

Abstract
In voltage-dependent channels, positive charges contained within the S4 domain are the voltage-sensing elements. The “voltage-sensor paddle” gating mechanism proposed for the KvAP K+ channel has been the subject of intense discussion regarding its general applicability to the family of voltage-gated channels. In this model, the voltage sensor composed of the S3b and the S4 segment shuttles across the lipid bilayer during channel activation. Guided by this mechanism, we assessed here the accessibility of residues in the S3 segment of the Shaker K+ channel by using cysteine-scanning mutagenesis. Mutants expressed robust K+ currents in Xenopus oocytes and reacted with methanethiosulfonate ethyltrimethylammonium in both closed and open conformations of the channel. Because Shaker has a long S3–S4 linker segment, we generated a deletion mutant with only three residues to emulate the KvAP structure. In this short linker mutant, all of the tested residues in the S3b were accessible to methanethiosulfonate ethyltrimethylammonium in both closed and open conformations. Because the S3b moves together with the S4 domain in the paddle model, we tested the effects of deleting two negative charges or adding a positive charge to this region of the channel. We found that altering the S3b net charge does not modify the total gating charge involved in channel activation. We conclude that the S3b segment is always exposed to the external milieu of the Shaker K+ channel. Our results are incompatible with any model involving a large membrane displacement of segment S3b.
Keywords: methanethiosulfonate ethyltrimethylammonium accessibility, paddle model, S3–S4 linker, gating charge
Voltage-dependent K+ channels are membrane proteins that are formed by four repeating monomers. The secondary structure of the monomers has six α-helix segments (S1–S6), which are divided into a voltage-sensing domain (S1–S4) and an ion-conducting pore domain (S5–S6). In Shaker K+ channels, four charged arginine residues belonging to the S4 segment are driven from the intracellular to the extracellular side of the structure upon membrane depolarization (1–5). This conformational change of the protein leads to the opening of the ion-conducting pore. Three molecular models of the mechanism of this charge movement have evolved based on various experimental approaches, including crystallography, biochemistry, and electrophysiology. First, in the “classic” model, the S4 segment is enclosed in the protein and its charged residues travel in and out, driven by the electric field (6–8). The S4 segment movement can be a transmembrane translation from extracellular to intracellular crevices through a “gating pore” (9–14). This variation of the classical model is the “sliding-helix” model. An alternative is a rotation of the S4 segment, with minimal transmembrane displacement. As the segment rotates, it exposes the charges residues to either the intracellular or the extracellular crevices. This is the “rotating helix” model (15, 16). Second, the “paddle” model proposes that the S4 segment is in the periphery of the protein and travels in and out through the lipid bilayer (17). The third model has been denominated “collapsing gating canal” because, in this model, the protein surrounding the S4 segment changes its conformation, and the S4 segment does not move at all with respect to the membrane plane (4, 10, 18). The participation of the S3 segment is different in these three models. For channels with a short S3–S4 linker, such as KvAP, the sliding-helix model predicts that the S3 segment is dragged into the protein by S4 when the channel undergoes a conformational change from activated to resting. However, S3b stays extracellular for long S3–S4 linker channels such as Shaker (6). In the paddle model, the C terminus half of S3 (S3b) shuttles in and out as a unit tightly bound to S4. In the collapsing gating canal model, the S3b does not have a role in charge movement. It is important to note that, of all of the models discussed above, the paddle model is the only one that makes strong predictions regarding what should happens to the S3b segment. In this study, we tested these predictions by using two different experimental strategies. First, we examined the accessibility of S3 residues by using cysteine-scanning mutagenesis and, second, we measured the contribution of the S3 segment to the gating charges of Shaker. Mutagenesis and accessibility studies on the Shaker channel demonstrated that S3b residues are exposed to extracellular thiol-reactive methanethiosulfonate ethyltrimethylammonium (MTSET) in the open and in the closed conformation. The same result holds for a Shaker mutant where the S3–S4 linker is deleted. However, we found that genetic manipulation of the charges in the S3b segment has no impact on the gating charge of these channels. We conclude that the S3b segment does not leave the extracellular side of the channel in its resting conformation.
Methods
Molecular Biology. Constructs used in this study did not have N-type inactivation ShakerH4Δ (6–46) (ShakerΔ). The ShakerΔ K+ channel cDNA was originally cloned into an engineered version of the pBSTA vector. The S3–S4 deletion mutants were prepared by PCR-based mutagenesis (Expand Hi Fidelity PCR System, Boehringer Mannheim; or Pfu DNA polymerase, Stratagene). Primer design, purification of the PCR products, transformation of the dh5α or xli blue-competent Escherichia coli cells, colony selection, DNA sequencing, subcloning, and transcription to cRNA were done as described in ref. 19. Splicing by overlap extension of two PCR products bearing overlapping complementary ends was used to construct the single-point-mutation cysteine mutants. The mutated constructs contained the flanking restriction sites XbaI and Eco91I for insertion as cassettes into the original plasmids for expression.
Electrophysiology. Xenopus oocytes were injected with 50 nl of cRNA solution (0.1–0.2 μg/μl). After 2–4 days of mRNA injection, macroscopic currents induced by the expression of Shaker channels in the membrane of the oocytes were recorded by using the cut-open oocyte voltage clamp (20) or the patch–clamp technique in cell-attached configuration (19, 21). The extracellular solution contained 110 mM K·Mes, 2 mM CaCl2, and 10 mM Hepes (pH 7.4). The intracellular solution contained 110 mM K·Mes, 2 mM EDTA, and 10 mM Hepes (pH 7.4). The microelectrode used to measure the membrane voltage contained 2.7 M Na·Mes, 10 mM NaCl, and 10 mM Hepes (pH 7.0). Microelectrode resistance was 100–300 kΩ. Ionic currents were usually acquired at 2.5 kHz and filtered at 500 Hz. Patch–clamp pipettes were 30–60 μm in diameter with a resistance of 400–600 kΩ. The conductance–voltage curve was obtained by using tail-current measurements. Initial current amplitude was calculated from the exponential fit to the tail-current time course. Normalized conductance, G/Gmax data were fitted by using the following Boltzmann function:
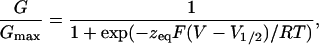 |
[1] |
where F is Faraday's constant, R is the universal gas constant, T is the absolute temperature, zeq is the equivalent gating charge per channel, V is the membrane voltage, and V1/2 is the voltage where the Boltzmann distribution is equal to 1/2.
MTSET Labeling. Labeling of the channel by MTSET was performed by superfusion of the oocyte under cut-open oocyte voltage clamp. The MTSET solution was freshly prepared by directly dissolving the compound in the external solution to a final concentration of 100 μM. Before the accessibility experiments, the oocytes were treated with the reducing agent DTT (50–100 μM) for 15 min. We followed the time course of channel modification by using a closed and a closed/open protocol (see Fig. 2 A and B). In the closed protocol, the membrane was kept at –110 mV for 10 s, and current was measured at the end of a 40- to 100-mV test pulse lasting for 40 ms. In the closed/open protocol, the membrane was kept at –110 mV for 1 s, and current was measured at the end of a 80- to 160-mV test pulse lasting for 400 ms. MTSET solutions were prepared just before the accessibility experiment was started. MTSET concentration was not corrected for hydrolysis because the total time of the experiment was never >2 min. In all of the cysteine-substituted channels, labeling followed a monoexponential function of the time-MTSET concentration coordinate (Ms). We obtained second-order rate constant (kclosed, M–1·s–1) for the closed conformation from the exponential-curve fitting on experiments applying the closed protocol. For experiments using the open/closed protocol, the result of the curve fitting, kopen/closed, contains the rate constant for the open and closed conformation: kopen and kclosed. The second-order rate constant kopen was calculated from kopen/closed by using the equation kopen/closed = (0.4/1.4)kopen + (1/1.4)kclosed because the membrane was depolarized during 0.4 s and hyperpolarized during 1 s. We used the average of kclosed obtained in a separate set of experiments. MTSET did not modify the electrophysiological characteristics of ShakerΔ channels.
Fig. 2.

Voltage protocols for the assay of MTSET modification. (A) Closed protocol. The oocyte was clamped to a holding potential of –110 mV, channels were opened by applying a test pulse to either 40 or 100 mV with a duration of 40 ms, and the repetition rate was a pulse every 10 s. The magnitude of the test pulse was chosen by considering the characteristics of the voltage activation curve of the mutant channel. MTSET was added to the external side to a final concentration of 100 μM. Currents were measured by using the cut-open oocyte voltage-clamp technique. (B) Open/closed protocol. Channels were kept open by applying an 80–120 mV pulse with a 400-ms duration from a holding potential of –110. The total time in the closed state was 1 s. MTSET was added to the external side to a final concentration of 100 μM. (C) External accessibility of cysteines to MTSET. Cysteines were in positions 328, 326, 324, and 321 in the S3b segment and position 315 in the C terminus of segment S3a. All tested positions were accessible to MTSET in both the open/closed and the closed states. Rate constants expressed in M–1·s–1 are given in the figure for a representative experiment of each of the mutants. Note that the control Shaker channel contains a cysteine in position 308 that is inaccessible to the thiol reagent.
Special care was taken to eliminate the possibility that the reaction between open channel and MTSET was just a consequence of accumulation of slow inactivation. Control experiments that omitted the addition of external MTSET were performed under the experimental conditions set by the open-pulse protocol. These experiments demonstrated that slow inactivation is either not developed or follows a time course, which is on a completely different temporal scale to that of the MTSET modification.
Limiting-Slope Analysis. The effective gating charge coupled to channel opening was obtained from the limiting-slope analysis (22–24) by using cell-attached patch clamp. The ionic current recorded during a voltage ramp was subtracted off line from the linear leakage and subsequently converted into conductance [G(V)] by dividing the ionic current by its driving force. The G–V plot was fitted to the following monoexponential approximation in the limit of very negative voltages:
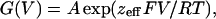 |
[2] |
where, zeff is the number of effective charges per channel, R is the universal gas constant, T is the absolute temperature, and F is Faraday's constant. The zeff value was obtained also from the following relationship:
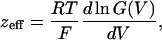 |
[3] |
where the derivative was calculated as ΔG(V)/G(V)ΔV by using ΔV = 1 mV.
As pointed out by Zagotta et al. (25), a plot of zeff as a function of conductance is a good test of having reached the true limiting slope. In this plot, at very negative voltages, zeff will asymptotically approach a saturating value that is equal to the total number of charges per channel.
Results
Accessibility of S3b Amino Acid Residues in a Native Length S3–S4 Linker K+ Channel. Here, we use the S3a and S3b segment definitions given by Jiang et al. (26), based on a sequence alignment with KvAP (Fig. 1A). To probe the accessibility of the S3b region of the protein, we mutated selected residues in the S3b domain to cysteines and probed their reactivity with MTSET. In control experiments, we found that MTSET exposure causes no change in channel properties; therefore, the six natural occurring cysteine residues, including Cys-308 on S3a, are not accessible to the reagent in either the open or closed configuration of the channel. As an additional control, we prepared Shaker mutation V363C on the S4 segment, and we found that MTSET reacts with cysteine in the open conformation only, as expected from previous reports on the reactivity of S4 segment residues (data not shown) (9, 10).
Fig. 1.

Shaker cysteine mutants express robust voltage-dependent currents. (A) Partial amino acid sequence of Shaker. Segments S3a, S3b, and S4 (bold) and the bar above the segments are assigned after Jiang et al. (26). Vertical green arrows show the native cysteines and the positions of the point mutations for cysteines. Horizontal double-headed arrows mark the four deleted regions. (B) Functional expression in Xenopus oocytes of cysteine mutants made in a ShakerΔ background. The holding potential was –100 mV, and the membrane was pulsed to voltages between –70 to +130 mV in 5 mV increments, followed by a step to –60 mV for ShakerΔ and the I315C, F324C, and T326C mutants. For the I321C and A328C, the holding potential was –100 mV and the membrane was pulsed to voltages between –100 to +10 mV in 5-mV increments, followed by a step to –100 mV. All current measurements were performed by using cell-attached patch–clamp technique. (C) Effects on the voltage activation induced by the mutation of S3 amino acid residues in a ShakerΔ background to cysteines. •, ShakerΔ WT; ○, I315C; ▴, I321C; ♦, F324C; +, T326C; □, A328C. Each point is the average of determinations on four to seven separate patches. Solid lines were drawn by using Eq. 1. V1/2 values were –29 ± 5 mV, –2 ± 16 mV, –47 ± 1 mV, 4 ± 15 mV, –17 ± 12 mV, and –36 ± 6 mV for ShakerΔ, I315C, I321C, F324C, T326C, and A328C, respectively. The equivalent numbers of gating charges, zeq, were 2.5 ± 1.2, 1.2 ± 0.9, 4.4 ± 0.1, 1.1 ± 0.2, 1.3 ± 0.6, and 4.5 ± 1.6 for ShakerΔ, I315C, I321C, F324C, T326C, and A328C, respectively.
The cysteine-scanning mutagenesis of S3 segment that we performed includes point mutations I315C on the C terminus of S3a, I321C, F324C, and T326C and A328C on S3b (Fig. 1A). Fig. 1B shows the macroscopic current records measured from channels expressed in Xenopus oocytes by using cell-attached patch–clamp technique. Shaker K+ channel cysteine mutants expressed robust currents with steady-state G–V curves comparable with those of the ShakerΔ control. Fig. 1C shows the open probability vs. voltage curves for the different mutants. Perturbations of the free-energy difference between the open and closed conformations are small (within ±2 kcal/mol; 1 cal = 4.18 J), indicating that electrophysiological characteristics of the mutant channels are similar to those of the parent channel. Cysteine-substituted residues on positions 315, 321, 324, 326, and 328 on the long-linker Shaker channel were accessible to externally added cysteine-modifying compound MTSET when tested under the closed- or open/closed-pulse protocol (Fig. 2C). Native Cys-308 is not accessible; therefore, the limit of MTSET accessibility is between positions 309 and 314.
Accessibility of S3b Amino Acid Residues in a Short Linker K+ Channel. Next, we investigated the accessibility of cysteine residues in a short-linker Shaker K+ channel. This Shaker K+ channel mutant termed LAI mutant contains only three residues in the S3–S4 linker: Leu-358, Ala-359, and Ile-369 [ShakerΔ (6–46), Δ (330–357)]. In the LAI mutant background, we constructed point mutants I315C, A321C, T324C, F326C, and I328C (Fig. 1A). Mutants expressed robust currents with steady-state G–V curves, maximum open probability, number of gating charges, and slow inactivation kinetics compared with those of the LAI mutant (Fig. 3A and data not shown). Perturbation of the free-energy difference between the open and closed conformations were within ±1 kcal/mol, indicating that the electrophysiological characteristics of the mutant channels were similar to those of the LAI parent channel. The cysteine-substituted residues in all of these mutants were accessible to externally added MTSET under the closed or open/closed-pulse protocol (Fig. 3B).
Fig. 3.

Accessibility of S3b residues of short S3–S4 linker mutants. (A) Effects on the voltage-dependent activation in the S3 cysteine mutants with short-linker (LAI) background. •, LAI (control); ○, I315C; ▴, I321C; ♦, F324C; +, T326C; □, A328C. Each point is the average of determinations on four to seven separate patches. Solid lines were drawn by using Eq. 1. V1/2 values were –15 ± 8 mV, 7 ± 2 mV, –19 ± 2 mV, 32 ± 1 mV, 26 ± 5 mV, and 22 ± 9 mV for LAI (control), I315C, I321C, F324C, T326C, and A328C, respectively. The equivalent number of gating charges, zeq, was 2.2 ± 0.5, 1.8 ± 0.4, 4.5 ± 1.3, 0.9 ± 0.04, 1.1 ± 0.4, and 0.8 ± 0.4 for LAI (control), I315C, I321C, F324C, T326C, and A328C, respectively. (B) External accessibility of cysteines to MTSET in a short linker Shaker channel (LAI). Cysteines were in positions 328, 326, 324, and 321 in the S3b segment and in position 315 in C-terminal end of S3a. All tested positions were accessible to MTSET in both the open/closed and the closed states. Rates expressed in M–1·s–1 are given for representative experiments on each of the mutants. Note that the control LAI channel contains a cysteine in position 308 that is inaccessible to the thiol reagent. (C) State-dependent accessibility of the different cysteine mutants. Second-order rate constants are plotted for cysteine modification in the closed (•) and open (○) states. Mutant labeled F324-C10aa is a shortened linker Shaker mutant containing a S3–S4 linker of 10 aa (SSNQAMSLAI; see Fig. 1 A) and a cysteine in position 324. Mutant labeled F324C-5aa is a short linker Shaker mutant containing a S3–S4 linker of 5 aa (MSLAI; see Fig. 1 A) and a cysteine in position 324. LAC, LCI, and CAI are short linker Shaker mutants containing a S3–S4 linker of 3 aa and cysteines in the indicated positions. Each point is the mean of three or more determinations, and rates are expressed in M–1·s–1.
In Fig. 3C, we show the results of the calculations of the second-order reaction rate constant of MTSET labeling. Calculation of the closed rate constant (filled symbols) is straightforward because the channel was closed during 10 s and open only 0.04 s under the closed-pulse protocol. In Fig. 3, we compare the MTSET reaction rate constants for the closed estate in all cysteine mutants in native and short linker backgrounds (10, 5, and 3 residues). We conclude that deletion of the S3–S4 linker has no impact on the accessibility of S3 segment residues from position 315 to 328 in the closed conformation. For the open/closed-pulse protocol, the channel is 1 s in the closed conformation and 0.4 s in the open conformation. Therefore, calculation of the open reaction-rate constants involves a correction for the time that the channel was exposed to MTSET while closed (see Methods). Because the closed reaction-rate constant of the closed conformation has to be measured on a separate experiment, the resulting kopen constants contain the error of two determinations. We found that the reaction-rate constant for the open estate is equal to or larger than the rate constant for the closed state on the 3-aa linker mutants (open symbols in Fig. 3C). For the mutants containing the 5-aa linker, the 10-aa linker, and the native S3–S4 linker, this rule is also true. We also tested the accessibility of the three residues on the linker of LAI deletion mutant: L358C, A359C, and I360C (Fig. 3C). Our results demonstrate that the short linker is exposed to extracellular MTSET for open and closed conformations.
Alteration of the Number of Charges on S3b. To investigate the involvement of S3b in voltage sensing, we removed a large part of the S3–S4 linker and the part of the C terminus of S3b, leaving out four consecutive negative charges. This deletion had no detectable effect on the gating charge measured by using the limiting-slope method. Gating charge of the WT Shaker channel is 12.2 ± 0.6 for the WT channel (19), 12.3 ± 0.9 for the MSLAI deletion mutant (19), 12.1 ± 0.4 for the LAI mutant (21), and 11.5 for the VVA mutant (21). These results demonstrate that residues E333, E334, E335, and D336 in S3b and S3–S4 linker do not contribute to the gating charge of the channel. To confirm this conclusion, we introduced a positive charge (T326R) on the S3b segment into shortened linker deletion mutants containing a 3-aa linker: LAI or VVA (see Fig. 1A). The VVA-T326R and the LAI-T326R linker mutants gave robust macroscopic currents (Fig. 4A). The VVA-T326R mutant has the advantage over the LAI-T326R mutant that its V1/2 is –81 mV, allowing better resolution of the limiting slope (Fig. 4B). We extracted zeff by using the limiting-slope method (19, 22, 24). The conductance–voltage curve for the VVA-T326R mutant obtained with the ramp method is shown in Fig. 4C. A fit to the data at very negative potentials (Po→0; Eq. 2) gave zeff = 12.2. To verify the validity of our assumption Po→0, we determined zeff from the slope of the logarithm of the G–V curve at low open probabilities (Eq. 3; see Methods). Fig. 4D shows that, when z is plotted as a function of voltage, it reaches a plateau at a value very close to that obtained by the exponential fit (horizontal line in Fig. 4D). For the WT Shaker K+ channels, we obtained zeff = 12.5 ± 0.62 (n = 6) by using the same procedure (19, 21). Therefore, we conclude that a positive charge added to segment S3b does not contribute to the total gating charge of the channel (27).
Fig. 4.

Mutation T326R does not increment gating charge. (A) Potassium current carried by T326R mutation in LAI and VVA S3–S4 linkers of mutant Shaker channels. Holding potentials were –100 and –130 mV for the LAI and VVA mutants, respectively. The membrane was pulsed to voltages between –70 to +30 mV in 5-mV increments, followed by a step to –60 mV for the LAI mutants and –120 to +80 mV in 5-mV increments and then by a step to –110 mV. All mutants were recorded in cell-attached patch–clamp configuration. (B) Relative conductance as a function of pulse potential of the mutants in A, measured from the tail currents. The solid lines were drawn by using Eq. 1. V1/2 were –29 ± 5 mV, –17 ± 9 mV, 13 ± 8 mV, –68 ± 14 mV, and –83 ± 16 mV for ShakerΔ, LAI, T326R-LAI, VVA, and T326R-VVA, respectively. Limiting-slope analysis in charged S3b short linker mutant channels. (C) Semilogarithmic plot of the G vs. V relationship of the VVA-T326R mutant. The solid line indicates the fitting of the low-probability data to determine the limiting value of zeff. (D) z vs. V plot. The circles correspond to the values of z calculated from Eq. 3. The solid line indicates the value of zeff obtained from the monoexponential fit of conductance vs. voltage relationship for conductance values <0.001 (Eq. 2). Average values of zeff for the VVA mutant was 11.5 ± 0.5 (5) and form the VVA-T326R mutant was 12.5 ± 0.5 (5).
The lack of a modification of the total gating charge in additional-charge mutant tested is interesting, especially considering that in the VVA-T326R mutant four negative charges have been removed and one positive charged added to the S3b segment (Fig. 1A). From our results it is clear that a S3–S4 linker consisting of three amino acids suffices to allow all of the structural changes the S4 domain undergoes during channel activation without involving modifications in the location of the S3b segment.
Discussion
Models of Voltage-Sensor Displacement. The classical model for voltage-dependent channels places the charged S4 segment into an aqueous channel formed through the protein structure of the gating pore (16, 27–29). This channel is hourglass-shaped, has water-filled vestibules on both intracellular and extracellular ends, and narrows in the middle to form a corset bracing the S4 segment. Cysteine- (9, 10) and histidine-scanning mutagenesis (3, 4) revealed that the positively charged S4 residues are accessible from either side of the gating pore, and that the reactivity is state-dependent. Electric field drives the positive charges trapped in the corset, inward or outward, setting S4 into motion. The details of the S4 motion relative to the electric field, and of the remaining voltage-sensing structure, have been the subject of discussion and several topologically equivalent models have emerged. State-dependent accessibility can be modeled by screw-symmetry operations on S4 (7, 29), a pure rotation without translation (15), tilting of the segment, or even collapsing of the vestibule walls (4, 18). The extent of the linear motion is 0–16 Å, depending on the model.
We have reported (19, 21) that a mutant of the Shaker K+ channel containing no S3–S4 linker was able to produce robust K+ currents, albeit less voltage-dependent and with much slower activation kinetics than the parent Shaker channels. This unexpected result prompted us to produce a series of Shaker channel mutants containing S3–S4 linkers of different lengths with the aim of determining the minimum linker length necessary to recover the gating kinetics and the voltage-dependent characteristics of the parent Shaker K+ channel. To our surprise, we found that a mutant retaining only three amino acid (LAI) residues in the linker was able to reproduce most of the characteristics of parent Shaker K+ channel. These data are compatible with models in which (i) S4 travels a short distance in response to voltage (15); (ii) the S4 segment undergoes a considerable displacement but drags the S3b with it as proposed by the paddle model (17); or (iii) the S4 does not move at all and what changes is the thickness of the dielectric surrounding the sensor (4, 18). With a paddle model being proposed for KvAP channels, it became essential to test whether S4 indeed drags along S3 during activation.
Testing the Paddle Model. To settle this point on the Shaker channel, we decided to answer the following question. Does the voltage sensor involve a movement of S3? If S3 moves with the voltage sensor, then its accessibility must be state-dependent, and electrical charges on S3 must contribute to the total gating charge of the sensor. The experiments described here take advantage of a mutant of the Shaker channel that, as in the KvAP channel, contains only three amino acid residues in the S3–S4 linker. Cysteine-scanning and deletion or addition of charged residues demonstrate that S3 transmembrane motion is not involved in voltage sensing. This result places S3 on the extracellular side independent of the state and is inconsistent with the paddle model. Also, we demonstrate that none of the three residues of the S3–S4 linker gets buried on the closed conformation, and this result also conflicts with the paddle model in its original formulation. One of these residues, A359, was found to be accessible to external MTSET for the open and closed state on the native linker Shaker channel (9). This result was later revised (12), and it was found to be inaccessible for the closed state. This discrepancy can be a consequence of a restricted motion of this residue in our short linker deletion mutants. Even though S3b residues never get completely protected from MTSET attack, its reactivity is consistently lower for the closed state than for the open state of the sensor. This result indicates that there is some displacement of this part of the molecule, such as a tilt, as suggested for the sodium (29) and KvAP (29, 30) channels. Gandhi et al. found no change in accessibility of the S3b segment residues associated with the voltage-sensor operation (7). They built a molecular model based on the KvAP crystal in which the motion of S4 alone is the basis of voltage sensing (7). S4 can move independently from S3b in Shaker because of the flexibility of the 26-aa-long S3–S4 linker of this channel. A similar gating molecular mechanism was recently presented for the Shaker channel built by using the known distance constraints for the open conformation (6). This latter molecular model predicts state-dependent changes in S3b residues accessibility, for short S3–S4 linker Shaker. Here, we demonstrate that accessibility of S3b is modestly state-dependent for native and short S3–S4 linker, a fact not explained by present detailed molecular models. Also, we found that residue 315, on the C terminus of S3a is also accessible from the external side, a result consistent with the accessibility of the equivalent position in the sodium channel (29). Current detailed molecular models (6, 31) place this residue away from the external surface and out of reach of external MTSET, because the structure is very compact. Our results suggest that the structure is more relaxed than presently pictured and allows the formation of a water-filled crevice to solvate the charged S4 residues and allow MTSET to penetrate deep into protein.
Acknowledgments
We thank Miss Luisa Soto for able technical assistance; Drs. Richard Horn and Chris Gandhi for the discussion of our results; and Drs. Chris Ptak, Sudha Chakrapani, and Luis Cuello for reading and commenting on the manuscript. This work was supported by Chilean Fondecyt Grant 103-0830. The Centro de Estudios Científicos is a Millennium Institute and is funded in part by a grant from the Fundación Andes.
Author contributions: C.G., O.A., and R.L. designed research; C.G., F.J.M., and E.R. performed research; C.G., F.J.M., E.R., O.A., and R.L. analyzed data; and C.G., O.A., and R.L. wrote the paper.
Abbreviations: MTSET, methanethiosulfonate ethyltrimethylammonium; ShakerΔ, ShakerH4Δ.
See Commentary on page 4929.
References
- 1.Aggarwal, S. K. & MacKinnon, R. (1996) Neuron 16, 1169–1177. [DOI] [PubMed] [Google Scholar]
- 2.Seoh, S. A., Sigg, D., Papazian, D. M. & Bezanilla, F. (1996) Neuron 16, 1159–1167. [DOI] [PubMed] [Google Scholar]
- 3.Starace, D. M. & Bezanilla, F. (2001) J. Gen. Physiol. 117, 469–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Starace, D. M. & Bezanilla, F. (2004) Nature 427, 548–553. [DOI] [PubMed] [Google Scholar]
- 5.Swartz, K. J. (2004) Nat. Rev. Neurosci. 5, 905–916. [DOI] [PubMed] [Google Scholar]
- 6.Durell, S. R., Shrivastava, I. H. & Guy, H. R. (2004) Biophys. J. 87, 2116–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gandhi, C. S., Clark, E., Loots, E., Pralle, A. & Isacoff, E. Y. (2003) Neuron 40, 515–525. [DOI] [PubMed] [Google Scholar]
- 8.Shrivastava, I. H., Durell, S. R. & Guy, H. R. (2004) Biophys. J. 87, 2255–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Larsson, H. P., Baker, O. S., Dhillon, D. S. & Isacoff, E. Y. (1996) Neuron 16, 387–397. [DOI] [PubMed] [Google Scholar]
- 10.Yang, N., George, A. L., Jr., & Horn, R. (1996) Neuron 16, 113–122. [DOI] [PubMed] [Google Scholar]
- 11.Yusaf, S. P., Wray, D. & Sivaprasadarao, A. (1996) Pflügers Arch. 433, 91–97. [DOI] [PubMed] [Google Scholar]
- 12.Baker, O. S., Larsson, H. P., Mannuzzu, L. M. & Isacoff, E. Y. (1998) Neuron 20, 1283–1294. [DOI] [PubMed] [Google Scholar]
- 13.Wang, M. H., Yusaf, S. P., Elliott, D. J., Wray, D. & Sivaprasadarao, A. (1999) J. Physiol. 521, 315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schonherr, R., Mannuzzu, L. M., Isacoff, E. Y. & Heinemann, S. H. (2002) Neuron 35, 935–949. [DOI] [PubMed] [Google Scholar]
- 15.Cha, A., Snyder, G. E., Selvin, P. R. & Bezanilla, F. (1999) Nature 402, 809–813. [DOI] [PubMed] [Google Scholar]
- 16.Bezanilla, F. (2002) J. Gen. Physiol. 120, 465–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang, Y., Ruta, V., Chen, J., Lee, A. & MacKinnon, R. (2003) Nature 423, 42–48. [DOI] [PubMed] [Google Scholar]
- 18.Bell, D. C., Yao, H., Saenger, R. C., Riley, J. H. & Siegelbaum, S. A. (2004) J. Gen. Physiol. 123, 5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gonzalez, C., Rosenman, E., Bezanilla, F., Alvarez, O. & Latorre, R. (2000) J. Gen. Physiol. 115, 193–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taglialatela, M., Toro, L. & Stefani, E. (1992) Biophys. J. 61, 78–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gonzalez, C., Rosenman, E., Bezanilla, F., Alvarez, O. & Latorre, R. (2001) Proc. Natl. Acad. Sci. USA 98, 9617–9623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Almers, W. (1978) Rev. Physiol. Biochem. Pharmacol. 82, 96–190. [DOI] [PubMed] [Google Scholar]
- 23.Noceti, F., Baldelli, P., Wei, X., Qin, N., Toro, L., Birnbaumer, L. & Stefani, E. (1996) J. Gen. Physiol. 108, 143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sigg, D. & Bezanilla, F. (1997) J. Gen. Physiol. 109, 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zagotta, W. N., Hoshi, T., Dittman, J. & Aldrich, R. W. (1994) J. Gen. Physiol. 103, 279–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang, Y., Lee, A., Chen, J., Ruta, V., Cadene, M., Chait, B. T. & MacKinnon, R. (2003) Nature 423, 33–41. [DOI] [PubMed] [Google Scholar]
- 27.Ahern, C. A. & Horn, R. (2004) J. Gen. Physiol. 123, 205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gandhi, C. S. & Isacoff, E. Y. (2002) J. Gen. Physiol. 120, 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen, T. P. & Horn, R. (2002) J. Gen. Physiol. 120, 419–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elliott, D. J., Neale, E. J., Aziz, Q., Dunham, J. P., Munsey, T. S., Hunter, M. & Sivaprasadarao, A. (2004) EMBO J. 24, 4717–4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laine, M., Lin, M. C., Bannister, J. P., Silverman, W. R., Mock, A. F., Roux, B. & Papazian, D. M. (2003) Neuron 39, 467–481. [DOI] [PubMed] [Google Scholar]