

Abstract
While limited drug loading continues to be problematic for chemotherapeutics formulated in nanoparticles, we found that we could take advantage of colloidal drug aggregation to achieve high loading when combined with polymeric excipients. We demonstrate this approach with two drugs, fulvestrant and pentyl-PABC doxazolidine (PPD; a prodrug of doxazolidine, which is a DNA cross-linking anthracycline), and two polymers, polysorbate 80 (UP80) and poly(d,l-lactide-co-2-methyl-2-carboxytrimethylene carbonate)-graft-poly(ethylene glycol) (PLAC–PEG; a custom-synthesized, self-assembling amphiphilic polymer). In both systems, drug-loaded nanoparticles had diameters < 200 nm and were stable for up to two days in buffered saline solution and for up to 24 h in serum-containing media at 37 °C. While colloidal drug aggregates alone are typically unstable in saline and serum-containing media, we attribute the colloid stability observed herein to the polymeric excipients and consequent decreased protein adsorption. We expect this strategy of polymer-stabilized colloidal drug aggregates to be broadly applicable in delivery formulations.
Keywords: drug delivery, self-assembly, colloids, polymers, solvent exchange
Graphical abstract
INTRODUCTION
The inefficient formulation of hydrophobic small molecule drugs continues to be a barrier between drug development and clinical use. Although excipients can solubilize drugs for in vivo delivery, the high excipient concentrations necessary are associated with dose-limiting adverse effects, such as hypersensitivity and hemolysis.1-3 While nanoparticle delivery systems have been developed to overcome this toxicity and to improve drug bioavailability and biodistribution,4-6 these strategies are themselves limited by low drug loading.7-11
To produce more drug-rich systems and to overcome the limitations of excipient toxicity, an alternative approach has emerged to exploit the intrinsic physicochemical properties of a drug directly in formulation. These formulations generally take advantage of the immiscibility of hydrophobic drugs in aqueous media, which results in self-aggregation to produce a particle core.12,13 More recently, coformulation strategies have been developed that use macromolecules, either during or after particle formation, to suppress Ostwald ripening through stabilization of the drug–particle surface.14 However, less attention has been given to the potential self-assembly parameters of the drugs themselves.
Over the past decade, many drugs have been shown to self-assemble into colloidal drug aggregates. In early drug discovery,15 this leads to artifacts including both false positives in biochemical assays16,17 and false negatives in cellular assays.18,19 Although it is hard to predict,20 the mechanism of self-assembly for these colloidal aggregators is governed by a critical aggregation concentration (CAC). At concentrations above their CAC, self-assembly of these compounds by solvent-exchange methods leads to the generation of amorphous liquid–liquid phase-separated particles rather than crystalline precipitate.15,21 While the assembling properties have been well-studied, the utility of these aggregates is hindered by their instability.19,22 We and others have attempted to stabilize colloidal drug aggregates to further study their biological implications.14,23 Previously, we demonstrated that coaggregation with azo-dyes can stabilize colloids, resulting in the maintenance of structural integrity in high ionic strength solutions and serum-containing media.23 The incorporation of polymeric excipients, such as pluronics and polysorbates, remains an attractive method to stabilize colloidal aggregates due to the chemical diversity of polymers available and their ubiquity in pharmaceutical formulations. Work by Taylor et al. has shown that polymeric excipients can modulate the colloidal properties of drug aggregates; however, only modest improvements in stability have been achieved thus far.24,25
Here, we investigate how small molecule colloidal drug aggregation properties can be combined with polymeric excipients to substantially improve particle stability. Using pharmaceutical excipients and biocompatible amphiphilic polymers, we demonstrate that colloidal drug aggregates can be formulated for multi-day stability in both buffered saline and serum-containing media. With this strategy, we not only stabilize colloidal drug aggregates but also overcome the low drug loading typically found in traditional polymeric nano-particle systems. Monodisperse and stable colloidal formulations are achieved using polymeric excipients of two chemotherapeutics: the estrogen receptor antagonist fulvestrant19,26 and the novel anthracycline-derived prodrug of doxazolidine, pentyloxycarbonyl-(p-aminobenzyl)doxazolidinylcarbamate (PPD).27 After screening a series of polymers, we found that the optimal polymer–colloid combination is specific to each drug; however, this approach should be broadly applicable to other colloidal drug aggregators. As a proof of concept for use in drug delivery, we investigate the stability in serum-containing media, variations in protein adsorption properties, and interactions with cancer cells of these colloidal formulations.
MATERIALS AND METHODS
Materials
PPD was synthesized from expired clinical samples of doxorubicin (FeRx Inc., Aurora, CO) as previously described.27 Fulvestrant was purchased from Selleckchem. Poly(d,l-lactide-co-2-methyl-2-carboxy-trimethylene carbonate)-graft-poly(ethylene glycol) (PLAC–PEG) was synthesized by ring-opening polymerization using a pyrenebutanol initiator to a molecular weight of 12 000 g/mol and conjugated with an average of three PEG chains/backbone (10 000 g/mol PEG) as previously described.28 Polysorbate 80 (H2X, UP80) was purchased from NOF America Corporation. Vitamin E-PEG 1000 (VitEPEG), Pluronic F68, Pluronic F127, Brij L23, and Brij 58 were purchased from Sigma-Aldrich. McCoy’s 5A cell culture media, CholEsteryl BODIPY 542/563 C11, and Hoechst 33342 were purchased from Thermo Fisher Scientific. The SKOV-3 cell line was purchased from ATCC. Charcoal stripped fetal bovine serum and Hank’s balanced salt solution were purchased from Wisent Bioproducts.
Colloid Formation
Colloids of both fulvestrant and PPD were formulated upon dilution of organic stock solutions into an aqueous phase. Fulvestrant colloids were prepared by adding double-distilled water (880 μL) to a DMSO stock solution (10 μL at 5 mM) followed by the addition of 10× PBS (100 μL). Final fulvestrant drug and organic concentrations were 50 μM and 1% (v/v), respectively. PPD colloids were prepared in a similar manner, with the drug stock solution at 12.5 mM in DMF leading to formulations with a final drug concentration of 500 μM and an organic concentration of 4% (v/v). Excipients were incorporated into formulations prior to colloid formation. Since the concentration of organic solvents is in the same range as that typically found in pharmaceutical formulations (up to 10%), we do not anticipate any toxicity. PLAC–PEG was added to the organic phase, whereas all other excipients studied were dissolved in the aqueous phase. The amounts of polymers were chosen based on the initial concentration of the drug being formulated. For fulvestrant colloids formulated at 50 μM, excipients were used at the following concentrations: 0.001% (w/v) UP80, 0.01% F127, 0.01% F68, 0.01% Brij L23, 0.01% Brij 58, 0.01% VitE-PEG, and 0.004% PLAC–PEG. For PPD colloids formulated at 500 μM, excipients were used at the following concentrations: 0.01% UP80, 0.05% F127, 0.05% F68, 0.01% Brij L23, 0.01% Brij 58, 0.01% VitE-PEG, and 0.04% PLAC–PEG. Precipitate formation was inspected visually during screening of excipients.
Colloid Characterization
Colloid diameter, polydispersity, and normalized scattering intensity were measured by dynamic light scattering (DLS) using a DynaPro Plate Reader II (Wyatt Technologies) with a laser width optimized for colloidal aggregate detection (i.e., particles in the 100–1000 nm radius range) by the manufacturer. Operating conditions were a 60 mW laser at an 830 nm wavelength and a detector angle of 158°. Samples were measured in a 96-well format with 100 μL and 20 acquisitions per sample.
Colloids (5 μL) were deposited from 50 and 500 μM solutions of fulvestrant and PPD, respectively, onto glow discharged transmission electron microscope (TEM) grids and allowed to adsorb for 5 min. The solution was then wicked away, and the grid was washed briefly with water (5 μL). Grids were then allowed to dry and negatively stained with either uranyl acetate (5 μL, 10 s, 2% solution, pH ~ 4) for PPD colloids or ammonium molybdate (5 μL, 30 s, 1% solution, pH 7) for fulvestrant colloids prior to imaging on a Hitachi H-7000 microscope operating at 75 kV.
In Vitro Serum Stability
The stability of fulvestrant and PPD colloids under serum conditions was determined using fast protein liquid chromatography (FPLC) using a previously established method.9,29 Colloids were incubated with 20% fetal bovine serum (FBS, Charcoal stripped) and 1% penicillin–streptomycin at 37 °C. At 0, 6, 12, 24, and 48 h, 500 μL aliquots were removed and injected onto a Superdex 200 gel filtration column. Samples were run with a flow rate of 1.5 mL/min and 1× PBS as the mobile phase. For fulvestrant, colloids were co-formulated with the FRET pair of CholEsteryl BODIPY FL and BODIPY 542/563 (500 nM), and fluorescent emission at 575 nm was determined using a Tecan plate reader followed by integration of colloid peak area using GraphPad software version 6.0. For PPD, elution peak areas at 480 nm were calculated using UNICORN software version 5.31. Peak area is used as a measurement of intact colloid population. Protein components of FBS, drugs, and polymer excipients will all contribute to the absorbance at 280 nm. Neither the polymer nor the FBS contribute to the absorbance at 480 nm or the fluorescence at 575 nm. Without polymer, PPD and fulvestrant colloids precipitate rapidly in PBS; thus, their stability could not be assessed by FPLC.
In Vitro Protein Adsorption
Fulvestrant and PPD colloids were prepared at 50 μM (1 mL) as before in the presence or absence of UP80 and PLAC–PEG, respectively. Colloids were then incubated with 50 nM bovine serum albumin (BSA), human immunoglobulin G (IgG), or fibrinogen for 10 min. Colloids were then pelleted by centrifugation for 1 h at 16 000g at 4 °C. After centrifugation, 950 μL of supernatant was removed and the pellet was resuspended in the remaining 50 μL. To prepare samples for sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), 10 μL of supernatant or 10 μL of resuspended pellet was mixed with 10% glycerol, 2% sodium dodecyl sulfate (SDS), and 100 mM β-mercaptoethanol. This process effectively breaks up colloids and releases any colloid-bound protein. Each sample was boiled at 100 °C for 5 min. Proteins (15 μL sample volume) were then separated by SDS-PAGE and identified using Coomassie blue G-250 staining. Protein band intensities were quantified using ImageJ software. Data are expressed as the relative protein band intensities of the pellet to that of the supernatant.
In Vitro Cell Uptake
SKOV-3 cells were maintained at 37 °C in 5% CO2 in McCoy’s 5A media supplemented with 10% FBS, 10 UI/mL penicillin, and 10 μg/mL streptomycin. Cells were seeded at 12 000 cells/well in 8-well borosilicate glass chamber slides and allowed to adhere overnight. Cells were incubated with 50 μM of appropriate formulations for 45 min in serum-free or 10% serum conditions. Fulvestrant colloids were co-formulated with CholEsteryl BODIPY 542/563 C11 (500 nM) for visualization. Following incubation, cells were rinsed and counterstained with Hoechst 33342. Cells were imaged on an Olympus FV1000 confocal laser-scanning microscope at 60× magnification under live-cell imaging conditions. Excitation and emission wavelengths were as follows: for Hoechst, excitation at 405 nm and emission at 460 nm; for fulvestrant colloids co-formulated with BODIPY, excitation at 559 nm and emission at 572; and for PPD colloids and doxorubicin (DOX) formulations, excitation at 488 nm and emission at 520 nm.
RESULTS
Polymer-Stabilized Colloids
Hydrophobic chemothera-peutics, such as fulvestrant (Figure 1A) and PPD (Figure 1B), form colloidal aggregates with CACs of 0.5 μM18 and 14 μM (Figure S1), respectively. Consistent with other colloidal drug aggregates,22 the addition of salt causes massive aggregation and precipitation of both drugs (Figure 1C,D, pink bars). In an effort to prevent colloid precipitation and improve stability in the presence of salts, each drug was co-formulated with one of seven different polymers ranging from clinically used excipients (UP80, Pluronics F68 and F127, and Brij 58 and L23) to amphiphilic polymers used in micelle systems (PLAC–PEG, VitE-PEG) as a screening tool to identify formulations warranting further investigation. Polymers were used at 0.001–0.05% (w/v), a low weight percent relative to that used in traditional drug formulations, which are orders of magnitude higher.30 All formulations in water had an initial diameter of <200 nm regardless of the presence of excipient or type of excipient used (Figure 1C,D). When formulated in PBS buffer, the addition of polymers prevented or reduced the aggregation of colloids. In contrast, the absence of polymers led to the formation of drug aggregates larger than 1 μm, which precipitated from solution within minutes.
Figure 1.
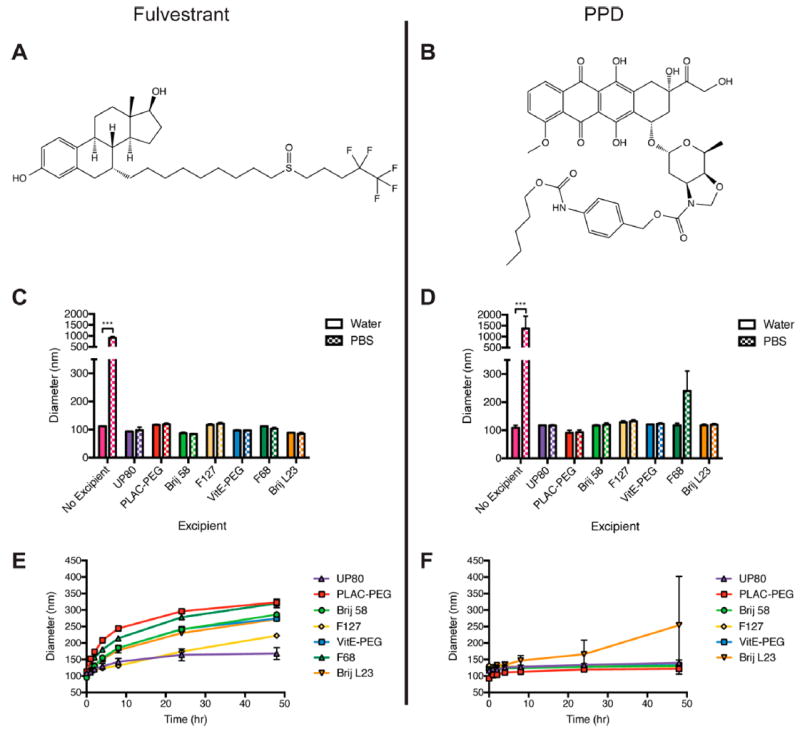
(A) Fulvestrant and (B) PPD were selected for their intrinsic chemotherapeutic efficacy and aggregation properties. Formulation of (C) fulvestrant and (D) PPD colloids in water or PBS in the presence of the following polymeric excipients: UP80, PLAC–PEG, Brij 58, Pluronic F127, VitE-PEG, Pluronic F68, and Brij L23. Initial diameters of the formulations are shown. Incubation of (E) fulvestrant and (F) PPD formulations at 37 °C, showing size changes over 48 h. UP80 was the optimal polymer to maintain the size of fulvestrant over time in buffered salt solution (PBS) compared to other polymers. Stability of PPD with F68 could not be assessed due to precipitation. PLAC–PEG was the optimal polymer to maintain the size of PPD, with the smallest nanoparticle size over the incubation period. (n = 3, mean + SD, *** p < 0.001.)
In the presence of polymeric excipients, colloids were stable over 48 h at 37 °C (Figure 1E,F). For fulvestrant, formulation with UP80 resulted in homogeneous colloids stable over 48 h. Fulvestrant–UP80 colloids had initial diameters of 109 ± 7 nm, which increased to 168 ± 18 nm over 48 h. Other polymers, such as PLAC–PEG, partially inhibited the growth rate of fulvestrant colloids in high salt buffer compared to the drug alone; however, the initial fulvestrant colloid diameter doubled over a 48 h period, demonstrating that UP80 was a more effective stabilizing agent. In contrast, PLAC–PEG was the optimal polymer to stabilize PPD colloids over 48 h. PPD–PLAC–PEG colloids had initial diameters of 93 ± 8 nm, which grew to 122 ± 6 nm. While other polymers stabilized PPD colloids, they were not as effective as PLAC–PEG. For example, Pluronic F68 showed a doubling in size within minutes of exposure to a high salt buffer (Figure 1D). Both fulvestrant–UP80 and PPD–PLAC–PEG maintained a narrow size distribution over the incubation period (PDI < 0.2). Fulvestrant–UP80 and PPD–PLAC–PEG formulations have drug loadings of 75 and 50 wt %, respectively, calculated as the absolute amount of drug per total mass (drug and excipient mass).
We characterized the morphology of our most stable and monodisperse formulations, fulvestrant–UP80 and PPD–PLAC–PEG, using TEM. Imaging confirmed the spherical morphology of the resulting particles, with multiple fields of view used to determine particle size distributions for each formulation (Figure S2); fulvestrant–UP80 colloids had diameters of 53 ± 15 nm, and PPD–PLAC–PEG had diameters of 60 ± 16 nm. Thus, we observed smaller diameters by TEM than those determined by DLS, which is consistent with the drying effects that occur in the vacuum environment used for TEM compared to the hydrated state in DLS. Even small amounts of polymer excipients (0.001% UP80 and 0.04% PLAC–PEG) for these two formulations significantly improved their stability in buffered aqueous solutions. This prompted us to investigate stability in more biologically relevant conditions, such as serum-containing media.
Serum Stability
Encouraged by the enhanced stability of fulvestrant–UP80 and PPD–PLAC–PEG colloids in buffered solutions, we sought to better assess the structural integrity and stability of these formulations over time in serum-containing media using both TEM imaging and FPLC separation. Representative fields of view from TEM imaging (Figure 2A,B and additional fields of view in Figure S3) show that both formulations are present in 10% serum over a 48 h time period; fulvestrant–UP80 colloids (Figure 2A,C) increased in size and dispersity during the incubation from an initial diameter of 67 ± 17 nm to a final diameter of 222 ± 77 nm (Figure 2C). PPD–PLAC–PEG colloids maintained their size and distribution over time (Figure 2B,D), with initial and final diameters of 36 ± 10 and 36 ± 11 nm, respectively. Importantly, the spherical morphology of the colloids was retained for both formulations over the incubation period.
Figure 2.
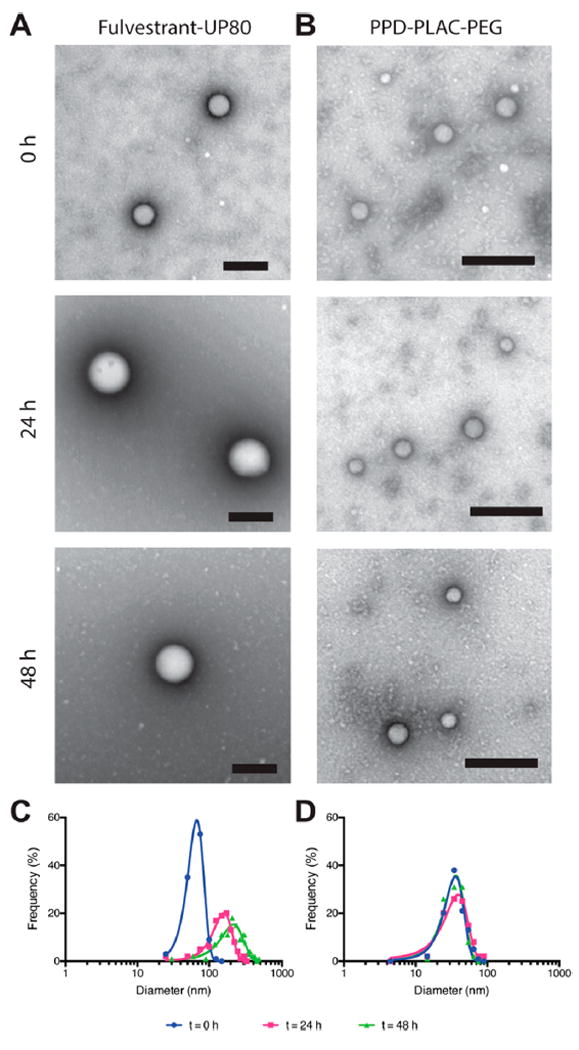
Fulvestrant–UP80 and PPD–PLAC–PEG characterization in 10% serum. (A, B) Representative TEM images of particles in serum at 0, 24, and 48 h. (A) Fulvestrant–UP80 colloids were stained with ammonium molybdate, and (B) PPD–PLAC–PEG colloids were stained with uranyl acetate; scale bars are 200 nm. (C, D) TEM frequency distribution showing a mean diameter increase and peak broadening of (C) fulvestrant–UP80 colloidal aggregates over time, whereas (D) PPD–PLAC–PEG maintains its size and distribution over time.
The protein corona that forms on particle surfaces can cause premature drug release due to partitioning of the drug into the hydrophobic pockets of proteins; thus, we investigated drug release in the stabilized colloidal formulations in serum. To quantify the drug release, our two formulations were incubated in 20% serum, which is representative of in vivo serum conditions and allows colloids to be separated and quantified by FPLC directly.28 At select time points, up to 48 h, the colloidal population was separated from serum proteins and free drug using size-exclusion chromatography (SEC) and the colloid peak area was used as a proxy for drug concentration (Figures 3A,B and S4). Notably, bare colloids could not be separated by this method due to their rapid precipitation under salt conditions. Fulvestrant–UP80 was co-formulated with a BODIPY FRET pair (Figure S5), enabling fluorescence emission detection, whereas PPD–PLAC–PEG colloids were quantified by absorbance at 480 nm. Both formulations showed little dissociation over 24 h (Figure 3C). Fulvestrant–UP80 colloidal aggregates began to dissociate after this time, with almost 50% of the drug released at 48 h. PPD–PLAC–PEG aggregates showed no dissociation over the 48 h time period. Encouragingly, the stability data obtained for the colloids by FPLC separation reflect the trends observed by TEM (Figure 2).
Figure 3.
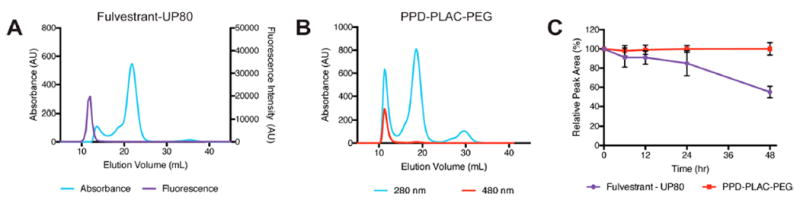
Serum stability of colloidal formulations by FPLC. Traces show separation between serum proteins (absorbance at 280 nm) and (A) fulvestrant–UP80 colloids (tracked by fluorescence using a BODIPY FRET pair) and (B) PPD–PLAC–PEG colloids (unique absorbance at 480 nm) at t = 0. (C) Comparison of the peak area under the colloid curve over time to the area at t = 0 h to determine colloid stability as a function of time. Both colloids are stable up to 24 h, with fulvestrant–UP80 colloids dissociating between 24 and 48 h and PPD–PLAC–PEG colloids showing no evidence of dissociation over 48 h.
Protein Adsorption
We hypothesized that the polymeric excipients used to stabilize the colloidal formulations reduced protein adsorption and thereby provided stability in serum. To test this hypothesis, we used a previously reported method of centrifugation and gel electrophoresis of colloidal formulations to identify surface-bound proteins.16 We studied the interaction of colloidal aggregates of fulvestrant vs fulvestrant–UP80 and PPD vs PPD–PLAC–PEG with the main serum proteins: BSA, IgG, and fibrinogen. Colloids were incubated with each protein and then pelleted by centrifugation. Proteins in the supernatant and those in the pelleted colloid fractions were identified by gel electrophoresis (Figure S6). All three proteins studied were concentrated (at a 5–15-fold increase) in the pelleted fraction when incubated with bare colloids, indicating significant adsorption to the colloid surface (Figure 4). In contrast, none of the three proteins studied was concentrated in the pelleted fraction when incubated with polymer-stabilized colloidal formulations of both fulvestrant–UP80 and PPD–PLAC–PEG, indicating minimal protein adsorption (Figure 4). These data are consistent with several other particle systems that use high-density PEG surfaces to reduce protein adsorption and particle opsonization.31-33
Figure 4.
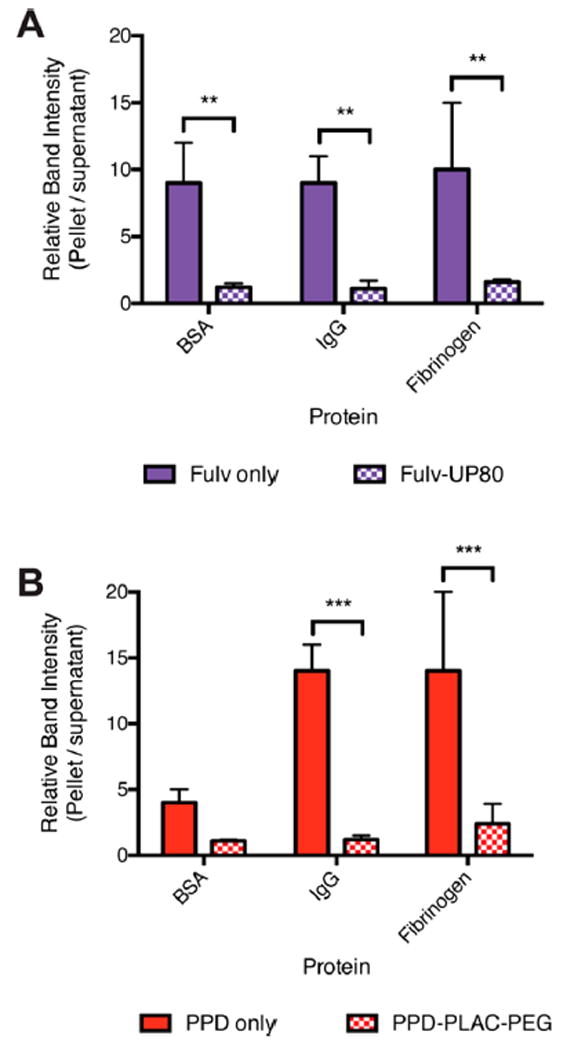
Formulation of colloids with excipient polymers reduces protein adsorption. BSA, IgG, and fibrinogen (50 nM) adsorption are significantly increased on the surface of bare colloids of (A) fulvestrant and (B) PPD (filled bars) compared to colloids stabilized with the appropriate polymer (checkered bars): fulvestrant–UP80 and PPD–PLAC–PEG (n = 3, mean + SD, **p < 0.01, ***p < 0.001).
Cellular Uptake
To investigate the cellular uptake of colloidal aggregates vs drug monomers, which typically diffuse across cell membranes, fulvestrant–UP80 and PPD–PLAC–PEG were incubated with the human epithelial ovarian cancer SKOV-3 cell line (Figure 5) in both the absence and presence of serum. DOX, an anthracycline chemotherapeutic from which PPD is derived, does not form colloidal aggregates and was used as a positive control that can freely permeate cell membranes. DOX and PPD were directly tracked by excitation at 488 nm, whereas fulvestrant–UP80 colloids were co-formulated with a BODIPY dye that was visualized by excitation at 559 nm. Fluorescence of the non-colloid-forming DOX was diffuse, colocalizing with cell nuclei under both serum-free and serum conditions. Conversely, intracellular fluorescence of fulvestrant–UP80 and PPD–PLAC–PEG colloids was observed only under serum-free conditions. Even then, the fluorescence was observed as punctate features within the cell body. In serum-containing media, little to no fluorescence was observed for the colloidal formulations.
Figure 5.
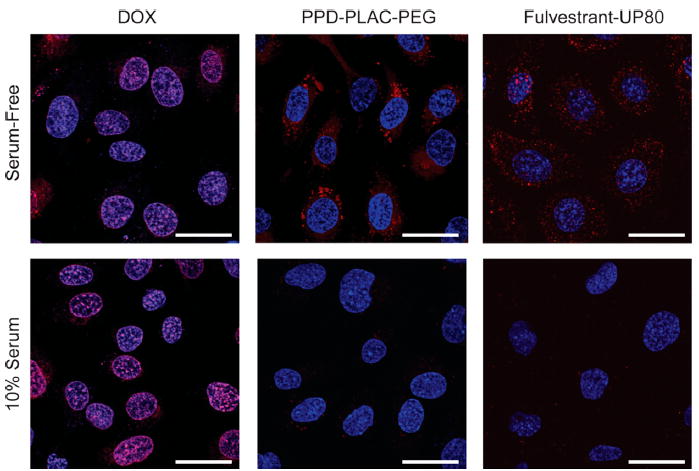
Representative images of the cell uptake of DOX (monomer) and the colloidal formulations of PPD–PLAC–PEG and fulvestrant–UP80 (tracked by BODIPY). SKOV-3 cells were used for all experiments. DOX monomer freely permeates the cell membrane, showing localized fluorescence within the cells’ nuclei. PPD and fulvestrant colloids show uptake only under serum-free conditions, with punctate fluorescence within the cell body. There is no evidence of cell uptake of colloids in serum-containing media. (Scale bar represents 30 μm.)
DISCUSSION
While the intrinsic colloidal aggregation properties of hydrophobic small molecule drugs are often unpredictable, this phenomenon can be exploited and controlled with the addition of excipients. Using a nanoprecipitation method to formulate colloidal aggregates with polymeric excipients, we can produce stable high drug loaded particles resistant to changes in salt and serum conditions. Absolute drug loadings of our two formulations, fulvestrant–UP80 (75 wt %) and PPD–PLAC–PEG (50 wt %), are an order of magnitude higher than conventional micelle formulations, which are typically <10 wt %.7 By incorporating polymeric excipients, the particles are stabilized and aggregation is prevented through steric repulsion between hydrophilic polymer chains.28,33 In the absence of polymer, salts in the solution lead to charge shielding at the surface of colloidal species, causing particle fusion and rapid aggregation and leading to eventual crystallization and precipitation.34 The use of amphiphilic polymers allows hydrophobic segments of the polymer to interact with the hydrophobic colloidal core and hydrophilic segments of the polymer to extend into the aqueous phase to provide steric stability. Due to mixing of the drug and polymer during colloid formation, the hydrophobic segments of the polymer may be entangled within the colloid.
At present, the limited number of colloid–polymer combinations studied here prevent general predictions on optimal drug–polymer pairs. We hypothesize that the solubility parameters play an important role in determining which polymer would be best suited to stabilize a drug colloid, as has been shown by computational approaches used in other studies.35,36 The concentration of polymer used in these formulations plays an important role in stabilizing colloidal species.37,38 If the polymer concentration is too low, then there is insufficient coverage of the colloidal surface to prevent aggregation and coalescence of the drug colloids. If the polymer concentration is too high and above the critical micelle concentration (CMC), then the polymers themselves form micelles and solubilize the drug rather than stabilize the colloid. Accordingly, polymer concentrations near their CMCs were chosen for this study (Table S1), providing adequate surface coverage without colloid disruption.
While the stability in salts is essential, we sought to characterize these colloidal aggregates in biologically relevant serum-containing media. Blood proteins destabilize particles, and premature drug release often results from drugs partitioning into the hydrophobic pockets of proteins adsorbed to the particle surface.39 In fact, the nonstabilized colloids could not be assayed in 20% serum due to their inherent instability and rapid precipitation. In contrast, we show that polymers stabilize these colloidal particles for at least 24 h in 20% FBS by monitoring the drugs using their spectral properties. We hypothesized that this stability is due to a reduction in protein adsorption to the colloidal surface, consistent with the use of hydrophilic polymers, such as PEG, in other particle platforms.31,40 To evaluate the interaction between the main components of serum—albumin, globulins, and fibrinogens—and the particle surface, we used a centrifugation method to identify surface-bound proteins, as has been previously used to study the inhibition of enzymes by colloid surface sequestration.16,41 This method is limited by the concentration of proteins that can be evaluated; however, nonstabilized colloids showed significant adsorption of proteins to their surface, whereas polymer stabilized colloids showed reduced protein adsorption. While colloidal drug aggregates have been previously defined by their ability to adsorb proteins, the use of polymers alters this property and yields formulations with increased stability.
To further probe the stability of the colloidal aggregates, we investigated their interactions with cells in vitro. Under serum-free conditions, distinct punctate fluorescence was observed intracellularly for both fulvestrant and PPD colloidal formulations, which is typical of internalized particles that are trafficked through the endolysosomal pathway.42,43 Corroborating previous literature, DOX, a compound that does not form colloidal aggregates, freely permeated lipid membranes and localized in the nucleus.44 In serum-containing media, while the cellular uptake of DOX was not significantly influenced, fulvestrant–UP80 and PPD–PLAC–PEG colloids were not internalized by cells. This is consistent with our previous observations that compounds in colloid form are not internalized by cells and thus lose their efficacy in cell culture.19 The presence of proteins precludes the nonspecific uptake of particles due to decreased adhesion of particles to the cell surface and supports the need for cellular targeting agents on the particles.8,45,46
It is clear from this study that the combination of hydrophobic drug and polymer strongly influences particle size and stability over time. This is consistent with other polymer-based nanoparticle formulations where similar drug and vehicle compatibility leads to optimized drug loading, stability, and in vivo circulation.47-51 Of the formulations tested here, the combinations of fulvestrant–UP80 and PPD–PLAC–PEG are optimal for colloidal stability in both high ionic strength aqueous and serum containing solutions. Chemical modifications of both the drug and the polymeric excipient have been used previously to enhance molecular interactions that provide improved particle stability.12,13,52 However, this strategy often requires revalidation of materials, especially with respect to the drug. Directly screening for and exploiting the colloidal aggregation properties of drugs, as demonstrated here, can provide a mechanism to significantly increase drug loading and stability, without the need for chemical modification. With a continued increase in chemical diversity of both colloid-forming drugs and polymeric excipients, the methods outlined here will find further application in formulating drug-rich nanoparticle delivery systems.
CONCLUSIONS
By incorporating polymeric excipients into colloidal formulations of two relevant chemotherapeutics, fulvestrant and PPD, we demonstrated stability in both salt and serum containing-media. This enhanced stability can be attributed to reduced adsorption of serum proteins to the surface of the particles. We anticipate that this strategy of using polymers to stabilize colloidal drug aggregates is broadly applicable, thereby opening up new opportunities to achieve high drug loadings for drug delivery applications.
Supplementary Material
Acknowledgments
This work was supported by grants from the Canadian Cancer Society Research Institute (to M.S.S. and B.K.S.), the US National Institutes of General Medical Sciences (GM71630 to B.K.S. and M.S.S.), and the US National Cancer Institute (CA143549 to T.H.K.). A.N.G. and J.L. were supported, in part, by the Natural Sciences and Engineering Research Council (NSERC) of Canada Postgraduate Research Scholarships, and C.K.M. was supported, in part, by an NSERC postdoctoral fellowship. We thank S. Boyle and B. Calvieri from the University of Toronto Microscopy Imaging Laboratory for assistance with TEM imaging and members of the Shoichet laboratories for thoughtful discussions.
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharma-ceut.6b01015.
Additional fields of view for TEM characterization, fluorescence properties of colloids, and SDS-PAGE gels after protein incubation (PDF)
References
- 1.Strickley RG. Solubilizing Excipients in Oral and Injectable Formulations. Pharm Res. 2004;21(2):201–230. doi: 10.1023/b:pham.0000016235.32639.23. [DOI] [PubMed] [Google Scholar]
- 2.Kiss L, Walter FR, Bocsik A, Veszelka S, Ózsvári B, Puskás LG, Szabó-Révész P, Deli MA. Kinetic Analysis of the Toxicity of Pharmaceutical Excipients Cremophor EL and RH40 on Endothelial and Epithelial Cells. J Pharm Sci. 2013;102(4):1173–1181. doi: 10.1002/jps.23458. [DOI] [PubMed] [Google Scholar]
- 3.Bravo Gonzalez RC, Huwyler J, Boess F, Walter I, Bittner B. In Vitro Investigation on the Impact of the Surface-Active Excipients Cremophor EL, Tween 80 and Solutol HS 15 on the Metabolism of Midazolam. Biopharm Drug Dispos. 2004;25(1):37–49. doi: 10.1002/bdd.383. [DOI] [PubMed] [Google Scholar]
- 4.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an Emerging Platform for Cancer Therapy. Nat Nanotechnol. 2007;2(12):751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 5.Cabral H, Kataoka K. Progress of Drug-Loaded Polymeric Micelles Into Clinical Studies. J Controlled Release. 2014;190:465–476. doi: 10.1016/j.jconrel.2014.06.042. [DOI] [PubMed] [Google Scholar]
- 6.Luque-Michel E, Imbuluzqueta E, Sebastián V, Blanco-Prieto MJ. Clinical Advances of Nanocarrier-Based Cancer Therapy and Diagnostics. Expert Opin Drug Delivery. 2017;14:75–92. doi: 10.1080/17425247.2016.1205585. [DOI] [PubMed] [Google Scholar]
- 7.Park K. Facing the Truth About Nanotechnology in Drug Delivery. ACS Nano. 2013;7(9):7442–7447. doi: 10.1021/nn404501g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors Affecting the Clearance and Biodistribution of Polymeric Nano-particles. Mol Pharmaceutics. 2008;5(4):505–515. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Logie J, McLaughlin CK, Tam RY, Shoichet MS. Innovative Use of the Taxol Binding Peptide Overcomes Key Challenges of Stable and High Drug Loading in Polymeric Nanomicelles. Chem Commun. 2015;51(60):12000–12003. doi: 10.1039/c5cc04282h. [DOI] [PubMed] [Google Scholar]
- 10.Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WCW. Analysis of Nanoparticle Delivery to Tumours. Nat Rev Mater. 2016;1(5):16014. [Google Scholar]
- 11.Eetezadi S, Ekdawi SN, Allen C. The Challenges Facing Block Copolymer Micelles for Cancer Therapy: in Vivo Barriers and Clinical Translation. Adv Drug Delivery Rev. 2015;91:7–22. doi: 10.1016/j.addr.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 12.Maksimenko A, Dosio F, Mougin J, Ferrero A, Wack S, Reddy LH, Weyn AA, Lepeltier E, Bourgaux C, Stella B, Cattel L, Couvreur PA. Unique Squalenoylated and Nonpegylated Doxorubicin Nanomedicine with Systemic Long-Circulating Properties and Anticancer Activity. Proc Natl Acad Sci U S A. 2014;111(2):E217–E226. doi: 10.1073/pnas.1313459110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma W, Cheetham AG, Cui H. Building Nanostructures with Drugs. Nano Today. 2016;11(1):13–30. doi: 10.1016/j.nantod.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen G, Xing R, Zhang N, Chen C, Ma G, Yan X. Interfacial Cohesion and Assembly of Bioadhesive Molecules for Design of Long-Term Stable Hydrophobic Nanodrugs Toward Effective Anticancer Therapy. ACS Nano. 2016;10(6):5720–5729. doi: 10.1021/acsnano.5b07276. [DOI] [PubMed] [Google Scholar]
- 15.Coan KED, Shoichet BK. Stoichiometry and Physical Chemistry of Promiscuous Aggregate-Based Inhibitors. J Am Chem Soc. 2008;130(29):9606–9612. doi: 10.1021/ja802977h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGovern SL, Helfand BT, Feng B, Shoichet BK. A Specific Mechanism of Nonspecific Inhibition. J Med Chem. 2003;46(20):4265–4272. doi: 10.1021/jm030266r. [DOI] [PubMed] [Google Scholar]
- 17.Sassano MF, Doak AK, Roth BL, Shoichet BK. Colloidal Aggregation Causes Inhibition of G Protein-Coupled Receptors. J Med Chem. 2013;56(6):2406–2414. doi: 10.1021/jm301749y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Owen SC, Doak AK, Wassam P, Shoichet MS, Shoichet BK. Colloidal Aggregation Affects the Efficacy of Anticancer Drugs in Cell Culture. ACS Chem Biol. 2012;7(8):1429–1435. doi: 10.1021/cb300189b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Owen SC, Doak AK, Ganesh AN, Nedyalkova L, McLaughlin CK, Shoichet BK, Shoichet MS. Colloidal Drug Formulations Can Explain “Bell-Shaped” Concentration-Response Curves. ACS Chem Biol. 2014;9(3):777–784. doi: 10.1021/cb4007584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Irwin JJ, Duan D, Torosyan H, Doak AK, Ziebart KT, Sterling T, Tumanian G, Shoichet BK. An Aggregation Advisor for Ligand Discovery. J Med Chem. 2015;58(17):7076–7087. doi: 10.1021/acs.jmedchem.5b01105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ilevbare GA, Taylor LS. Liquid–Liquid Phase Separation in Highly Supersaturated Aqueous Solutions of Poorly Water-Soluble Drugs: Implications for Solubility Enhancing Formulations. Cryst Growth Des. 2013;13(4):1497–1509. [Google Scholar]
- 22.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A Common Mechanism Underlying Promiscuous Inhibitors From Virtual and High-Throughput Screening. J Med Chem. 2002;45(8):1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 23.McLaughlin CK, Duan D, Ganesh AN, Torosyan H, Shoichet BK, Shoichet MS. Stable Colloidal Drug Aggregates Catch and Release Active Enzymes. ACS Chem Biol. 2016;11(4):992–1000. doi: 10.1021/acschembio.5b00806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ilevbare GA, Liu H, Pereira J, Edgar KJ, Taylor LS. Influence of Additives on the Properties of Nanodroplets Formed in Highly Supersaturated Aqueous Solutions of Ritonavir. Mol Pharmaceutics. 2013;10(9):3392–3403. doi: 10.1021/mp400228x. [DOI] [PubMed] [Google Scholar]
- 25.Jackson MJ, Toth SJ, Kestur US, Huang J, Qian F, Hussain MA, Simpson GJ, Taylor LS. Impact of Polymers on the Precipitation Behavior of Highly Supersaturated Aqueous Danazol Solutions. Mol Pharmaceutics. 2014;11(9):3027–3038. doi: 10.1021/mp500201s. [DOI] [PubMed] [Google Scholar]
- 26.Wakeling AE, Dukes M, Bowler J. A Potent Specific Pure Antiestrogen with Clinical Potential. Cancer Res. 1991;51(15):3867–3873. [PubMed] [Google Scholar]
- 27.Barthel BL, Zhang Z, Rudnicki DL, Coldren CD, Polinkovsky M, Sun H, Koch GG, Chan DCF, Koch TH. Preclinical Efficacy of a Carboxylesterase 2-Activated Prodrug of Doxazolidine. J Med Chem. 2009;52(23):7678–7688. doi: 10.1021/jm900694z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Logie J, Owen SC, McLaughlin CK, Shoichet MS. PEG-Graft Density Controls Polymeric Nanoparticle Micelle Stability. Chem Mater. 2014;26(9):2847–2855. [Google Scholar]
- 29.Zhao X, Poon Z, Engler AC, Bonner DK, Hammond PT. Enhanced Stability of Polymeric Micelles Based on Postfunctionalized Poly(Ethylene Glycol)- B-Poly(Γ-Propargyl L-Glutamate): the Substituent Effect. Biomacromolecules. 2012;13(5):1315–1322. doi: 10.1021/bm201873u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalepu S, Nekkanti V. Insoluble Drug Delivery Strategies: Review of Recent Advances and Business Prospects. Acta Pharm Sin B. 2015;5(5):442–453. doi: 10.1016/j.apsb.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walkey CD, Olsen JB, Guo H, Emili A, Chan WCW. Nanoparticle Size and Surface Chemistry Determine Serum Protein Adsorption and Macrophage Uptake. J Am Chem Soc. 2012;134(4):2139–2147. doi: 10.1021/ja2084338. [DOI] [PubMed] [Google Scholar]
- 32.Liu Y, Hu Y, Huang L. Influence of Polyethylene Glycol Density and Surface Lipid on Pharmacokinetics and Biodistribution of Lipid-Calcium-Phosphate Nanoparticles. Biomaterials. 2014;35(9):3027–3034. doi: 10.1016/j.biomaterials.2013.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.OWENS D, III, PEPPAS N. Opsonization, Biodistribution, and Pharmacokinetics of Polymeric Nanoparticles. Int J Pharm. 2006;307(1):93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 34.Overbeek JTG. Recent Developments in the Understanding of Colloid Stability. J Colloid Interface Sci. 1977;58(2):408–422. [Google Scholar]
- 35.Latere Dwan’Isa JP, Rouxhet L, Préat V, Brewster ME, Ariën A. Prediction of Drug Solubility in Amphiphilic Di-Block Copolymer Micelles: the Role of Polymer-Drug Compatibility. Pharmazie. 2007;62:499–504. [PubMed] [Google Scholar]
- 36.Marsac PJ, Shamblin SL, Taylor LS. Theoretical and Practical Approaches for Prediction of Drug-Polymer Miscibility and Solubility. Pharm Res. 2006;23(10):2417–2426. doi: 10.1007/s11095-006-9063-9. [DOI] [PubMed] [Google Scholar]
- 37.Zaccone A, Wu H, Lattuada M, Morbidelli M. Correlation Between Colloidal Stability and Surfactant Adsorption/Association Phenomena Studied by Light Scattering. J Phys Chem B. 2008;112(7):1976–1986. doi: 10.1021/jp0776210. [DOI] [PubMed] [Google Scholar]
- 38.Santander-Ortega MJ, Jodar-Reyes AB, Csaba N, Bastos-Gonzalez D, Ortega-Vinuesa JL. Colloidal Stability of Pluronic F68-Coated PLGA Nanoparticles: a Variety of Stabilisation Mechanisms. J Colloid Interface Sci. 2006;302(2):522–529. doi: 10.1016/j.jcis.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 39.Liu J, Zeng F, Allen C. Influence of Serum Protein on Polycarbonate-Based Copolymer Micelles as a Delivery System for a Hydrophobic Anti-Cancer Agent. J Controlled Release. 2005;103(2):481–497. doi: 10.1016/j.jconrel.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 40.Gref R, Lück M, Quellec P, Marchand M, Dellacherie E, Harnisch S, Blunk T, Müller RH. Stealth” Corona-Core Nanoparticles Surface Modified by Polyethylene Glycol (PEG): Influences of the Corona (PEG Chain Length and Surface Density) and of the Core Composition on Phagocytic Uptake and Plasma Protein Adsorption. Colloids Surf B. 2000;18(3-4):301–313. doi: 10.1016/s0927-7765(99)00156-3. [DOI] [PubMed] [Google Scholar]
- 41.Coan KED, Maltby DA, Burlingame AL, Shoichet BK. Promiscuous Aggregate-Based Inhibitors Promote Enzyme Unfolding. J Med Chem. 2009;52(7):2067–2075. doi: 10.1021/jm801605r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yameen B, Choi WI, Vilos C, Swami A, Shi J, Farokhzad OC. Insight Into Nanoparticle Cellular Uptake and Intracellular Targeting. J Controlled Release. 2014;190:485–499. doi: 10.1016/j.jconrel.2014.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gaudin A, Tagit O, Sobot D, Lepetre-Mouelhi S, Mougin J, Martens TF, Braeckmans K, Nicolas V, Desmaële D, De Smedt SC, Hildebrandt N, Couvreur P, Andrieux K. Transport Mechanisms of Squalenoyl-Adenosine Nanoparticles Across the Blood-Brain Barrier. Chem Mater. 2015;27(10):3636–3647. [Google Scholar]
- 44.Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an Update on Anticancer Molecular Action, Toxicity and Novel Drug Delivery Systems. J Pharm Pharmacol. 2013;65(2):157–170. doi: 10.1111/j.2042-7158.2012.01567.x. [DOI] [PubMed] [Google Scholar]
- 45.Walkey CD, Olsen JB, Song F, Liu R, Guo H, Olsen DWH, Cohen Y, Emili A, Chan WCW. Protein Corona Fingerprinting Predicts the Cellular Interaction of Gold and Silver Nanoparticles. ACS Nano. 2014;8(3):2439–2455. doi: 10.1021/nn406018q. [DOI] [PubMed] [Google Scholar]
- 46.Lesniak A, Fenaroli F, Monopoli MP, Åberg C, Dawson KA, Salvati A. Effects of the Presence or Absence of a Protein Corona on Silica Nanoparticle Uptake and Impact on Cells. ACS Nano. 2012;6(7):5845–5857. doi: 10.1021/nn300223w. [DOI] [PubMed] [Google Scholar]
- 47.Letchford K, Liggins R, Burt H. Solubilization of Hydrophobic Drugs by Methoxy Poly(Ethylene Glycol)-Block-Polycaprolactone Diblock Copolymer Micelles: Theoretical and Experimental Data and Correlations. J Pharm Sci. 2008;97(3):1179–1190. doi: 10.1002/jps.21037. [DOI] [PubMed] [Google Scholar]
- 48.Liu J, Xiao Y, Allen C. Polymer–Drug Compatibility: a Guide to the Development of Delivery Systems for the Anticancer Agent, Ellipticine. J Pharm Sci. 2004;93(1):132–143. doi: 10.1002/jps.10533. [DOI] [PubMed] [Google Scholar]
- 49.Yang C, Tan JPK, Cheng W, Attia ABE, Ting CTY, Nelson A, Hedrick JL, Yang YY. Supramolecular Nanostructures Designed for High Cargo Loading Capacity and Kinetic Stability. Nano Today. 2010;5(6):515–523. [Google Scholar]
- 50.Gou J, Feng S, Xu H, Fang G, Chao Y, Zhang Y, Xu H, Tang X. Decreased Core Crystallinity Facilitated Drug Loading in Polymeric Micelles Without Affecting Their Biological Performances. Biomacromolecules. 2015;16(9):2920–2929. doi: 10.1021/acs.biomac.5b00826. [DOI] [PubMed] [Google Scholar]
- 51.Zhao Y, Fay F, Hak S, Manuel Perez-Aguilar J, Sanchez-Gaytan BL, Goode B, Duivenvoorden R, de Lange Davies C, Bjørkøy A, Weinstein H, Fayad ZA, Pérez-Medina C, Mulder WJM. Augmenting Drug–Carrier Compatibility Improves Tumour Nanotherapy Efficacy. Nat Commun. 2016;7:11221. doi: 10.1038/ncomms11221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang P, Wang D, Su Y, Huang W, Zhou Y, Cui D, Zhu X, Yan D. Combination of Small Molecule Prodrug and Nanodrug Delivery: Amphiphilic Drug–Drug Conjugate for Cancer Therapy. J Am Chem Soc. 2014;136(33):11748–11756. doi: 10.1021/ja505212y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.