKN1-deficient or RPH3A-deficient mice. However, we did not observe obvious neutrophil retraction defect in our intravital microscopy examination. We also performed in vitro transendothelial cell migration assay using transwells covered with mouse endothelial cells. No significant differences were observed between wildtype and PKN1-null or RPH3A-null neutrophils (Fig. S3D). Thus, it is very unlikely that PKN1- or RPH3A-deficiency causes strong uropod retraction defects that would affect neutrophil transendothelial migration.
PKN1-mediated phosphorylation of RPH3A regulates the polarization
We next investigated whether RPH3A is an actual substrate for PKN1. We found that PKN1 phosphorylated RPH3A in an in vitro kinase assay, and the phosphorylation could be detected by an anti-phospho-AKT substrate motif antibody (Fig. 6A). Both PKN1 and AKT belong to the AGC protein kinase superfamily, and some of these kinases share similar substrate specificity. We mapped RPH3A residues phosphorylated by PKN1 by mutating the Ser or Thr residues within each of the putative AKT substrate motifs (RXXS/T) found in RPH3A. When RPH3A residues Ser-234 and 271 were both mutated, the protein could no longer be phosphorylated by PKN1 in an in vitro kinase assay (Fig. 6B). Of note, residue Ser-234 was actually recognized by the anti-AKT substrate motif antibody because when it was mutated the protein could not be detected by the antibody in Western blot analysis (data not shown). Furthermore, an antibody specific to phosphorylated Ser-234 of RPH3A failed to detect RPH3A in PKN1-deficienct neutrophils (Fig. 6C), suggesting that PKN1 is the key kinase that phosphorylates RPH3A in neutrophils.
Figure 6. PKN1-mediated phosphorylation of RPH3A is required for polarization.

A. PKN1 phosphorylates RPH3A. Phosphorylation of recombinant RPH3A protein by recombinant PKN1 protein were performed in an in vitro kinase assay, and RPH3A phosphorylation was detected by Western blot analysis using an anti-phospho-AKT substrate motif antibody.
B. RPH3A residues S234 and S271 are phosphorylated by PKN1. WT RPH3A and its mutants SA1 (S234A) and SA (S234, 271A) were subjected to an in vitro kinase assay using [32P]ATP. Phosphorylated RPH3A ([32P]RPH3A) was detected by a phosphoimager, while the inputs were detected by Western blot analysis.
C. RPH3A phosphorylation at Ser 234 depends on PKN1. WT and PKN1-null neutrophils were analyzed by Western blot analysis using an anti-phospho-Ser234 RPH3A, anti-RPH3A, PKN1 or actin antibody.
D,E. WT RPH3A, but not its phosphorylation-defective mutant, can rescue RAB21 polarization in RPH3A-null cells. RPH3A-null neutrophils were transfected with RPH3A-HA (D) or RPH3A-SA-HA (E). The cells were then stained with an anti-HA antibody and anti-RAB21 antibody. Only anti-HA staining positive cells were examined.
F. Phosphorylated RPH3A shows enhanced binding to RAB21. Purified RPH3A protein was first incubated with PKN1 in the presence or absence of ATP in a kinase assay buffer for 30 min at 37 °C. Phosphorylated RPH3A (pRPH3A) is evidenced by its upshift in the blot compared to unphosphorylated RPH3A (RPH3A). These RPH3A proteins were then subjected to a pulldown assay with GTPγS-bound RAB21. The proteins were detected by Western blot analysis.
We next tested the importance of RPH3A phosphorylation in neutrophil polarization. We found that expression of WT RPH3A in RPH3A-null neutrophils could rescue RAB21 polarization in response to Fb stimulation (Fig. 6D). In addition, RPH3A was also polarized (Fig. S4A, B, Movie 2) together with RAB21 (Fig. 6D) and PIP5K1C90 (Fig. S4C), and overexpressing RAB21DN can impair the polarization of RPH3A (Fig. S4D, E). However, the RPH3A mutant with the phosphorylation sites mutated failed to polarize or rescue RAB21 polarization in RPH3A-null neutrophils upon Fb stimulation (Fig. 6E). Instead, the mutant protein is localized in the vicinity of RAB21 (Fig. 6E) in these RPH3A-null neutrophils, suggesting that PKN1-mediated phosphorylation may be required for polarized trafficking of RAB21 and its associated cargo PIP5K1C90. To understand why the phosphorylation is important, we examined the effect of the phosphorylation on RAB21 and RPH3A interaction. RPH3A that was pre-phosphorylated by PKN1 showed enhanced interaction with RAB21 compared to non-phosphorylated RPH3A in the absence of calcium (Fig. 6F). We have tested and found that phosphorylated RPH3A can bypass the need for calcium to interact with RAB21 (data not shown). We also examined localization of PKN1 in neutrophils and found that it showed polarized localization together with RPH3A in stimulated WT neutrophils (Fig. S4F). Similar to RAB21, RPH3A, and PIP5K1C90, PKN1 shows no polarization in RPH3A-null neutrophils (Fig. S4G) and is localized in the proximity to the phosphorylation-defective RPH3A mutant (Fig. S4G) and probably RAB21 by inference (Fig. 6E). We have previously shown that β2-integrin exhibited polarized colocalization with PIP5K1C90 in Fb-stimulated neutrophils (Xu et al., 2010), but in PKN1 or RPH3A-deficient neutrophils β2-integrin polarization was also impaired (Fig. S4H). These results together suggest a model as depicted in Fig. 7A; PKN1-mediated phosphorylation of RPH3A facilitates the recruitment of RAPH3A by RAB21, which is required for polarized transport of the vesicles and their associated cargos from endosomes to their polarized destinations in neutrophils. Disruption of any step of this mechanism, either by expression of a DN RAB21, genetic inactivation of PKN1 or RPH3A, or substitution of a phosphorylation-defective mutant for WT RPH3A would halt the polarized trafficking process and cause localization of these proteins at or near the endosomes. In addition, this pathway is independent of chemoattractant-induced neutrophil polarization epitomized by leading edge F-actin polarization as neither PKN1 nor RPH3A deficiency affects fMLP-induced F-actin polarization (Fig. S4I).
Figure 7. PKN1-deficiency attenuates renal ischemia-reperfusion injury.

A. Schematic presentation of a model for RPH3A and its phosphorylation to regulate directional vesicle trafficking and PIP5K1C90 polarization. Integrin engagement triggers endocytosis of PIP5K1C90, which traffics through EEA1-positive endosomes and emerges in RAB21-positive recycling vesicles. GTP-bound RAB21 exits from the endosomes and recruits PKN1-phosphorylated RHP3A. This process is required for polarized RAB21 vesicle trafficking and PIP5K1C90 polarization.
B,C. Loss of PKN1 reduces renal neutrophil infiltration. Neutrophil infiltration into reperfused kidneys was evaluated by flow cytometry after dissociated kidney cells were stained with CD11b and Ly6G (B) and by immunostaining of renal tissue sections with anti-Ly6B (C). Representative flowcytometric charts are shown in Fig. S5. Data in B are presented as means ± SEM (**, p<0.01, Student’s t-Test, n=5).
D. Blood creatinine contents from mice that were subjected to the renal ischemia-reperfusion procedure. Data are presented as means ± SEM (**, p<0.01, Student’s t-Test, n=5).
E. Histological examination of renal tissue injury. Renal histological sections from mice that were subjected to the renal ischemia-reperfusion procedure were stained with Periodic acid-Schiff (PAS) dye, and the representative sections are shown.
PKN1-deficiency ameliorates renal ischemia-reperfusion injury
Neutrophils play important roles in reperfusion-induced renal tissue damage (Heinzelmann et al., 1999). Thus, we examined the effects of myeloid-specific PKN1 deficiency on neutrophil recruitment and tissue damage using the renal ischemia-reperfusion injury model in an attempt to provide additional in vivo validation of the PIP5K1C90 polarization pathway we characterized in this study. Reperfusion after renal arterial ischemia resulted in marked infiltration of neutrophils detected by flow cytometry (Fig. 7B & S5) and immunohistostaining (Fig. 7C) of kidney samples. Accompanied by neutrophil infiltration, there was renal damage evidenced by the increases in serum creatinine content (Fig. 7D) and the number of coagulative necrotic renal tubules (Fig. 7E). Both neutrophil infiltration and renal damage were significantly reduced in Pkn1m/m mice subjected to the same ischemia-reperfusion procedure (Fig. 7B–E & S5). Thus, PKN1 in myeloid cells plays an important role in renal ischemia-reperfusion injury.
DISCUSSION
In this study, we characterized a phosphorylation-regulated polarization mechanism in mouse neutrophils. This mechanism involves polarization of RAB21 most likely through RAB21-associated vesicles in a manner depending on PKN1-mediated phosphorylation of a newly identified RAB21 effector RPH3A. This mechanism is important for integrin activation, endothelial cell adhesion, and neutrophil infiltration in inflammation models.
We previously showed that β2 integrin activation triggers polarization of PIP5K1C90 in neutrophils via vesicle transport (Xu et al., 2010). In this study, we show that PKN1-phosphorylation-regulated RAB21 polarization underlies PIP5K1C90 polarization in neutrophils. These data together allow us to propose a model as depicted in Fig. 7A, in which integrin engagement triggers endocytosis of PIP5K1C90, a plasma membrane-associated protein, which interacts with talin and endocytic structural protein adaptin-2 (AP2) (Di Paolo et al., 2002; Lee et al., 2005; Ling et al., 2002; Thieman et al., 2009; Xu et al., 2010). PIP5K1C90 likely traffics through EEA1-positive endosomes and emerges largely in RAB21-positive recycling vesicles. GTP-bound RAB21 exits from the endosomes (Jean et al., 2012) and preferentially recruits phosphorylated RHP3A as PKN1-mediated RPH3A phosphorylation enhances RPH3A interaction with RAB21. We believe that the interaction has an important role in transport of RAB21-vesicles carrying PIP5K1C90 from endosomes to its polarized destination on the plasma membrane.
While RAB21 and PKN1-mediated phosphorylation of RPH3A are involved in trafficking of PIP5K1C90 away from the endosomes, additional mechanisms are likely to be needed for PIP5K1C90 to reach its final destination on plasma membranes. One reason for this assertion is that expression of the constitutively active mutant of RAB21, like the RAB21DN mutant, impaired PIP5k1C90 polarization in contrast to the expression of the WT RAB21 (Fig. 3C), suggesting that GTP/GDP cycling is required for the polarization. In addition, the phosphor-mimetic mutant of RPH3A, unlike wildtype RPH3A, failed to rescue the polarization defect of RPH3A-deficient neutrophils and it cannot rescue the polarization defect of PKN1-deficient neutrophils, either (data not shown). These suggest a possibility that subsequent dephosphorylation of RPH3A may be needed for the vesicles targeted to their final destination. Moreover, in order for PIP5K1C90 to be targeted to its polarized final destination on the plasma membranes, something on the plasma membrane has to mark the destination. Future studies will address these questions.
PIP5K1C90 promotes RHOA activation and increases integrin affinity and neutrophil adherence to endothelial cells (Xu et al., 2010). Polarized PIP5K1C90 leads to polarized activation of RHOA, thus increasing the local active RHOA concentration that would result in greater integrin activation and stronger adhesion, which is needed by the inflamed endothelium to capture and hold on neutrophils under the shear flow of blood stream. Given that PKN1 and RPH3A-deficiency shows neutrophil phenotypes, including reduced integrin binding, endothelium adhesion, and neutrophil infiltration, consistent with PIP5K1C-deficiency or loss of PIP5K1C90 polarization (Xu et al., 2010), PKN1 and RPH3A-mediated PIP5K1C90 polarization may largely be accounted for these neutrophil phenotypes. RAB21 is also known to be involved in integrin recycling (Alanko et al., 2015; Mai et al., 2011; Pellinen et al., 2006; Pellinen et al., 2008; Stenmark, 2009). We previously showed that β2-integrin also showed posterior polarization in activated neutrophils (Xu et al., 2010). The localization of β2-integrin is more diffused than PIP5K1C90, and there is partial co-localization with PIP5K1C90 (Xu et al., 2010). Thus, there is a possibility that the directional vesicle trafficking mechanism characterized in this study may also have a role in integrin recycling. This will be further investigated in the future.
Ischemia-reperfusion injury occurs following myocardial infarction, transplantation, stroke, and trauma. It can lead to organ failure and remains the major cause of death in critically ill patients. There is a lack of effective therapeutic strategies for combating Ischemia-reperfusion injury. Neutrophils are the primary responders following ischemia and reperfusion, and their infiltration is positively correlated to the inflammatory reaction and injury severity associated with Ischemia-reperfusion. In this study, we show that PKN1-deficiency significantly ameliorates tissue injury in a renal ischemia-reperfusion model. PKN1 is a serine/threonine protein kinase, which is a druggable target. In addition, global knockout of PKN1 shows no developmental or gross phenotypes (Quetier et al., 2016; Yasui et al., 2012)(data not shown), suggesting low likelihood of severe on-target side effects from its inhibition. Thus, PKN1 may be a potential therapeutic target for combating ischemia-reperfusion injury. However, because mice lacking its close homolog PKN2 are embryonically lethal (Quetier et al., 2016), specific inhibitors of PKN1 may be needed.
EXPERIMENTAL PROCEDURES
Reagents and constructs
Antibodies to the following antigens were acquired commercially: GST (2624, Cell Signaling), His (2366, Cell Signaling), HA (MMS-101R, Covance), RAB21 (R4405, Sigma), RAB5 (D-11, Santa Cruz), EEA1 (612006, BD), p-S234-RPH3A (Thermo Fisher, PA1-4693), RPH3A (Aviva System Biology, ARP59498_P050), and PIP5K1C90 ([Di Paolo et al., 2002] kindly provided by Dr. Pietro De Camilli). Protein A/G-PLUS-agarose beads were acquired from Santa Cruz Biotechnology (Santa Cruz, CA). Formyl-Met-Leu-Phe (fMLP) was purchased from Sigma (St. Louis, MO). The cDNAs for RAB21, and RPH3A, were acquired from Open Biosystems (Lafayette, CO).
Mice
The floxed PKN1 mice were generated using a gene targeting construct as depicted in Fig. S1A. The construct was transfected in the ES cells and cells in which desirable homologous recombination has occurred were selected and used for blastocyst injection to produce chimeric mice, which were backcrossed into C57BL background for at least 5 generations. The floxed mice were then crossed with the B6.129-Lyzstm1(cre)Ifo/J mice from Jackson Lab. The floxed PKN1 mice is available at the Jackson Lab now. The RPH3A-null mice were obtained from the Jackson Lab. Wildtype C57BL mice were purchased from Jackson Lab and used for neutrophil isolation. All animal studies were approved by the institutional animal care and use committees of Yale University.
Neutrophil preparation and transfection
Murine neutrophils were purified from bone marrows as previously described (Zhang et al., 2010). Briefly, bone marrow cells collected from mice (8–10 weeks old) were treated with the ACK buffer (155 mM NH4Cl, 10 mM KHCO3 and 127 µM EDTA) for red blood cell lysis, followed by a discontinuous Percoll density gradient centrifugation. Neutrophils were collected from the band located between 81% and 62% of Percoll. For transient transfection of primary mouse neutrophils, three million neutrophils were electroporated with 1.6 µg endotoxin-free plasmids or 300 nM of siRNA using the human monocyte nucleofection kit (Lonza, Switzerland) with an Amaxa electroporation system. The cells were then cultured for overnight in the medium supplied with the kit containing 10% FBS and 25 ng/ml recombinant GM-CSF (PeproTech, Rocky Hill, NJ). Cell sorting was done by a FACS Aria sorter (BD, San Jose, CA). We have used this method to overexpress protein in primary neutrophils for many years and electroporated neutrophils have good response to chemoattractant stimulation (Gao et al., 2015; Tang et al., 2011; Xu et al., 2010; Zhang et al., 2013). And this method is also widely used by other groups (Loison et al., 2014; Magalhaes et al., 2007; Sun et al., 2007).
Immunostaining and observation of neutrophils
Neutrophils were fixed with 4% Para-formaldehyde for 10 min and permeabilized with 0.01% saponin for 5 min and blocked with 2% BSA in PBS for 1hr. Cells were then incubated with 1:100 diluted primary antibodies in blocking buffer at 4°C for overnight. Next day, secondary antibodies with conjugated fluorescent probes (Alexa488 colored in green and Alexa633 colored in red in the figures) were 1:200 in blocking buffer and incubated with cells for 1hr at room temperature. Mounted slides were observed using a Leica SP5 confocal microscopy. Because the fluorescence of the GFP protein expressed in neutrophils quenches after the fixation, permeabilization, overnight primary antibody staining, and repeated washes, the presence of GFP does not interfere with Alexa488 signals.
For quantification of colocalization of Rab21 with PIP5K1C-90, Z stack images of consecutive optical planes spaced by 0.2 µm were acquired to cover the whole cell volume using confocal microscopy. Pearson’s coefficient was determined using Imaris 7.2.3. For quantification of polarized distribution, target proteins were stained with appropriate antibodies and imaged under Leica SP5 confocal microscopy. Cell peripheries were traced using the ImageJ Segment line tool with a width of 5 (examples are shown in Fig. S6). Florescence intensities were plotted along the lines (Fig. S6). The ratios of florescence intensities of 25% cell periphery surrounding the highest florescence intensity to total florescence intensities were calculated (see examples in Fig. S6). We arbitrarily take cells with the ratios over 0.5 as being polarized and below 0.5 as being non-polarized. In other word, if the florescence intensity in 25% of the cell periphery is accounted more than 50% of the total florescence intensity of the cell periphery, we call the cell polarized.
Preparation and purification of recombinant proteins and pulldown assay
Full-length RPH3A and its mutants and segments as well as RAB21 were expressed in E coli and purified by affinity purification. For the pulldown assays, recombinant GST-GTPase RAB21 protein (0.2 mg/ml in 10 µl dissolved in Buffer A containing 10 mM phosphate buffer pH 7.4, 10% glycerol, and 0.2 mM EDTA) was added with 1 µl of 10 mM GDP or GTPγS stock (dissolved in 50 mM Tris-HCl, pH 7.8) for a final concentration of 1 mM guanine nucleotides and incubated on ice for 15 min. Then, RPH3A (0.2 mg/ml in 10 µl dissolved in Buffer A) and 180 µl Buffer B (10 mM phosphate buffer pH 7.4, 10% glycerol, 1% Triton X-100, 0.1% SDS, 3 mM DTT, protease inhibitor cocktail, 10 mM MgCl2, 0.1 mM GTPγS or GDP, with or without 0.1 mM CaCl2) were added to RAB21 protein and incubated at 4 °C on a rotating platform for overnight. The protein mixture was then added to glutathione beads pre-blocked with 1% BSA. After the beads were washed 6 times with Buffer B, proteins bound to the beads were analyzed by Western blotting.
Neutrophil infiltration into inflamed peritonea and flow chamber adhesion assay
For the peritonitis infiltration model, purified wild type and mutant neutrophils were labeled with 2.5 µM CFSE [5-(and −6)-carboxyfluorescein diacetate succinimidyl esters] and 2.5 µM Far-Red DDAO SE, respectively, and vice versa. The WT and mutant cells with different fluorescence labels were mixed at a 1:1 ratio and injected into retro-orbital venous sinus of wildtype littermates (8–10 weeks old, female), which were injected with 2ml of 3% Thioglycolate (TG) two hours earlier. The mice were euthanized one and half hour later. Cells in their peritonea were collected and analyzed by cell counting and flow cytometry. The data presented are the combination of the experiments with reciprocal fluorescence labeling.
To examine neutrophil adherence to endothelial cells under shear stress, mouse endothelial cells (Wang et al., 2008) were cultured to confluency on 10µg/ml fibronectin coated coverslips and treated with 50 ng/ml TNFα for 4 hours. The coverslips containing the endothelial cell layer were washed with PBS and placed in a flow chamber apparatus (GlycoTech). The WT and mutant cells labeled different fluorescence labels as described above were mixed at a 1:1 ratio and flowed into the chamber at a shear flow rate of 1 dyn/cm2. The adherent cells were then examined and counted under a fluorescence microscope.
ICAM-binding assay
The assay was carried out as previously described (Konstandin et al., 2006). The ICAM-1-Fc-F(ab’)2 complexes was generated by incubating Cy5-conjugated AffiniPure goat anti-human Fcγ fragment-specific IgG F(ab')2 fragments (Jackson Immunobiology) and ICAM-1-Fc (100 µg/ml, R&D) at 4 °C for 30 min in PBS. Neutrophils, which were resuspended at 0.5 × 106 cells/ml in PBS containing 0.5% BSA, 0.5 mM Mg2+ and 0.9 mM Ca2+, were mixed with the ICAM-1-Fc-F(ab’)2 complexes in the presence or absence of fMLP for durations specified in the figure legends. The reactions were terminated by adding 4% paraformaldehyde. After 5 min, fixation was stopped by adding 3-ml ice-cold FACS buffer. Cells were pelleted, resuspended in 300 µl of FACS buffer, and analyzed on a flow cytometer.
Bone marrow transplantation and intravital microscopy
Bone marrows from RPH3A-deficient mice and their littermates were transplanted into wildtype recipient mice (C57/BL, 8 weeks old, male) that had been subjected to 1000cGy X-Ray irradiation. Eight weeks later, the transplanted mice were used for intravital observation.
For intravital observations, mice were injected intrascrotally with recombinant mouse TNFα (0.5µg; R&D Systems; MN, USA) in 200 µl of saline for 4h prior to recording. To visualize neutrophils, rat monoclonal antibody RB6-8C5 against mouse Ly-6G (Gr-1) conjugated to Alexa 647 (2µg in 200ul saline; eBioscience San Diego, CA, USA) was injected intravenously into the tail vein after TNFα stimulation. A mixture of 10 mg/kg xylazine (Bayer Inc., Animal Health, Toronto, ON, Canada) and 200 mg/kg ketamine hydrochloride (Rogar/STB Inc., Montreal, QC, Canada) was injected intraperitoneally to anesthetize mice. In all protocols, the left jugular vein was cannulated to administer additional anesthetics or antibodies. To visualize the vasculature of the cremaster muscle, mice were injected with 50µg rat mAb against mouse PECAM-1 [eBioscience San Diego, CA, USA Clone 390, which has been previously reported not to interfere with leukocyte recruitment (Christofidou-Solomidou et al., 1997; Tasaka et al., 2003)] conjugated to Alexa 594 (Invitrogen, Eugene, OR, USA). The mouse cremaster muscle was used to study neutrophil recruitment as previously described (Liu et al., 2005) with the exception that neutrophil extravasation was visualized with spinning-disk confocal microscopy using a 20x water/0.95 immersion objective to focus the excitation onto the sample (Olympus, Center Valley, PA, USA). A 512×512 pixel back-thinned electron-multiplying charge-coupled device camera (C9100-13, Hamamatsu) was used for fluorescence detection. Volocity software (Improvision) was used to acquire and analyze images. A neutrophil was considered to be adherent if it remained stationary for more than 30 s, and total leukocyte adhesion was quantified as the number of adherent cells within a 100 µm length of venule during 5 min. Leukocyte emigration was defined as the number of cells in the extravascular space within a 200×300 µm area (0.06 mm2) adjacent to the observed venule and was normalized against the number of adherent cells.
In vitro chemotaxis assay in a Dunn chamber
The chemotaxis assay using a Dunn chamber was carried out as previously described (Zicha et al., 1997) with some modifications as detailed in (Zhang et al., 2010). We analyzed wildtype and mutant neutrophils simultaneously by labeling the cells with different tracing dyes. We alternated the labeled group in the study to completely eliminate the possibility of any influence from the dye. Time-lapse image series were acquired at 30-second intervals for 30 mins and were analyzed using the MetaMorph image analysis software as described in (Zhang et al., 2010). We normally obtain two parameters to quantify neutrophil chemotaxis: average directional errors and motility. The average directional error measures the angle between the cell migration direction and the gradient direction and reflects how well a cell follows the gradient. Motility is cell migration speed.
Transendothelial migration assay
Mouse endothelial cells (Wang et al., 2008) were seeded into the upper well of a 24-well Boyden chamber until confluent, followed by incubation with 10ng/ml TNF-α for 4h. The WT or mutant neutrophils were plated in the upper well and medium with or without 5µM fMLP was added to the lower chamber. Neutrophils that migrate across the endothelial cells were recovered from the lower chamber and quantified using ATPlite system (PerkinElmer).
Murine kidney ischemia/reperfusion models
The left and right renal arteries of mutant and their littermate control mice (9–10 weeks old, male) were clamped for 30 min using vascular clamps (Fine Science Tools, CA) through a midline abdominal incision under general anesthesia. After ischemia, the clamps were removed, the wounds were sutured, and the animals were allowed to recover for 24 h before euthanization. Blood was collected by cardiac puncture in heparin-containing tube, and creatinine levels were determined using the DetectX serum creatinine kit (Arbor Assays, Ann Arbor, MI). Kidneys were dissected from mice. One kidney of a mouse was fixed in 4% paraformaldehyde, embedded in paraffin, and stained for the Periodic acid-Schiff (PAS) dye and a Ly6B antibody. The other kidney from the same mouse were digested with Collagenase I (300 U) and analyzed by flow cytometry.
Statistical Analysis
Statistical analyses were performed using a two-tailed Student’s t test. The results are indicated as mean ± SD or mean ± SEM. A p<0.05 was considered statistically significant.
Supplementary Material
Movie 1: Three-dimensional visualization of Rab21 polarization, related to Figure S2G.
Movie 2: Three dimensional visualization of RPH3A polarization, related to Figure S4B.
Acknowledgments
We thank Michelle Orsulak for technical assistance and the Microsurgery Core of Yale Cardiovascular Research Center for performing renal ischemic procedures. The work is supported by NIH grants to D.W. (HL108430 and HL120465), AHA grant to W.T. (14SDG20490020), Yale/NIDA Neuroproteomics Center (DA018343) and the Snyder Mouse Phenomics Resources Laboratory and Live Cell Imaging Facility, both of which were funded by the Snyder Institute for Chronic Diseases at the University of Calgary.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions: D.W. and W.T. supervised the project and developed the concepts. D.W., W.T., Q.Y., C.R., B.P. and P.K. designed the experiments. W.T., Q.Y., C.R., W.X., B.P., J.Z. and Y.Z. performed the experiments. D.W., W.T., Q.Y., C.R. and B.P. analyzed the data. D.W. and W.T. wrote the manuscript. All the authors contributed to the manuscript and approved the final version.
References
- Abram CL, Lowell CA. Convergence of immunoreceptor and integrin signaling. Immunol Rev. 2007;218:29–44. doi: 10.1111/j.1600-065X.2007.00531.x. [DOI] [PubMed] [Google Scholar]
- Afonso PV, Parent CA. PI3K and chemotaxis: a priming issue? Science signaling. 2011;4:e22. doi: 10.1126/scisignal.2002019. [DOI] [PubMed] [Google Scholar]
- Alanko J, Mai A, Jacquemet G, Schauer K, Kaukonen R, Saari M, Goud B, Ivaska J. Integrin endosomal signalling suppresses anoikis. Nat Cell Biol. 2015;17:1412–1421. doi: 10.1038/ncb3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano M, Tsumura Y, Taki K, Harada H, Mori K, Nishioka T, Kato K, Suzuki T, Nishioka Y, Iwamatsu A, et al. A proteomic approach for comprehensively screening substrates of protein kinases such as Rho-kinase. PloS one. 2010;5:e8704. doi: 10.1371/journal.pone.0008704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear JE, Haugh JM. Directed migration of mesenchymal cells: where signaling and the cytoskeleton meet. Current opinion in cell biology. 2014;30:74–82. doi: 10.1016/j.ceb.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll KS, Hanna J, Simon I, Krise J, Barbero P, Pfeffer SR. Role of Rab9 GTPase in facilitating receptor recruitment by TIP47. Science. 2001;292:1373–1376. doi: 10.1126/science.1056791. [DOI] [PubMed] [Google Scholar]
- Caswell PT, Vadrevu S, Norman JC. Integrins: masters and slaves of endocytic transport. Nature reviews Molecular cell biology. 2009;10:843–853. doi: 10.1038/nrm2799. [DOI] [PubMed] [Google Scholar]
- Christofidou-Solomidou M, Nakada MT, Williams J, Muller WA, DeLisser HM. Neutrophil platelet endothelial cell adhesion molecule-1 participates in neutrophil recruitment at inflammatory sites and is down-regulated after leukocyte extravasation. J Immunol. 1997;158:4872–4878. [PubMed] [Google Scholar]
- Di Paolo G, Pellegrini L, Letinic K, Cestra G, Zoncu R, Voronov S, Chang S, Guo J, Wenk MR, De Camilli P. Recruitment and regulation of phosphatidylinositol phosphate kinase type 1 gamma by the FERM domain of talin. Nature. 2002;420:85–89. doi: 10.1038/nature01147. [DOI] [PubMed] [Google Scholar]
- Fletcher SJ, Rappoport JZ. Moving forward: polarised trafficking in cell migration. Trends in cell biology. 2010;20:71–78. doi: 10.1016/j.tcb.2009.11.006. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Kanno E, Yamamoto A. Rabphilin and Noc2 are recruited to dense-core vesicles through specific interaction with Rab27A in PC12 cells. J Biol Chem. 2004;279:13065–13075. doi: 10.1074/jbc.M306812200. [DOI] [PubMed] [Google Scholar]
- Gao K, Tang W, Li Y, Zhang P, Wang D, Yu L, Wang C, Wu D. Front-signal-dependent accumulation of the RHOA inhibitor FAM65B at leading edges polarizes neutrophils. J Cell Sci. 2015;128:992–1000. doi: 10.1242/jcs.161497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt T, Ley K. Monocyte trafficking across the vessel wall. Cardiovasc Res. 2015 doi: 10.1093/cvr/cvv147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez L, Jr, Scheller RH. Regulation of membrane trafficking: structural insights from a Rab/effector complex. Cell. 1999;96:755–758. doi: 10.1016/s0092-8674(00)80585-1. [DOI] [PubMed] [Google Scholar]
- Graziano BR, Weiner OD. Self-organization of protrusions and polarity during eukaryotic chemotaxis. Current opinion in cell biology. 2014;30C:60–67. doi: 10.1016/j.ceb.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Guo W. The exocyst complex in polarized exocytosis. Current opinion in cell biology. 2009;21:537–542. doi: 10.1016/j.ceb.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzelmann M, Mercer-Jones MA, Passmore JC. Neutrophils and renal failure. American journal of kidney diseases : the official journal of the National Kidney Foundation. 1999;34:384–399. doi: 10.1016/s0272-6386(99)70375-6. [DOI] [PubMed] [Google Scholar]
- Imhof BA, Aurrand-Lions M. Adhesion mechanisms regulating the migration of monocytes. Nature reviews Immunology. 2004;4:432–444. doi: 10.1038/nri1375. [DOI] [PubMed] [Google Scholar]
- Jean S, Cox S, Schmidt EJ, Robinson FL, Kiger A. Sbf/MTMR13 coordinates PI(3)P and Rab21 regulation in endocytic control of cellular remodeling. Molecular biology of the cell. 2012;23:2723–2740. doi: 10.1091/mbc.E12-05-0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimoto K, Shao D, Takagi H, Maceri G, Zablocki D, Mukai H, Ono Y, Sadoshima J. Hypotonic swelling-induced activation of PKN1 mediates cell survival in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2011;300:H191–200. doi: 10.1152/ajpheart.00232.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppi M, Simonsen A, Bremnes B, Vieira A, Callaghan J, Stenmark H, Olkkonen VM. The small GTPase Rab22 interacts with EEA1 and controls endosomal membrane trafficking. J Cell Sci. 2002;115:899–911. doi: 10.1242/jcs.115.5.899. [DOI] [PubMed] [Google Scholar]
- Klinkert K, Echard A. Rab35 GTPase: A Central Regulator of Phosphoinositides and F-actin in Endocytic Recycling and Beyond. Traffic. 2016;17:1063–1077. doi: 10.1111/tra.12422. [DOI] [PubMed] [Google Scholar]
- Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nature reviews Immunology. 2013;13:159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- Kolsch V, Charest PG, Firtel RA. The regulation of cell motility and chemotaxis by phospholipid signaling. J Cell Sci. 2008;121:551–559. doi: 10.1242/jcs.023333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstandin MH, Sester U, Klemke M, Weschenfelder T, Wabnitz GH, Samstag Y. A novel flow-cytometry-based assay for quantification of affinity and avidity changes of integrins. J Immunol Methods. 2006;310:67–77. doi: 10.1016/j.jim.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Lee SY, Voronov S, Letinic K, Nairn AC, Di Paolo G, De Camilli P. Regulation of the interaction between PIPKI gamma and talin by proline-directed protein kinases. J Cell Biol. 2005;168:789–799. doi: 10.1083/jcb.200409028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling K, Doughman RL, Firestone AJ, Bunce MW, Anderson RA. Type I gamma phosphatidylinositol phosphate kinase targets and regulates focal adhesions. Nature. 2002;420:89–93. doi: 10.1038/nature01082. [DOI] [PubMed] [Google Scholar]
- Liu L, Cara DC, Kaur J, Raharjo E, Mullaly SC, Jongstra-Bilen J, Jongstra J, Kubes P. LSP1 is an endothelial gatekeeper of leukocyte transendothelial migration. J Exp Med. 2005;201:409–418. doi: 10.1084/jem.20040830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loison F, Zhu H, Karatepe K, Kasorn A, Liu P, Ye K, Zhou J, Cao S, Gong H, Jenne DE, et al. Proteinase 3-dependent caspase-3 cleavage modulates neutrophil death and inflammation. The Journal of clinical investigation. 2014;124:4445–4458. doi: 10.1172/JCI76246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokuta MA, Senetar MA, Bennin DA, Nuzzi PA, Chan KT, Ott VL, Huttenlocher A. Type Igamma PIP kinase is a novel uropod component that regulates rear retraction during neutrophil chemotaxis. Molecular biology of the cell. 2007;18:5069–5080. doi: 10.1091/mbc.E07-05-0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Wilson JM. Rab14 specifies the apical membrane through Arf6-mediated regulation of lipid domains and Cdc42. Sci Rep. 2016;6:38249. doi: 10.1038/srep38249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magalhaes MA, Zhu F, Sarantis H, Gray-Owen SD, Ellen RP, Glogauer M. Expression and translocation of fluorescent-tagged p21-activated kinase-binding domain and PH domain of protein kinase B during murine neutrophil chemotaxis. J Leukoc Biol. 2007;82:559–566. doi: 10.1189/jlb.0207126. [DOI] [PubMed] [Google Scholar]
- Mai A, Veltel S, Pellinen T, Padzik A, Coffey E, Marjomaki V, Ivaska J. Competitive binding of Rab21 and p120RasGAP to integrins regulates receptor traffic and migration. J Cell Biol. 2011;194:291–306. doi: 10.1083/jcb.201012126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor H, Flynn P, Nobes CD, Hall A, Parker PJ. PRK1 is targeted to endosomes by the small GTPase, RhoB. J Biol Chem. 1998;273:4811–4814. doi: 10.1074/jbc.273.9.4811. [DOI] [PubMed] [Google Scholar]
- Millar AL, Pavios NJ, Xu J, Zheng MH. Rab3D: a regulator of exocytosis in non-neuronal cells. Histol Histopathol. 2002;17:929–936. doi: 10.14670/HH-17.929. [DOI] [PubMed] [Google Scholar]
- Mukai H. The structure and function of PKN, a protein kinase having a catalytic domain homologous to that of PKC. Journal of biochemistry. 2003;133:17–27. doi: 10.1093/jb/mvg019. [DOI] [PubMed] [Google Scholar]
- Mukherjee D, Sen A, Aguilar RC. RhoGTPase-binding proteins, the exocyst complex and polarized vesicle trafficking. Small GTPases. 2014;5:e28453. doi: 10.4161/sgtp.28453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauseef WM, Borregaard N. Neutrophils at work. Nature Immunology. 2014;15:602–611. doi: 10.1038/ni.2921. [DOI] [PubMed] [Google Scholar]
- Nourshargh S, Alon R. Leukocyte Migration into Inflamed Tissues. Immunity. 2014;41:694–707. doi: 10.1016/j.immuni.2014.10.008. [DOI] [PubMed] [Google Scholar]
- Orlando K, Guo W. Membrane organization and dynamics in cell polarity. Cold Spring Harb Perspect Biol. 2009;1:a001321. doi: 10.1101/cshperspect.a001321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz Sandoval C, Simmen T. Rab proteins of the endoplasmic reticulum: functions and interactors. Biochem Soc Trans. 2012;40:1426–1432. doi: 10.1042/BST20120158. [DOI] [PubMed] [Google Scholar]
- Pellinen T, Arjonen A, Vuoriluoto K, Kallio K, Fransen JA, Ivaska J. Small GTPase Rab21 regulates cell adhesion and controls endosomal traffic of beta1-integrins. J Cell Biol. 2006;173:767–780. doi: 10.1083/jcb.200509019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellinen T, Tuomi S, Arjonen A, Wolf M, Edgren H, Meyer H, Grosse R, Kitzing T, Rantala JK, Kallioniemi O, et al. Integrin trafficking regulated by Rab21 is necessary for cytokinesis. Developmental cell. 2008;15:371–385. doi: 10.1016/j.devcel.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Quetier I, Marshall JJ, Spencer-Dene B, Lachmann S, Casamassima A, Franco C, Escuin S, Worrall JT, Baskaran P, Rajeeve V, et al. Knockout of the PKN Family of Rho Effector Kinases Reveals a Non-redundant Role for PKN2 in Developmental Mesoderm Expansion. Cell Rep. 2016;14:440–448. doi: 10.1016/j.celrep.2015.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ. Life at the leading edge. Cell. 2011;145:1012–1022. doi: 10.1016/j.cell.2011.06.010. [DOI] [PubMed] [Google Scholar]
- Rose DM, Alon R, Ginsberg MH. Integrin modulation and signaling in leukocyte adhesion and migration. Immunol Rev. 2007;218:126–134. doi: 10.1111/j.1600-065X.2007.00536.x. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Shirataki H, Nakanishi H, Takai Y. Rab3A–rabphilin-3A system in neurotransmitter release. Advances in second messenger and phosphoprotein research. 1997;31:279–294. doi: 10.1016/s1040-7952(97)80025-0. [DOI] [PubMed] [Google Scholar]
- Schluter OM, Schnell E, Verhage M, Tzonopoulos T, Nicoll RA, Janz R, Malenka RC, Geppert M, Sudhof TC. Rabphilin knock-out mice reveal that rabphilin is not required for rab3 function in regulating neurotransmitter release. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1999;19:5834–5846. doi: 10.1523/JNEUROSCI.19-14-05834.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelef MA, Tauzin S, Huttenlocher A. Neutrophil migration: moving from zebrafish models to human autoimmunity. Immunol Rev. 2013;256:269–281. doi: 10.1111/imr.12124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JC, Griffiths G, Wessling-Resnick M, Fransen JA, Bennett H, Jones AT. A role for the small GTPase Rab21 in the early endocytic pathway. J Cell Sci. 2004;117:6297–6311. doi: 10.1242/jcs.01560. [DOI] [PubMed] [Google Scholar]
- Singh NK, Kundumani-Sridharan V, Kumar S, Verma SK, Kotla S, Mukai H, Heckle MR, Rao GN. Protein kinase N1 is a novel substrate of NFATc1-mediated cyclin D1-CDK6 activity and modulates vascular smooth muscle cell division and migration leading to inward blood vessel wall remodeling. J Biol Chem. 2012;287:36291–36304. doi: 10.1074/jbc.M112.361220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nature reviews Molecular cell biology. 2009;10:513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- Sun CX, Magalhaes MA, Glogauer M. Rac1 and Rac2 differentially regulate actin free barbed end formation downstream of the fMLP receptor. J Cell Biol. 2007;179:239–245. doi: 10.1083/jcb.200705122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaney KF, Huang CH, Devreotes PN. Eukaryotic chemotaxis: a network of signaling pathways controls motility, directional sensing, and polarity. Annu Rev Biophys. 2010;39:265–289. doi: 10.1146/annurev.biophys.093008.131228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Zhang Y, Xu W, Harden TK, Sondek J, Sun L, Li L, Wu D. A PLCbeta/PI3Kgamma-GSK3 signaling pathway regulates cofilin phosphatase slingshot2 and neutrophil polarization and chemotaxis. Developmental cell. 2011;21:1038–1050. doi: 10.1016/j.devcel.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaka S, Qin L, Saijo A, Albelda SM, DeLisser HM, Doerschuk CM. Platelet endothelial cell adhesion molecule-1 in neutrophil emigration during acute bacterial pneumonia in mice and rats. Am J Respir Crit Care Med. 2003;167:164–170. doi: 10.1164/rccm.2202011. [DOI] [PubMed] [Google Scholar]
- Thieman JR, Mishra SK, Ling K, Doray B, Anderson RA, Traub LM. Clathrin regulates the association of PIPKIgamma661 with the AP-2 adaptor beta2 appendage. J Biol Chem. 2009;284:13924–13939. doi: 10.1074/jbc.M901017200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich F, Heisenberg CP. Trafficking and cell migration. Traffic. 2009;10:811–818. doi: 10.1111/j.1600-0854.2009.00929.x. [DOI] [PubMed] [Google Scholar]
- Wang Z, Liu B, Wang P, Dong X, Fernandez-Hernando C, Li Z, Hla T, Claffey K, Smith JD, Wu D. Phospholipase C beta3 deficiency leads to macrophage hypersensitivity to apoptotic induction and reduction of atherosclerosis in mice. The Journal of clinical investigation. 2008;118:195–204. doi: 10.1172/JCI33139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weninger W, Biro M, Jain R. Leukocyte migration in the interstitial space of non-lymphoid organs. Nature reviews Immunology. 2014;14:232–246. doi: 10.1038/nri3641. [DOI] [PubMed] [Google Scholar]
- Williams MR, Azcutia V, Newton G, Alcaide P, Luscinskas FW. Emerging mechanisms of neutrophil recruitment across endothelium. Trends in immunology. 2011;32:461–469. doi: 10.1016/j.it.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Wang P, Petri B, Zhang Y, Tang W, Sun L, Kress H, Mann T, Shi Y, Kubes P, et al. Integrin-induced PIP5K1C kinase polarization regulates neutrophil polarization, directionality, and in vivo infiltration. Immunity. 2010;33:340–350. doi: 10.1016/j.immuni.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui T, Sakakibara-Yada K, Nishimura T, Morita K, Tada S, Mosialos G, Kieff E, Kikutani H. Protein kinase N1, a cell inhibitor of Akt kinase, has a central role in quality control of germinal center formation. Proc Natl Acad Sci U S A. 2012;109:21022–21027. doi: 10.1073/pnas.1218925110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Tang W, Jones MC, Xu W, Halene S, Wu D. Different roles of G protein subunits beta1 and beta2 in neutrophil function revealed by gene expression silencing in primary mouse neutrophils. J Biol Chem. 2010;285:24805–24814. doi: 10.1074/jbc.M110.142885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Tang W, Zhang H, Niu X, Xu Y, Zhang J, Gao K, Pan W, Boggon TJ, Toomre D, et al. A network of interactions enables CCM3 and STK24 to coordinate UNC13D–driven vesicle exocytosis in neutrophils. Developmental cell. 2013;27:215–226. doi: 10.1016/j.devcel.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zicha D, Dunn G, Jones G. Analyzing chemotaxis using the Dunn direct-viewing chamber. Methods Mol Biol. 1997;75:449–457. doi: 10.1385/0-89603-441-0:449. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie 1: Three-dimensional visualization of Rab21 polarization, related to Figure S2G.
Movie 2: Three dimensional visualization of RPH3A polarization, related to Figure S4B.