

Abstract
Objectives
Niacin has been used for seven decades to modulate plasma lipids, but its mechanism of action is still unclear. We sought to determine whether variants in the niacin receptor gene, hydroxycarboxylic receptor 2 (HCAR2) are associated with lipid response to treatment.
Methods
Coding variants, rs7314976 (p.R311C) and rs2454727 (p.M317I), were genotyped in 2067 participants from the Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides and Impact on Global Health Outcomes (AIM-HIGH) trial. AIM-HIGH was a randomized, placebo controlled trial to assess the effect of extended release (ER) niacin in patients with cardiovascular disease aggressively treated with low density lipoprotein (LDL-C) lowering therapy.
Results
We did not find an association of p.R311C or p.M317I with change in LDL-C, triglycerides (TG) or high-density lipoprotein cholesterol (HDL-C) at one year in groups receiving placebo or ER niacin. In white subjects, the reduction in lipoprotein(a) [Lp(a) ] in response to niacin was greater in homozygous carriers of the major 317M allele (-22.7%; p=0.005) compared with minor allele carriers (-15.3%). This was directionally consistent in the black participants. Upon combining both groups, the reduction in Lp(a) in response to niacin was significantly greater in the homozygous major allele carriers (-23.0%; p=0.003) compared with minor allele carriers (-15.2%).
Conclusions
Understanding the genetic contribution to variation in response to niacin therapy, including Lp(a) reduction, could uncover mechanisms by which niacin decreases Lp(a), an important independent risk factor for cardiovascular disease.
Keywords: Niacin, dyslipidemia, pharmacogenetics, lipoprotein (a), lipids, statins, HCAR2, GPR109A
Introduction
Niacin's potent lipid-modifying properties were first described in normal and hypercholesterolemic subjects in 1955 by Altschul and colleagues [1]. Niacin favorably modulates the lipoprotein profile by reducing total cholesterol (TC), low density lipoprotein cholesterol (LDL-C), triglycerides (TG) and raising high density lipoprotein cholesterol (HDL-C) [2]. Niacin is one of few drugs that also significantly lowers lipoprotein (a) (Lp(a))[3], identified as an independent risk factor for cardiovascular disease [4,5]. In the pre-statin era, niacin was the first pharmacologic agent shown to reduce the incidence of nonfatal myocardial infarction and cardiac death [6,7]. Niacin has also demonstrated beneficial effects on arterial plaque regression in combination with statin therapy [8,9]. However in recent clinical outcomes trials, including The Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides and Impact on Global Health Outcomes (AIM-HIGH) trial, the addition of extended release (ER) niacin to intensive LDL-C lowering therapy did not reduce atherothrombotic events compared with LDL-C lowering therapy alone [10,11].
Pharmacological effects of niacin are not well understood. Most prominently, niacin stimulates the hydroxyl-carboxylic acid receptor 2 (HCAR2, also known as GPR109A and the niacin receptor) located on the surface of adipocytes, resulting in the anti-lipolytic effect of the drug [12]. The reduction in free fatty acids returning to the liver may reduce the assembly and secretion of very low density lipoproteins (VLDL),[2] however additional mechanisms may be operative [13]. Since HCAR2 is not expressed in the liver, in vitro studies suggest that niacin may inhibit TG synthesis by directly inhibiting diacylglyercol acyltransferase-2, the key enzyme for TG synthesis [14]. Niacin may increase HDL-C via reduction of HDL-apoA-I catabolism [15] and inhibition of cholesteryl ester transfer protein (CETP) [16]. The mechanism of Lp(a)-lowering with niacin is unknown.
There is a growing body of literature regarding additional anti-inflammatory effects of niacin that are both dependent [17-20] and independent [21] of HCAR2. This receptor is expressed in various immune cells of the body such as macrophages, neutrophils, and epidermal Langerhans cells [17,18]. In an experimental model of mice deficient of HCAR2, Lukasova showed that niacin reduced the progression of atherosclerosis by a HCAR2 mediated effect on bone-marrow derived cells, independent of its lipid lowering effect [17].
The effects of niacin on lipids and cardiovascular risk are variable. Genetic variation in HCAR2 could influence niacin's effects on plasma lipids, Lp(a), or cardiovascular events, but this has not been tested [22]. First we sequenced HCAR2 in 294 healthy subjects to discover novel coding variants. We identified two commonly occurring coding single nucleotide polymorphisms (SNPs): rs7314976 (C931T, p.R311C) and rs2454727 (G951A, p.M317I) occurring in the c-terminal tail. However, the functionality of these SNPs is unknown. The primary objective of this study was to determine whether these variants were associated with the change in LDL-C, HDL-C, TG and Lp(a) following treatment with ER Niacin in the AIM-HIGH study. The secondary objective was to determine whether the HCAR2 variants were associated with cardiovascular events in the trial and differentially modulated by treatment group.
Methods
Ethics Statement
AIM-HIGH participants provided written informed consent and all research as conducted according to the principles outlines in the Declaration of Helsinki. The protocol was approved by the Institutional Review Boards (IRB) at all participating clinical sites. The genetic sub-study of AIM-HIGH was approved by the IRB at the University of Pennsylvania.
Healthy cohort study
This preliminary study recruited healthy volunteers to a University of Pennsylvania inpatient Clinical and Translational Research Center protocol to investigate the genomic basis of the flushing and metabolic responses to pharmacological doses of niacin. Healthy men and non-pregnant, non-lactating women age 18-45, with BMI 18-30 kg/m2, and of self-reported white or black race were included. Here we report the results of the HCAR2 gene sequencing in 294 subjects. The study was approved by the University of Pennsylvania Institutional Review Board (NCT00953667). All subjects provided written informed consent.
AIM-HIGH cohort
The AIM-HIGH study (NCT00120289) design and baseline characteristics of the participants have been previously published [10]. Briefly, the trial tested whether ER niacin 1500-2000 mg per day added to intensive statin therapy, as compared with statin therapy alone, would reduce the risk of cardiovascular events in patients with established atherosclerotic cardiovascular disease and atherogenic dyslipidemia (low levels of HDL-C, elevated TG, and small dense particles of LDL-C). Of the total 3414 AIM-HIGH participants randomized into the trial, 2067 had provided DNA and had complete phenotype data to perform the current analysis.
Laboratory Methods
Analytical Measurements
Lipid and Lp(a) measurements were performed at the Northwest Lipid Metabolism and Diabetes Research Laboratory at the University of Washington as previously described [10]. Analysis of Lp(a) was performed by a monoclonal anti-body-based enzyme-linked immunoadsorbent assay (ELISA) as previously published [23].
Sequencing of HCAR2 in the healthy cohort study
DNA was extracted from buffy coat samples using a QIA Symphony SP instrument (Qiagen, Inc. Valencia, CA). Due to the high degree of homology of HCAR2 and HCA3, being 95% identical on the protein level [24], we employed a two-step process for sequencing using a nested primer approach. We sequenced the 5′ untranslated region (UTR) and the coding sequence (Figure 1). Polymerase chain reaction (PCR) amplification and sequencing of the single exon region of HCAR2 were carried out with the following primers for amplification: F, ACATGACATAAAGGCAGGCGT; R, TTCTTGCGATGGTTATTTAAGGAG. PCR reactions were amplified in a total volume of 25 μL using 100 ng of genomic DNA, 0.4 μM of each forward and reverse primer, ready-to-go PCR beads (Amersham) while undergoing denaturation at 94° C for 2 minutes, followed by 35 cycles (94° C for 30 seconds, 62° C for 30 seconds, and 72° C for 90 seconds), and an extension at 72° C for 7 minutes. The unincorporated nucleotides and primers were removed by incubation with ExoSAP-IT (USB Corp, Cleveland, OH) for 15 minutes at 37°C followed by enzyme inactivation at 80°C for 15 minutes prior to sequencing. Sequencing was performed at GENEWIZ, Inc. (South Plainfield, NJ) with the following primers: F, GATGCCGATCCAGAATGGCGG; F, CACCACACAGACACACACCTCC; R, CCGCCATTCTGGATCGGCATC; R, TTCTTGCGATGGTTATTTAAGGA. Sequences were aligned using Sequencher software (GeneCodes Corp., Ann Arbor, MI). Allelic variations noted from multiple alignments were verified by inspecting chromatograms. Linkage disequilibrium blocks in each race group were identified using Haploview 4.2 [25].
Figure 1.

Schematic of HCAR2 gene structure. The 5′untranslated region (UTR) and all 1092 nucleotides of the coding region were sequenced in healthy subjects. The location of nonsynonomous single nucleotide polymorphisms (SNPs) identified in this study is denoted by diamonds, while the synonymous SNPs are denoted by the triangles. The transmembrane domains of the translated protein are depicted by dark grey boxes.
Genotyping in AIM-HIGH
DNA was extracted from buffy coat samples using the FlexiGene DNA Kit (Qiagen Inc, Valencia, CA). We employed a two-step process for genotyping using a nested primer approach. First we amplified the HCAR2 gene region with the primers described above. Single nucleotide polymorphisms in HCAR2, rs7314976 and rs2454727, were genotyped in AIM-HIGH participants using by TaqMan Allelic discrimination Assay (Applied Biosystems, Foster City, CA, USA). Subjects were excluded from the analysis if they reported race other than white or black (n=27). The HCAR2 p.R311C and p.M317I variants were successfully genotyped in 2050 and 2067 individuals, respectively. The TaqMan genotyping call rate was 96% with an error rate of 8% for p.R311C and 6% for p.M317I as determined on 51 DNA samples genotyped in duplicate. In all cases, discordant calls were caused on the pair being classified as a genotype call and the other member of the pair being classified as failure to genotype. All SNPS were consistent with Hardy-Weinberg equilibrium (p>0.2). Subjects were excluded from the analysis if they were missing outcome data leaving 1994 white and 73 black individuals for the baseline lipoprotein analysis and 1818 white and 58 black individuals for the 1 year lipoprotein analysis. The CV event analysis was performed in 1994 white and 73 black individuals for whom event data were available.
Statistical analysis
The primary outcomes were defined as the percent change in plasma concentrations of LDL-C, HDL-C, TG and Lp(a) from baseline to 1 year after treatment with either statin + placebo or statin + ER niacin. Secondarily, we also examined the baseline pre-treatment plasma concentrations of the lipid traits. Baseline TG and Lp(a) were log-transformed before analysis because of non-normal distribution. The percent changes in lipids and Lp(a) at 1 year were normally distributed. We employed linear regression analysis to test the dominant effect [26] of HCAR2 genotype on baseline lipid and Lp(a) values and on the percent change in lipids and Lp(a) at one year after randomization, adjusting for age, sex, body mass index (BMI) and baseline lipid values. The one year lipid and Lp(a) outcomes were evaluated separately in the niacin and placebo groups. The analyses were first performed separately for each race group to avoid concerns of population stratification. Since there was no race × treatment interaction in the percent change in lipid and Lp(a), we also reported the combined results. Cox regression models were fit to determine the hazard of experiencing a cardiovascular event including HCAR2 genotype, age, sex, race and BMI in the model. Kaplan-Meier survival curves for time to CV event were created for the HCAR2 M317I variant. Cardiovascular events were defined as the composite of death from coronary heart disease, nonfatal myocardial infarction, ischemic stroke, hospitalization for an acute coronary syndrome, or symptom-driven coronary or cerebral revascularization [10]. For the primary outcome, the Bonferroni correction was applied to account for multiple comparisons, based on 8 tests (2 SNPs × 4 lipid traits), so that each association was claimed at the level of 0.006. A post-hoc power analysis was performed for the change in Lp(a) outcome in the niacin group. With 916 patients, we had 74% power to detect the differences in means we observed by M317I genotype at an alpha level of 0.006. Analysis was performed using STATA version 13.1 (College Station, TX).
Results
We sequenced the single exon HCAR2 gene in 294 healthy subjects. The summary of the variation is listed in Table 1. Figure 1 provides a schematic of the HCAR2 gene structure and the location of the SNPs identified by the sequencing study. Six polymorphic sites were identified, two of these resulted in amino acid changes. The two most common coding variants C931T (p.R311C) and G951A (p.M317I) occurred at lower frequency in the black subjects (Table 1). These variants were predicted to be benign by SIFT and PolyPhen [27]. The coding variants were in linkage disequilibrium (LD) (r2=0.96 for whites and r2=1.0 for blacks). One major LD block was identified in each race group (Figure S1). To determine whether variation in HCAR2 was associated with the lipid response to niacin, we chose to genotype these two commonly occurring coding SNPs in the AIM-HIGH cohort.
Table 1. Summary of variation identified in HCAR2 by sequencing the healthy cohort (n=294).
| Rs. No. | Chr: bp1 | Base Position | Alleles | AA position | AA change | MAF EA | MAF AA | SIFT2 | PolyPhen2 |
|---|---|---|---|---|---|---|---|---|---|
| rs115977736 | 12:122702960 | 324 | T/C | 108 | Ala-Ala | 0.000 | 0.039 | - | - |
| rs137895931 | 12:122702726 | 558 | C/T | 186 | Phe-Phe | 0.005 | 0.000 | - | - |
| rs142740966 | 12:122702531 | 753 | G/C | 251 | Arg-Arg | 0.000 | 0.005 | - | - |
| rs7314976 | 12:122702353 | 931 | C/T | 311 | Arg-Cys | 0.161 | 0.023 | 0.26 | 0.015 |
| rs2454727 | 12:122702333 | 951 | G/A | 317 | Met-Ile | 0.385 | 0.147 | 0.19 | 0 |
| rs142025654 | 12:122702303 | 981 | G/A | 327 | Thr-Thr | 0.000 | 0.025 | - | - |
Build GRCh38.p10
SIFT and PolyPhen scores obtained from www.ensembl.org; both of these variants were predicted to be tolerated by SIFT and benign by PolyPhen
MAF= minor allele frequency
EA= European Americans
AA= African-Americans
The clinical and demographic characteristics for the AIM-HIGH population that provided DNA during the course of the study (n=2067) as compared with the whole cohort are provided in Table 2. There was no difference between baseline lipid values seen in the genetic subgroup compared to the larger AIM-HIGH study cohort [10,23]. In the complete cohort, as previously published, the addition of ER niacin to statin treatment resulted in a 25% increase in HDL-C compared with 10% in the placebo group at two years (p<0.001). Niacin further decreased LDL-C by 12.0% and triglyceride concentrations by 28.6% as compared to 5.5% and 8.1% in the placebo group, respectively [10]. In a separate analysis of AIM-HIGH, niacin significantly decreased Lp(a) levels by 21% in the statin + ER niacin group compared with 5.9% in the statin + placebo group (p<0.05) [23].
Table 2. Clinical and Demographic Characteristics of the AIM-HIGH participants.
| Genetic subgroup | Total AIM-HIGH study | |||
|---|---|---|---|---|
| mean± sd median (IQR) | Statin + Placebo (n=1020) | Statin + ER Niacin (n=1047) | Statin + Placebo (n=1696) | Statin + ER Niacin (n=1718) |
| Age (years) mean± sd | 64.0 ± 8.7 | 64.6 ± 8.7 | 63.7 ± 8.7 | 63.7 ± 8.8 |
| Sex, n (%) female | 164 (16.1) | 158 (15.1) | 251 (14.8) | 253 (14.7) |
| Race, n (%) Black | 35 (3.4) | 38 (3.6) | 49 (2.9) | 68 (4.0) |
| Baseline LDL-C (mg/dL) | 74.6 ± 22.2 | 73.5 ± 22.0 | 75.8 ± 24.3 | 76.2 ± 25.7 |
| Baseline HDL-C (mg/dL) | 35.1 ± 5.6 | 34.6 ± 5.6* | 35.3 ± 5.9 | 34.8 ± 5.9* |
| Baseline TG (mg/dL) | 162 (133-215) | 166 (131-217) | 162 (128-218) | 164 (127-218) |
| Baseline Lp(a) (nmol/L) | 32 (13-118) | 36 (14- 132) | 32.7 (13.1-122.6) | 36.1 (13.5-126.6) |
Lipid traits reported as mean (sd); Triglycerides and Lp(a) reported as median (interquartile range).
HDL-C levels were nominally lower in the statin + ER niacin group at baseline p=0.04 in the genetic subgroup and in the whole AIM-HIGH cohort as previously reported.
LDL-c= low-density lipoprotein cholesterol, HDL-c= high-density lipoprotein cholesterol, TG=triglycerides, Lp(a)= lipoprotein (a)
Table 3 provides associations of HCAR2 p.R311C and p.M317I with baseline LDL-C, HDL-C, TG and Lp(a) in the AIM-HIGH cohort combined. The minor allele frequency of HCAR2 SNPs in the white subjects was p.R311C (MAF 0.17) and p.M317I (MAF 0.39), consistent with our sequencing study in the healthy cohort and to what has been previously reported in dbSNP. The frequency of these variants was lower in the black subjects; p.R311C (MAF 0.06) and p.M317I (MAF 0.16). There were no significant associations with baseline lipid values.
Table 3. Association of HCAR2 variants with baseline lipid values in participants from the AIM-HIGH study.
| White Ancestry (MAF 0.17) | Black Ancestry (MAF 0.06) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| SNP | Trait | ArgArg (n=1359) | ArgCys (n=553) | CysCys (n=65) | p-value | ArgArg (n=64) | ArgCys (n=9) | CysCys (n=0) | p-value |
| p.R311C | LDL-C (mg/dL) | 74.1 ± 22.2 | 74.0 ± 21.7 | 70.0 ± 21.3 | 0.49 | 82.2 ± 24.5 | 70.6 ± 19.9 | n/a | 0.15 |
| HDL-C (mg/dL) | 34.7 ± 5.5 | 35.0 ± 5.6 | 35.3 ± 5.9 | 0.81 | 35.7 ± 6.0 | 35.0 ± 3.8 | n/a | 0.97 | |
| TG (mg/dL) | 162 (131-216) | 167 (131-218) | 169 (145-211) | 0.46 | 157 (120-205) | 148 (120-182) | n/a | 0.24 | |
| Lp(a)(nmol/l) | 33 (13-123) | 32 (13-135) | 31 (15-74) | 0.88 | 84 (41-148) | 51 (40-67) | n/a | 0.54 | |
| White Ancestry (MAF 0.39) | Black Ancestry (MAF 0.16) | ||||||||
| SNP | Trait | MetMet (n=737) | MetIle (n=928) | IleIle (n=329) | p-value | MetMet (n=50) | MetIle (n=22) | IleIle (n=1) | p-value |
| p.M317I | LDL-C (mg/dL) | 74.0 ± 22.6 | 74.0 ± 21.4 | 73.4 ± 22.3 | 0.74 | 81.9 ± 24.7 | 76.8 ± 22.8 | 110.0 | 0.47 |
| HDL-C (mg/dL) | 34.7 ± 5.4 | 34.8 ± 5.7 | 35.1 ± 5.6 | 0.50 | 36.4 ± 5.7 | 33.7 ± 5.5 | 40.0 | 0.24 | |
| TG (mg/dL) | 163 (132-222) | 164 (131-215) | 168 (134-211) | 0.67 | 155± (125-203) | 172 (116-204) | 122 | 0.59 | |
| Lp(a)(nmol/l) | 32 (13-107) | 32 (13-140) | 31 (13-116) | 0.31 | 65 (37-135) | 97 (51-203) | 30 | 0.14 | |
Lipid traits reported as mean ± sd; TG and Lp(a) reported as median (interquartile range). TG and Lp(a) were log-transformed prior to analysis. P-values were calculated using a dominant model adjusting for age, sex, race and body mass index.
LDL-c= low-density lipoprotein cholesterol, HDL-c= high-density lipoprotein cholesterol, TG=triglycerides, Lp(a)= lipoprotein (a), MAF= minor allele frequency
We then tested whether each SNP was associated with the change in lipids from baseline to 1 year after randomization. The percent changes in LDL-C, HDL-C and TG were not significantly associated with HCAR2 variants in either persons of white (Table 4) or black (Table 5) ancestry. Interestingly, the mean reduction in Lp(a) in response to niacin was significantly greater in homozygous carriers of the 317M major allele (-22.7 ± 35%; p=0.005) than in carriers of the minor allele (-15.3 ± 39.4%) (Table 4, Figure 2). This was directionally consistent in the small number of black participants (Table 5), and upon combining both race groups the robust response of Lp(a) to niacin in the 317M homozygous major allele carriers was statistically significant (p=0.003). Additionally, the reduction in Lp(a) and 317M genotype was evaluated in an interaction model and there was a significant treatment × genotype effect (p-interaction=0.005). The Lp(a) lowering effect due to niacin did not follow an additive model, since a similar decrease was observed in 317I minor allele carriers. There were minimal changes in Lp(a) in the placebo group and no differences by HCAR2 genotype (Tables 4,5).
Table 4. Association of HCAR2 variants with percent change in lipid values at one year by treatment in the White Participants.
| Statin + Placebo | Statin + ER Niacin | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| SNP | Trait | ArgArg (n=614) | ArgCys (n=253) | CysCys (n=32) | p-value | ArgArg (n=624) | ArgCys (n=256) | CysCys (n=24) | p-value |
| p.R311C | LDL-C (mg/dL) | -0.8 ± 32.8 | -0.4 ± 28.2 | 6.3 ± 28.3 | 0.96 | -3.5 ± 36.7 | -2.1 ± 41.0 | -1.8 ± 28.5 | 0.55 |
| HDL-C (mg/dL) | 9.3 ± 15.4 | 11.9 ± 16.0 | 11.9 ± 16.9 | 0.049 | 28.2 ± 24.9 | 26.4 ± 21.8 | 27.9 ± 21.1 | 0.23 | |
| TG (mg/dL) | 2.4 ± 40.2 | 0.7 ± 38.3 | 2.4 ± 51.7 | 0.98 | -21.4 ±38.7 | -24.0 ± 37.8 | -24.0 ±30.5 | 0.49 | |
| Lp(a)(nmol/l) | -2.9 ± 41.5 | -6.7 ± 41.2 | -15.2 ± 33.5 | 0.069 | -19.7± 37.7 | -13.8 ± 39.3 | -20.4 ±34.2 | 0.049 | |
| SNP | Trait | MetMet (n=344) | MetIle (n=412) | IleIle (n=146) | p-value | MetMet (n=327) | MetIle (n=441) | IleIle (n=148) | p-value |
| p.M317I | LDL-C (mg/dL) | -2.4 ± 31.9 | 0.06 ± 31.9 | 3.1 ± 29.1 | 0.23 | -3.7 ± 39.1 | -2.6 ± 37.4 | -3.5 ± 35.2 | 0.58 |
| HDL-C (mg/dL) | 9.7 ± 15.3 | 9.8 ± 16.2 | 11.4 ± 14.5 | 0.47 | 28.2 ± 25.4 | 27.9 ± 23.5 | 26.1 ± 22.8 | 0.62 | |
| TG (mg/dL) | 2.0 ± 39.1 | 1.8 ± 38.5 | 3.3 ± 46.2 | 0.96 | -20.9 ±38.4 | -23.0± 38.6 | -22.5 ±36.2 | 0.50 | |
| Lp(a)(nmol/l) | -3.1 ± 37.2 | -5.0 ± 44.0 | -5.8 ± 41.1 | 0.37 | -22.7 ±35.2 | -15.2 ± 40.1 | -15.8 ±37.3 | 0.0053 | |
Lipid traits reported as mean (sd) percent change from baseline to one year. P-values were calculated from a dominant model adjusting for age, sex, body mass index and baseline lipid levels.
Table 5. Association of HCAR2 variants with percent change in lipid values at one year by treatment in the Black Participants.
| Statin + Placebo | Statin + ER Niacin | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| SNP | Trait | ArgArg (n=26) | ArgCys (n=1) | CysCys (n=0) | p-value | ArgArg (n=25) | ArgCys (n=6) | CysCys (n=0) | p-value |
| p.R311C | LDL-C (mg/dL) | 2.0 ± 36.0 | 56.6 | n/a | 0.29 | -20.0 ± 29.8 | -6.1 ± 25.4 | n/a | 0.39 |
| HDL-C (mg/dL) | 5.6 ± 17.0 | 14.8 | n/a | 0.81 | 24.2± 24.5 | 28.8 ± 19.1 | n/a | 0.76 | |
| TG (mg/dL) | -3.8 ± 35.0 | 4.1 | n/a | 0.91 | -18.2 ± 45.6 | -42.7 ± 27.3 | n/a | 0.18 | |
| Lp(a)(nmol/l) | -2.8 ± 29.1 | -8.0 | n/a | 0.85 | -17.9 ± 48.2 | -28.8 ± 29.1 | n/a | 0.63 | |
| SNP | Trait | MetMet (n=20) | MetIle (n=7) | IleIle (n=0) | p-value | MetMet (n=18) | MetIle (n=12) | IleIle (n=1) | p-value |
| p.M317I | LDL-C (mg/dL) | 2.7 ± 37.4 | 7.9 ± 37.7 | n/a | 0.30 | -15.7 ± 32.4 | -18.5 ± 25.9 | -31.8 | 0.57 |
| HDL-C (mg/dL) | 6.7 ± 17.9 | 3.7 ± 13.8 | n/a | 0.65 | 23.8 ± 28.0 | 26.8 ± 16.5 | 27.5 | 0.55 | |
| TG (mg/dL) | -3.9 ± 37.3 | -2.6 ± 26.2 | n/a | 0.86 | -14.9 ± 52.9 | -33.4 ± 23.6 | -41.0 | 0.21 | |
| Lp(a)(nmol/l) | -2.0 ± 32.9 | -5.7 ± 11.3 | n/a | 0.93 | -28.5 ± 30.9 | -1.2 ± 57.8 | -77.6 | 0.11 | |
Lipid traits reported as mean (sd) percent change from baseline to one year. P-values were calculated from a co-dominant model adjusting for age, sex and body mass index.
Figure 2.
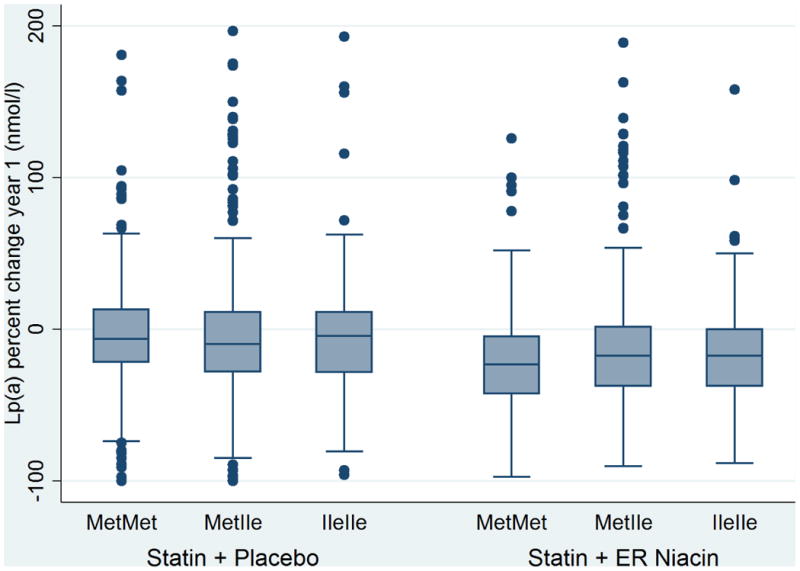
Change in Lipoprotein (a) levels by HCAR2 (rs2454727) genotype in white subject from the AIM-HIGH trial. The reduction in Lp(a) in response to niacin was greater in homozygous 317M carriers compared with 317I carriers (p=0.0053 in the niacin group; p=0.37 in the placebo group; p-interaction =0.005). The boxes on the boxplots represent the 75th percentile, median, and 25th percentile.
We then tested whether the cardiovascular event rate was different by HCAR2 genotype by Cox regression modeling. HCAR2 genotype did not increase the hazard of a CV event in either the placebo or niacin group (Table 6, Figure S2).
Table 6. Cardiovascular events by HCAR2 genotype in AIM-HIGH participants.
| SNP | Genotype | Statin + placebo (n=1,020) | HR | 95% CI | p-value | Statin + ER Niacin (n=1,047) | HR | 95% CI | p-value |
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| p.R311C | Arg/Arg | 121 (17.3%) | 0.87 | 0.65, 1.17 | 0.38 | 114 (15.7%) | 0.99 | 0.74, 1.32 | 0.94 |
| Arg/Cys | 41 (14.6%) | 49 (17.3%) | |||||||
| Cys/Cys | 5 (13.5%) | 2 (7.1%) | |||||||
|
| |||||||||
| p.M317I | Met/Met | 69 (17.8%) | 0.91 | 0.73, 1.14 | 0.42 | 65 (16.8%) | 1.06 | 0.85, 1.32 | 0.61 |
| Met/Ile | 71 (14.3%) | 72 (14.5%) | |||||||
| Ile/Ile | 24 (14.4%) | 34 (20.7%) | |||||||
HR= hazard ratio from Cox regression adjusted for age, sex, race, BMI
Discussion
In this study we evaluated whether two coding variants in HCAR2 (p.R311C and p.M317I) were associated with lipid and Lp(a) response to niacin in the AIM-HIGH trial. Polymorphisms in HCAR2 did not predict the change in LDL-C, HDL-C or TG after one year of niacin therapy. The reduction in Lp(a) in response to niacin was significantly greater in the homozygous carriers of the 317M major allele compared with minor allele carriers. Our findings confirm a previous report suggesting that niacin's lipid effect is independent of the HCAR2 receptor.[13] However, this is the first study to suggest that activation of the HCAR2 receptor by niacin may mediate Lp(a) lowering. Neither HCAR2 variant was associated with cardiovascular events in our analysis. This study is the first large association study of these variants and the first pharmacogenetic study of niacin therapy.
The functionality of the p.R311C and p.M317I variants in HCAR2 is not well known. The first SNP results in an arginine to cysteine substitution at amino acid 311, and the latter SNP results in a methionine to isoleucine substitution at amino acid 317. Prediction tools [27] to identify pathogenic variants such as Polyphen and SIFT have categorized these variants as benign. However, these tools rely heavily on sequence conservations among species and may not result in reliable predictions for variants located in a residue that is not present in the majority of species, as is the case for HCAR2. In addition, many functional prediction tools do not account for the location of the variants, which may have a substantial impact if they occur in a critical region of the protein or are involved with protein-protein interactions [27]. The p.R311C and p.M317I variants are not located in the transmembrane regions of the receptor and therefore are not involved with niacin binding [28]. Rather, they are found in the c-terminal tail, which has been demonstrated to be functionally important for facilitating the interactions with various cellular proteins regulating receptor desensitization and endocytosis in several GPCR family members [29]. In vitro experiments involving mutations of structural motifs in the c-terminal region of HCAR2 found that this region plays an important role in the regulation of the receptor surface localization and specifically the Δ315-328 mutant, the region of the M317I variant, exhibited a significant decrease in niacin-mediated internalization and arrestin3 translocation [30]. While mutagenesis experiments for the specific coding variants we describe have not been published in the peer-reviewed literature, patent literature from Arena Pharmaceuticals has shown that the 317I variant results in reduced ERK 1/2 phosphorylation and mitogen activated protein kinase (MAPK) activation in cell based assays [31]. If this is the case, individuals with this polymorphism would be expected to have reduced ERK 1/2 phosphorylation in response to niacin therapy.
Lipoprotein (a) is an atherogenic particle that has been associated with increased risk of coronary disease and stroke [4,5]. Lp(a) consists of an LDL-like particle bound to a plasminogen-like glycoprotein, apolipoprotein (a) [32]. Lp(a) levels are highly genetically influenced, with copy-number variation within the kringle IV- type 2 repeat of apolipoprotein (a) explaining 40% of the variation [33]. The number of kringle IV-2 repeats are inversely correlated with Lp(a) levels [33]. The contribution of Lp(a) on CVD risk is not well understood, but due to its structural composition it may contribute to both atherosclerosis and thrombosis [32].
In this study we found significantly greater reduction in Lp(a) in response to niacin in homozygous carriers of the 317M major allele compared with carriers of the minor allele. It is unknown how signaling through HCAR2 receptor may influence plasma Lp(a) concentrations. Lp(a) is found in atherosclerotic plaques and involved with macrophage foam cell formation similar to LDL [32]. The HCAR2 receptor is also found on macrophage and niacin acting via HCAR2 has been shown to stimulate cholesterol efflux by increasing ABCA1, ABCG1 and CD36 expression [17,34,35]. Carriers of the major allele may have enhanced ability to efflux lipid from foam cells. Alternatively, niacin may also work to lower Lp(a) via anti-inflammatory mechanisms. Lp(a) via its apo(a) component has be shown to promote oxidative and inflammatory actions on the vascular wall [32,36]. The apo(a) domain was found to increase IL-8 mRNA and protein expression in macrophage via the oxidized phospholipid (oxPL) modification of Lp(a) which facilitated the interaction of Lp(a) with CD36 and macrophage toll-like receptor [37]. Niacin also has anti-inflammatory effects that have been shown to be dependent on HCAR2 receptor activation on immune cells including macrophages, monocytes, neutrophils and dermal dendritic cells [17,20,38,39]. Niacin activation of HCAR2 inhibited monocyte chemotactic protein-1 (MCP-1) induced macrophage recruitment into atherosclerotic plaques; this effect was abolished in HCAR2-deficient mice [17]. Chemokine signaling via MCP-1 is a key component of immune cell recruitment into the atherosclerotic vessel wall, and niacin may reduce atherosclerosis by interfering with intracellular signaling pathways induced through chemokine receptors, which are also mediated through Gi-type G-proteins [17,40]. Since the HCAR2 317I variant causes reduced intracellular signaling as shown by diminished ERK1/2 phosphorylation and MAPK activation, it is conceivable that this variant cannot activate the necessary signaling pathways to interfere with macrophage chemotaxis and therefore would exhibit a reduced anti-inflammatory and anti-atherosclerotic response when exposed to niacin. However, we were not able to show a difference in cardiovascular events rates by HCAR2 genotype, likely due to the low number of events in the overall trial.
While we performed the genetic evaluation within the context of a prospective, randomized placebo-controlled design with longitudinal follow-up of lipids and cardiovascular events, this study has several limitations. We did not have access to another cohort of chronic niacin treatment to replicate our findings, thus our findings would require replication. It would be worth investigating whether these findings could be replicated in the HPS2-THRIVE study which employed a similar trial design [11]. The overall number of cardiovascular events was low in this study, limiting our ability to evaluate the effect of HCAR2 variants on these outcomes. The two coding variants we investigated are not found on genome wide arrays, likely due to 95% sequence homology of HCAR2 with HCAR3[24], and therefore have not been associated with lipid traits in prior genome-wide association studies [41,42]. We employed a two -step process in order to successfully amplify the SNP region, and this added step may limit high throughput genotyping in larger cohorts. Despite high homology to HCAR2, HCAR3 has ∼1000-fold less affinity for nicotinic acid and has been identified as a receptor for 3-OH-octanoic acid [43,44]. Additionally further work in required to clarify the role of Lp(a) levels as a contributor to cardiovascular risk in the black population. It is known that blacks have a higher Lp(a) levels than the white population [45], but these higher levels have not been consistently shown to translate into a higher risk of cardiovascular events in blacks [46,47]. Consensus statements from the European Atherosclerosis Society states that individuals are at increased risk for cardiovascular disease at levels >30 mg/dL (approximately equivalent to 75 mmol/L by newer assays)[48], but these cut-points may not apply to individuals of black or Asian ancestry [49]. We also observed higher Lp(a) levels at baseline in the AIM-HIGH trial, but the greatest benefit in terms of Lp(a) lowering due to niacin was observed in homozygous carriers of the 317M allele in both race groups. It is still unclear from the literature what is the optimal level of Lp(a) reduction required in black individuals to achieve cardiovascular risk reduction.
In conclusion, we observed differential Lp(a) lowering based on genotype at the M317I variant in response to niacin. We did not find an association, however, between coding variants in HCAR2 and the change in plasma LDL-C, HDL-C or TG with niacin treatment. Understanding the genetic contribution to variation in response to niacin therapy, including Lp(a) reduction, could uncover mechanisms by which niacin decreases Lp(a), an important independent risk factor for cardiovascular disease.
Supplementary Material
Supplemental Figure 1. Linkage Disequilibrium Plots obtained from the sequencing study in healthy subjects. One major LD block was identified in the (A) white and (B) black subjects. The two nonsynonymous variants, rs7314976 and rs2454727 were in strong LD.
Supplemental Figure 2. Kaplan-Meier Survival Curves for Cardiovascular Events. Time to event by HCAR2 M317I variants in white participants shown by treatment (A) statin + placebo and (B) statin + ER niacin. There was no difference in time to event by genotype.
Acknowledgments
Funding: This work has been funded by NHLBI grant R01HL086864 and a NIH-NHLBI SCCOR Project grant (P50-HL-083799) to DJR. ST was supported by a Clinical Research Program grant from the American Heart Association (11CRP7610016). AIM-HIGH was supported by the National Heart, Lung, and Blood Institute (U01 HL081616 and U01 HL081649) and by an unrestricted grant from AbbVie, Inc. AbbVie donated the extended release niacin, the matching placebo, and the ezetimibe; Merck donated the simvastatin. Neither of these companies had any role in the oversight or design of the study or in the analysis or interpretation of the data.
Footnotes
Conflict of Interest: RLD has consulted for Catabasis Pharmaceuticals, Inc., the maker of two novel niacin pro-drugs. All other authors have no conflicts.
References
- 1.Altschul R, Hoffer A, Stephen JD. Influence of nicotinic acid on serum cholesterol in man. Arch Biochem. 1955;54(2):558–559. doi: 10.1016/0003-9861(55)90070-9. [DOI] [PubMed] [Google Scholar]
- 2.Carlson LA. Nicotinic acid: the broad-spectrum lipid drug. A 50th anniversary review. J Intern Med. 2005;258(2):94–114. doi: 10.1111/j.1365-2796.2005.01528.x. [DOI] [PubMed] [Google Scholar]
- 3.Carlson LA, Hamsten A, Asplund A. Pronounced lowering of serum levels of lipoprotein Lp(a) in hyperlipidaemic subjects treated with nicotinic acid. J Intern Med. 1989;226(4):271–276. doi: 10.1111/j.1365-2796.1989.tb01393.x. [DOI] [PubMed] [Google Scholar]
- 4.Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301(22):2331–2339. doi: 10.1001/jama.2009.801. [DOI] [PubMed] [Google Scholar]
- 5.Clarke R, Peden JF, Hopewell JC, Kyriakou T, Goel A, Heath SC, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361(26):2518–2528. doi: 10.1056/NEJMoa0902604. [DOI] [PubMed] [Google Scholar]
- 6.Clofibrate and niacin in coronary heart disease. JAMA. 1975;231(4):360–381. [PubMed] [Google Scholar]
- 7.Canner PL, Berge KG, Wenger NK, Stamler J, Friedman L, Prineas RJ, et al. Fifteen year mortality in Coronary Drug Project patients: long-term benefit with niacin. J Am Coll Cardiol. 1986;8(6):1245–1255. doi: 10.1016/s0735-1097(86)80293-5. [DOI] [PubMed] [Google Scholar]
- 8.Taylor AJ, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER) 2: a double-blind, placebo-controlled study of extended-release niacin on atherosclerosis progression in secondary prevention patients treated with statins. Circulation. 2004;110(23):3512–3517. doi: 10.1161/01.CIR.0000148955.19792.8D. [DOI] [PubMed] [Google Scholar]
- 9.Taylor AJ, Villines TC, Stanek EJ, Devine PJ, Griffen L, Miller M, et al. Extended-release niacin or ezetimibe and carotid intima-media thickness. N Engl J Med. 2009;361(22):2113–2122. doi: 10.1056/NEJMoa0907569. [DOI] [PubMed] [Google Scholar]
- 10.Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365(24):2255–2267. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- 11.Landray MJ, Haynes R, Hopewell JC, Parish S, Aung T, Tomson J, et al. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371(3):203–212. doi: 10.1056/NEJMoa1300955. [DOI] [PubMed] [Google Scholar]
- 12.Tunaru S, Kero J, Schaub A, Wufka C, Blaukat A, Pfeffer K, et al. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat Med. 2003;9(3):352–355. doi: 10.1038/nm824. [DOI] [PubMed] [Google Scholar]
- 13.Lauring B, Taggart AKP, Tata JR, Dunbar R, Caro L, Cheng K, et al. Niacin Lipid Efficacy Is Independent of Both the Niacin Receptor GPR109A and Free Fatty Acid Suppression. Science Translational Medicine. 2012;4(148):148ra115. doi: 10.1126/scitranslmed.3003877. [DOI] [PubMed] [Google Scholar]
- 14.Kamanna VS, Kashyap ML. Mechanism of action of niacin. Am J Cardiol. 2008;101(8A):20B–26B. doi: 10.1016/j.amjcard.2008.02.029. [DOI] [PubMed] [Google Scholar]
- 15.Jin FY, Kamanna VS, Kashyap ML. Niacin decreases removal of high-density lipoprotein apolipoprotein A-I but not cholesterol ester by Hep G2 cells. Implication for reverse cholesterol transport. Arterioscler Thromb Vasc Biol. 1997;17(10):2020–2028. doi: 10.1161/01.atv.17.10.2020. [DOI] [PubMed] [Google Scholar]
- 16.van der Hoorn JW, de Haan W, Berbee JF, Havekes LM, Jukema JW, Rensen PC, et al. Niacin increases HDL by reducing hepatic expression and plasma levels of cholesteryl ester transfer protein in APOE*3Leiden.CETP mice. Arterioscler Thromb Vasc Biol. 2008;28(11):2016–2022. doi: 10.1161/ATVBAHA.108.171363. [DOI] [PubMed] [Google Scholar]
- 17.Lukasova M, Malaval C, Gille A, Kero J, Offermanns S. Nicotinic acid inhibits progression of atherosclerosis in mice through its receptor GPR109A expressed by immune cells. J Clin Invest. 2011;121(3):1163–1173. doi: 10.1172/JCI41651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanson J, Gille A, Zwykiel S, Lukasova M, Clausen BE, Ahmed K, et al. Nicotinic acid- and monomethyl fumarate-induced flushing involves GPR109A expressed by keratinocytes and COX-2-dependent prostanoid formation in mice. J Clin Invest. 2010;120(8):2910–2919. doi: 10.1172/JCI42273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plaisance EP, Lukasova M, Offermanns S, Zhang Y, Cao G, Judd RL. Niacin stimulates adiponectin secretion through the GPR109A receptor. American Journal of Physiology - Endocrinology And Metabolism. 2009;296(3):E549–E558. doi: 10.1152/ajpendo.91004.2008. [DOI] [PubMed] [Google Scholar]
- 20.Digby JE, Martinez F, Jefferson A, Ruparelia N, Chai J, Wamil M, et al. Anti-Inflammatory Effects of Nicotinic Acid in Human Monocytes Are Mediated by GPR109A Dependent Mechanisms. Arteriosclerosis, Thrombosis, and Vascular Biology. 2012;32(3):669–676. doi: 10.1161/ATVBAHA.111.241836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu BJ, Yan L, Charlton F, Witting P, Barter PJ, Rye KA. Evidence That Niacin Inhibits Acute Vascular Inflammation and Improves Endothelial Dysfunction Independent of Changes in Plasma Lipids. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30(5):968–975. doi: 10.1161/ATVBAHA.109.201129. [DOI] [PubMed] [Google Scholar]
- 22.Aslibekyan S, Straka RJ, Irvin MR, Claas SA, Arnett DK. Pharmacogenomics of high-density lipoprotein-cholesterol-raising therapies. Expert Rev Cardiovasc Ther. 2013;11(3):355–364. doi: 10.1586/erc.12.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Albers JJ, Slee A, O'Brien KD, Robinson JG, Kashyap ML, Kwiterovich PO, Jr, et al. Relationship of Apolipoproteins A-1 and B, and Lipoprotein(a) to Cardiovascular Outcomes: The AIM-HIGH Trial (Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglyceride and Impact on Global Health Outcomes) J Am Coll Cardiol. 2013;62(17):1575–1579. doi: 10.1016/j.jacc.2013.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zellner C, Pullinger CR, Aouizerat BE, Frost PH, Kwok PY, Malloy MJ, et al. Variations in human HM74 (GPR109B) and HM74A (GPR109A) niacin receptors. Hum Mutat. 2005;25(1):18–21. doi: 10.1002/humu.20121. [DOI] [PubMed] [Google Scholar]
- 25.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 26.Lettre G, Lange C, Hirschhorn JN. Genetic model testing and statistical power in population-based association studies of quantitative traits. Genet Epidemiol. 2007;31(4):358–362. doi: 10.1002/gepi.20217. [DOI] [PubMed] [Google Scholar]
- 27.Ohanian M, Otway R, Fatkin D. Heuristic methods for finding pathogenic variants in gene coding sequences. J Am Heart Assoc. 2012;1(5):e002642. doi: 10.1161/JAHA.112.002642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tunaru S, Lattig J, Kero J, Krause G, Offermanns S. Characterization of determinants of ligand binding to the nicotinic acid receptor GPR109A (HM74A/PUMA-G) Mol Pharmacol. 2005;68(5):1271–1280. doi: 10.1124/mol.105.015750. [DOI] [PubMed] [Google Scholar]
- 29.Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev. 2001;53(1):1–24. [PubMed] [Google Scholar]
- 30.Li G, Zhou Q, Yu Y, Chen L, Shi Y, Luo J, et al. Identification and Characterization of Distinct C-Terminal Domains of the Human Hydroxycarboxylic Acid Receptor-2 That Are Essential for Receptor Export, Constitutive Activity, Desensitization, and Internalization. Molecular Pharmacology. 2012;82(6):1150–1161. doi: 10.1124/mol.112.081307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liaw C, Kanemitsu-Parks M, Richman J, Maciejewski-Lenoir D, Connolly D. Methods for determining probability of an adverse or favorable reaction to a niacin receptor agonist. United States. 2009 [Google Scholar]
- 32.Tsimikas S, Hall JL. Lipoprotein(a) as a potential causal genetic risk factor of cardiovascular disease: a rationale for increased efforts to understand its pathophysiology and develop targeted therapies. J Am Coll Cardiol. 2012;60(8):716–721. doi: 10.1016/j.jacc.2012.04.038. [DOI] [PubMed] [Google Scholar]
- 33.Gavish D, Azrolan N, Breslow JL. Plasma Ip(a) concentration is inversely correlated with the ratio of Kringle IV/Kringle V encoding domains in the apo(a) gene. J Clin Invest. 1989;84(6):2021–2027. doi: 10.1172/JCI114395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaidarov I, Chen X, Anthony T, Maciejewski-Lenoir D, Liaw C, Unett DJ. Differential tissue and ligand-dependent signaling of GPR109A receptor: Implications for anti-atherosclerotic therapeutic potential. Cellular Signalling. 2013;25(10):2003–2016. doi: 10.1016/j.cellsig.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 35.Rubic T, Trottmann M, Lorenz RL. Stimulation of CD36 and the key effector of reverse cholesterol transport ATP-binding cassette A1 in monocytoid cells by niacin. Biochem Pharmacol. 2004;67(3):411–419. doi: 10.1016/j.bcp.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 36.Sotiriou SN, Orlova VV, Al-Fakhri N, Ihanus E, Economopoulou M, Isermann B, et al. Lipoprotein(a) in atherosclerotic plaques recruits inflammatory cells through interaction with Mac-1 integrin. FASEB J. 2006;20(3):559–561. doi: 10.1096/fj.05-4857fje. [DOI] [PubMed] [Google Scholar]
- 37.Scipione CA, Sayegh SE, Romagnuolo R, Tsimikas S, Marcovina SM, Boffa MB, et al. Mechanistic insights into Lp(a)-induced IL-8 expression: a role for oxidized phospholipid modification of apo(a) J Lipid Res. 2015;56(12):2273–2285. doi: 10.1194/jlr.M060210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zandi-Nejad K, Takakura A, Jurewicz M, Chandraker AK, Offermanns S, Mount D, et al. The role of HCA2 (GPR109A) in regulating macrophage function. The FASEB Journal. 2013 doi: 10.1096/fj.12-223933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Graff EC, Fang H, Wanders D, Judd RL. Anti-inflammatory effects of the hydroxycarboxylic acid receptor 2. Metabolism. 2016;65(2):102–113. doi: 10.1016/j.metabol.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 40.Offermanns S. The nicotinic acid receptor GPR109A (HM74A or PUMA-G) as a new therapeutic target. Trends Pharmacol Sci. 2006;27(7):384–390. doi: 10.1016/j.tips.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 41.Postmus I, Trompet S, Deshmukh HA, Barnes MR, Li X, Warren HR, et al. Pharmacogenetic meta-analysis of genome-wide association studies of LDL cholesterol response to statins. Nature communications. 2014;5:5068. doi: 10.1038/ncomms6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45(11):1274–1283. doi: 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taggart AKP, Kero J, Gan X, Cai TQ, Cheng K, Ippolito M, et al. (d)-β-Hydroxybutyrate Inhibits Adipocyte Lipolysis via the Nicotinic Acid Receptor PUMA-G. Journal of Biological Chemistry. 2005;280(29):26649–26652. doi: 10.1074/jbc.C500213200. [DOI] [PubMed] [Google Scholar]
- 44.Ahmed K, Tunaru S, Langhans CD, Hanson J, Michalski CW, Kölker S, et al. Deorphanization of GPR109B as a Receptor for the β-Oxidation Intermediate 3-OH-octanoic Acid and Its Role in the Regulation of Lipolysis. Journal of Biological Chemistry. 2009;284(33):21928–21933. doi: 10.1074/jbc.M109.019455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marcovina SM, Albers JJ, Wijsman E, Zhang Z, Chapman NH, Kennedy H. Differences in Lp[a] concentrations and apo[a] polymorphs between black and white Americans. J Lipid Res. 1996;37(12):2569–2585. [PubMed] [Google Scholar]
- 46.Collaboration TERF. Lipoprotein(a) Concentration and the Risk of Coronary Heart Disease, Stroke, and Nonvascular Mortality. JAMA: The Journal of the American Medical Association. 2009;302(4):412–423. doi: 10.1001/jama.2009.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Virani SS, Brautbar A, Davis BC, Nambi V, Hoogeveen RC, Sharrett AR, et al. Associations between lipoprotein(a) levels and cardiovascular outcomes in black and white subjects: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2012;125(2):241–249. doi: 10.1161/CIRCULATIONAHA.111.045120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nordestgaard BG, Chapman MJ, Ray K, Boren J, Andreotti F, Watts GF, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31(23):2844–2853. doi: 10.1093/eurheartj/ehq386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marcovina SM, Albers JJ. Lp(a) Measurements for Clinical Application. Journal of Lipid Research. 2015 doi: 10.1194/jlr.R061648. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Linkage Disequilibrium Plots obtained from the sequencing study in healthy subjects. One major LD block was identified in the (A) white and (B) black subjects. The two nonsynonymous variants, rs7314976 and rs2454727 were in strong LD.
Supplemental Figure 2. Kaplan-Meier Survival Curves for Cardiovascular Events. Time to event by HCAR2 M317I variants in white participants shown by treatment (A) statin + placebo and (B) statin + ER niacin. There was no difference in time to event by genotype.