

SUMMARY
RAD51 promotes homology-directed repair (HDR), replication fork reversal, and stalled fork protection. Defects in these functions cause genomic instability and tumorigenesis, but also generate hypersensitivity to cancer therapeutics. Here we describe the identification of RADX as an RPA-like, single-strand DNA binding protein. RADX is recruited to replication forks where it prevents fork collapse by regulating RAD51. When RADX is inactivated, excessive RAD51 activity slows replication elongation and causes double-strand breaks. In cancer cells lacking BRCA2, RADX deletion restores fork protection without restoring HDR. Furthermore, RADX inactivation confers chemotherapy and PARP inhibitor resistance to cancer cells with reduced BRCA2/RAD51 pathway function. By antagonizing RAD51 at forks, RADX allows cells to maintain a high capacity for HDR while ensuring that replication functions of RAD51 are properly regulated. Thus, RADX is essential to achieve the proper balance of RAD51 activity to maintain genome stability.
Keywords: replication stress, CXorf57, RAD51, BRCA2, SMARCAL1, ZRANB3, fork reversal, PARP inhibitor, RPA, double-strand break, homologous recombination
eTOC
Dungrawala et al. identify RADX as an RPA-like single strand DNA binding protein enriched at replication forks. RADX antagonizes RAD51 accumulation at forks, thereby preventing aberrant fork remodeling. RADX deletion confers fork protection and chemoresistance to BRCA2 mutant cells without affecting homologous recombination repair of double-strand breaks.
INTRODUCTION
Single-strand DNA (ssDNA)-binding proteins (SSBs) are essential regulators of DNA metabolic processes including replication, recombination, and repair. SSBs in eukaryotic cells include Replication Protein A (RPA), hSSB1, POT1, and RAD51. These proteins protect ssDNA, recruit other proteins, and regulate enzymatic activities in a variety of genomic contexts including at replication forks, sites of DNA damage, and at telomeres (Flynn and Zou, 2010; Oakley and Patrick, 2010; Richard et al., 2009). Their function is essential for genome duplication and stability.
RPA and RAD51 promote replication fork stability, especially in the context of replication stress. RPA is a trimer of three subunits that binds ssDNA using four oligonucleotide/oligosaccharide-binding (OB)-fold domains. RPA dynamically associates with the replication fork to facilitate lagging strand DNA synthesis, and binds stalled forks to regulate the replication checkpoint and prevent fork collapse. In this context, RPA recruits DNA damage response proteins including ATRIP and ETAA1 to activate ATR signaling, and also regulates fork processing enzymes like SMARCAL1 (Ball et al., 2005; Bass et al., 2016; Bétous et al., 2013; Bhat et al., 2015; Duursma et al., 2013; Haahr et al., 2016; Xu et al., 2008; Zou, 2017; Zou and Elledge, 2003). RPA exhaustion caused by defects in ATR signaling causes aberrant fork processing and fork collapse (Toledo et al., 2013).
RAD51 is best known for its ability to form filaments on ssDNA generated at resected double-strand breaks (DSBs) where it catalyzes a strand exchange reaction to initiate homology-directed repair (HDR) (Kowalczykowski, 2015; Symington, 2014). It also has at least two functions at stalled replication forks. First, it cooperates with motor proteins like SMARCAL1 and ZRANB3 to promote fork reversal as a mechanism to stabilize and repair stalled forks (Bétous et al., 2012, 2013; Ciccia et al., 2012; Zellweger et al., 2015). Second, RAD51 protects forks from nuclease-catalyzed degradation of nascent DNA strands (Hashimoto et al., 2010; Schlacher et al., 2011). It may also promote strand invasion to restart a replication fork after cleavage by structure-specific nucleases like MUS81 (Sarbajna and West, 2014). The fork protection and HDR functions of RAD51 depend on BRCA2, which assists in replacing RPA with RAD51 at resected DSBs and stabilizes RAD51 at stalled forks (Kolinjivadi et al., 2017). Thus, BRCA2-deficiency causes genomic instability and cancer predisposition due to a failure of HDR and fork degradation (Kass et al., 2016).
The break repair and fork protection defects in BRCA2-mutant cancer cells make them hypersensitive to DNA damaging agents used in cancer therapy and to drugs that target DNA repair like PARP inhibitors (Lord and Ashworth, 2012; O’Connor, 2015). However, many patients develop drug resistance. One resistance mechanism is the acquisition of secondary BRCA2 mutations that result in a functional protein (Edwards et al., 2008; Norquist et al., 2011; Sakai et al., 2008). In addition, re-acquisition of fork protection even in the absence of HDR restoration was recently demonstrated to yield PARP inhibitor resistance (Chaudhuri et al., 2016). The mechanisms by which this happen include inactivation of PTIP, PARP1, and other unidentified genetic alterations (Chaudhuri et al., 2016; Ding et al., 2016).
The HDR and replication fork protection functions of RAD51 are evolutionarily conserved in all kingdoms of life. For example, the bacterial RAD51 orthologue RecA also promotes replication fork reversal and protects newly synthesized DNA from being degraded by nucleases (Horii and Suzuki, 1968; Robu et al., 2001; Satta et al., 1979). RecA is both positively and negatively regulated to promote the proper balance between replication, recombination, and repair activities (Cox, 2007). This balance is partly achieved through the action of the RecX protein, a RecA antagonist (Drees et al., 2004; Stohl et al., 2003; Venkatesh et al., 2002; Vierling et al., 2000). No sequence homolog of RecX has been described in eukaryotic cells.
Here, we report the identification of a new replication stress response protein, RADX (RPA-related, RAD51-antagonist on X-chromosome) that has sequence similarity to RPA. RADX is recruited to replication forks and binds ssDNA via RPA-like OB-folds. RADX antagonizes the accumulation of RAD51 at forks. When RADX is inactivated, excessive RAD51 activity causes fork collapse in otherwise unstressed cells. In cells with reduced RAD51 function such as BRCA2-deficient cancer cells, RADX inactivation is sufficient to restore fork protection and confer chemoresistance. Thus, RADX is a novel regulator of RAD51 that functions at replication forks to maintain genome stability and may be an important determinant of chemosensitivity in cancer.
RESULTS
RADX is recruited to stalled replication forks
We recently utilized iPOND (Isolation of Proteins on Nascent DNA) coupled with quantitative SILAC (stable isotope labeling of amino acids in cell culture) mass-spectrometry to identify proteins recruited to stalled replication forks (Dungrawala et al., 2015). Replication fork proteomes of cells treated with hydroxyurea (HU) for increasing amounts of time (0.5 hours to 24 hours) were compared to untreated cells (Figure 1A). In these analyses, the abundance of most chromatin proteins, like histones, are essentially unchanged through the replication stress time course (Dungrawala et al., 2015); replication proteins, like the MCM2-7 complex, decrease in abundance due to the slow completion of DNA synthesis and termination events in the absence of new origin firing; and replication stress response proteins like RPA and ATR are enriched at stalled forks (Dungrawala et al., 2015). We identified CXorf57 (Chromosome X open reading frame 57) as one the most highly enriched proteins at stalled forks (Figure 1B). We then performed iPOND-SILAC experiments comparing cells treated with EdU for 10 minutes (pulse) and cells treated and then incubated without EdU for an hour (chase) as described previously (Dungrawala et al., 2015) to identify proteins enriched at elongating replication forks (Figure 1C). We found that CXorf57 is modestly enriched even at unstressed replication forks (Figure 1D). Based on our functional analyses we have named this protein RADX.
Figure 1. RADX is recruited to stalled replication forks.
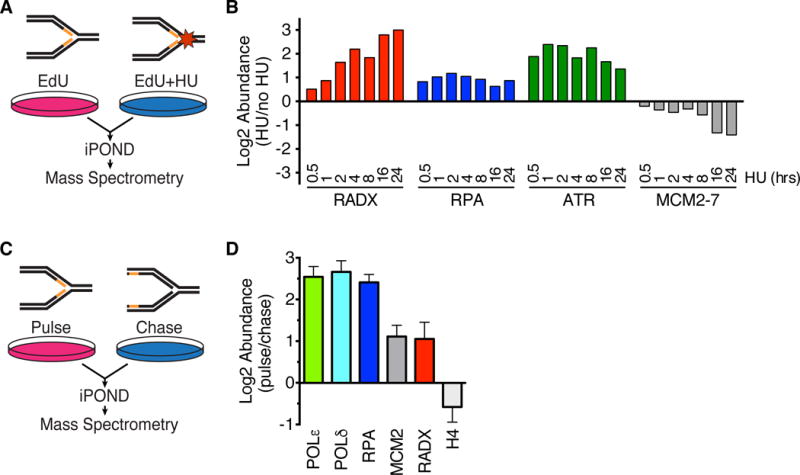
(A) iPOND-SILAC-MS was used to identify proteins enriched at forks in HU-treated cells. (B) Log2 abundance ratios for selected proteins or complexes are depicted. P-values for RADX range from 0.04 to 10−14 at different time points (see (Dungrawala et al., 2015) for original data, n=12 total). (C) iPOND-SILAC-MS identified proteins enriched at elongating forks in the absence of exogenous stress. (D) Log2 abundance ratios for selected proteins is depicted as mean +/− SEM, n=4 (n=2 each for HEK293T and HCT116).
RADX prevents replication fork collapse
To investigate its function, we utilized four siRNAs to deplete RADX (Figure S1A). RADX knockdown causes elevated levels of the DNA damage marker γH2AX specifically in S-phase cells, both in the absence (Figure 2A) and presence of HU (Figure 2B, Figure S1B). RADX depletion, even in the absence of added replication stress, caused an increase in DSBs as measured by a neutral comet assay in both cancer and non-cancer human cell lines (Figure 2C, Figure S1C). These effects are not due to large changes in the percentage of cells in S-phase (Figure S1D). RADXΔ cells derived using CRISPR-Cas9 targeting of the RADX gene in U2OS cells also displayed an increase in DSBs that could be rescued by complementation with an exogenously expressed RADX cDNA (Figure 2D, S1E, and S1F). We noticed in this complementation experiment that RADX cDNA expression did not fully complement the breaks caused by RADXΔ and modestly increased the level of DSBs in control U2OS cells, suggesting that overexpression of RADX also causes DNA damage. See below for a further exploration of overexpression phenotypes. Co-depletion of the structure-specific endonuclease MUS81 reduced both the DSBs and S-phase γH2AX phosphorylation caused by RADX silencing (Figure 2E,F and S1G) or RADX deletion (Figure S1H), suggesting that RADX prevents MUS81-catalyzed fork cleavage.
Figure 2. RADX prevents replication fork collapse. See also Figure S1.
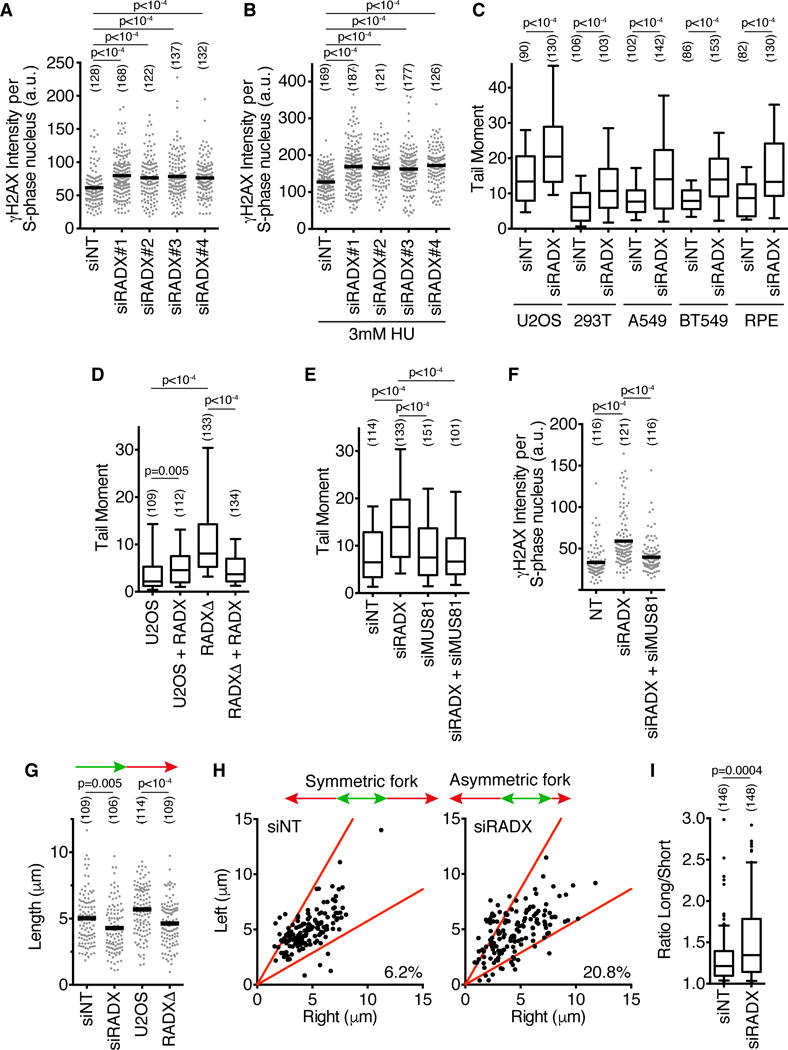
(A–B) U2OS cells were transfected with siRNAs (siNT=non-targeting), labeled with EdU for 10 minutes, and γH2AX intensity in EdU-positive cells was measured by quantitative imaging. In (B) cells were treated with 3mM HU for four hours. (C–E) DSBs were measured by neutral comet assay. For all box and whisker plots, the box depicts 25–75%, whiskers are 10–90%, and median is indicated. (F) γH2AX intensity in S-phase U2OS cells 72 hours post siRNA transfection. (G) U2OS cells transfected with siRNAs, or wild-type and RADXΔ cells, were labeled with IdU followed by CldU. CldU fiber length is plotted. (H and I) CldU lengths from left and right moving forks from the same origin were measured. In (I), the ratio of the sister fork lengths is plotted. The number of nuclei (A–F) or fibers (G–I) analyzed is indicated in parentheses. Black bars in A, B, F, and G indicate the mean. P-values were calculated using a Kruskal-Wallis ANOVA with Dunn’s posttest except in C and I, where a Mann-Whitney test was used. All experiments are representative of at least two replicates.
Finally, we utilized single molecule analysis of replicated DNA fibers to test if the increased DSBs in RADX-deficient cells affected replication fork progression. Indeed, both RADX siRNA and gene deletion decreased fork elongation rates (Figure 2G). In addition, RADX silencing also increased the frequency of asymmetric sister replication forks, confirming that RADX-deficient cells exhibit elevated rates of replication fork collapse (Figure 2H and I).
RADX binds DNA to maintain fork stability
An insight into the mechanism of RADX activity was obtained from analysis of the primary amino acid sequence and structural modeling, which predicts five structured domains, three of which are OB-folds (Figure 3A). The organization of the three OB-fold domains is reminiscent of the large subunit of RPA (RPA70). In addition, there is significant sequence similarity between RADX and RPA70 (Figure S2) leading us to hypothesize that RADX may bind ssDNA. To test this hypothesis, we purified a Flag-RADX recombinant protein from baculovirus-infected insect cells (Figure S3A). Pull-down experiments of the purified protein with DNA conjugated to magnetic beads confirmed that RADX does bind ssDNA (Figure 3B). RADX also binds double-stranded DNA (dsDNA), although the interaction with dsDNA is significantly weaker (Figure 3B). We confirmed this observation using electrophoretic mobility shift assays which indicated that RADX has approximately 75-fold higher affinity for ssDNA than dsDNA (Figure 3C).
Figure 3. RADX binds DNA using its OB-fold domains. See also Figure S2 and S3.
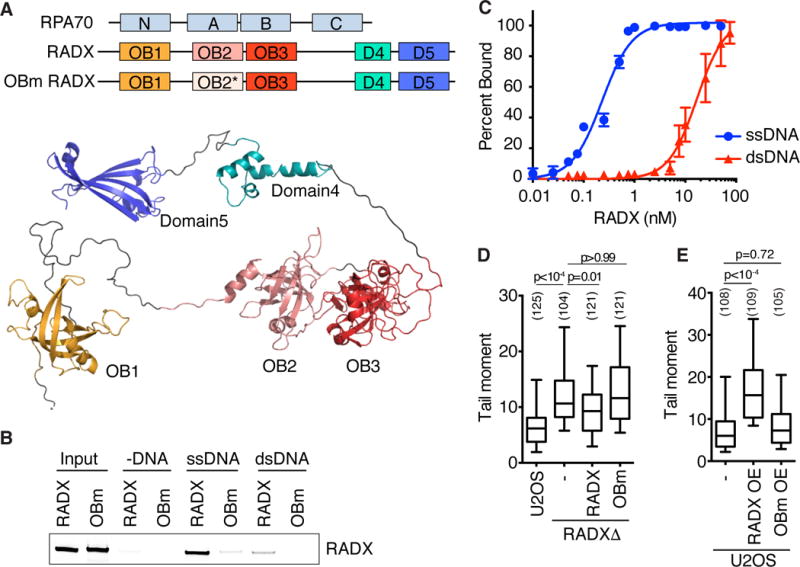
(A) Schematic of RADX and RPA70 depicting the OB-fold domains with a homology model of RADX generated using RaptorX. (B) DNA pull-down assays of purified RADX using ssDNA or dsDNA coupled to magnetic beads. (WT- wild type, OBm- OB mutant) (C) Electrophoretic mobility shift assays of RADX binding to ssDNA and dsDNA (mean+/−SD, n=3). (D) DSBs measured by neutral comet assay in RADXΔ cells expressing either wild-type or OBm RADX. The DSB level in parental U2OS cells is shown for comparison. (E) DSBs were measured after overexpression of wild-type or OBm RADX in U2OS cells. (−, mock). The number of nuclei analyzed is indicated. P-values were calculated by Kruskal-Wallis ANOVA with Dunn’s posttest. Experiments are representative of at least two replicates.
Sequence alignments indicate that the OB-1, OB-2, and OB-3 domains of RADX are most similar to the RPA70N protein recruitment domain, the RPA70A high-affinity ssDNA binding domain, and the telomeric ssDNA binding domain of POT1, respectively. Using the similarity of the RADX OB-2 domain to RPA70A and evolutionary conservation as guides, we designed mutations on surface amino acids of the OB-2 domain to determine if it is necessary to bind DNA (Figure 3A, S3A, S3C and S3D). Indeed, the OB-2 domain mutant (OBm) RADX has reduced affinity for both ssDNA as well as dsDNA (Figure 3B). The OBm RADX protein has some residual ssDNA binding suggesting that like RPA, RADX likely contains multiple DNA binding domains.
We then tested whether DNA binding is required to protect replication forks from collapse. As observed previously, reconstituting RADXΔ cells with wild-type RADX reduces DSB formation (Figure 3D). In contrast, OBm RADX does not rescue the RADXΔ cells indicating that DNA binding is essential to maintain fork stability (Figure 3D). As noted previously, expression of wild-type RADX cannot fully complement the elevated DSB phenotype of RADXΔ cells, perhaps because it is overexpressed (Figure S3B). When tested directly in U2OS cells, high levels of overexpressed wild-type RADX, but not the OBm RADX protein, caused an increase in DSB formation (Figure 3E and S3B). We conclude that DNA binding is important for RADX function, and that either too much or too little RADX expression causes the accumulation of DSBs in otherwise unstressed cells.
RADX reduces RAD51 association with replication forks
To understand how RADX maintains replication fork stability, we performed iPOND-SILAC-mass spectrometry analyses to compare the stalled fork proteomes of RADXΔ cells to parental wild-type cells (Figure 4A). This experiment was completed in four different clones of HEK293T RADXΔ cells, and in one clone of U2OS RADXΔ cells. The median enrichment value of each protein observed in the datasets is depicted in Figure 4B and the full dataset is presented in Supplemental Table S1. Strikingly, of the 1060 proteins quantified in all five experiments, RAD51 was the highest enriched at stalled forks in the RADXΔ cells as compared to the parental cells (Figure 4B). In contrast, many other DNA damage response proteins like BRCA2, RPA, and the MRN complex were neither more nor less abundant at stalled forks in RADXΔ cells compared to control cells.
Figure 4. RADX competes with RAD51 to localize to stalled forks. See also Figure S4 and Table S1.
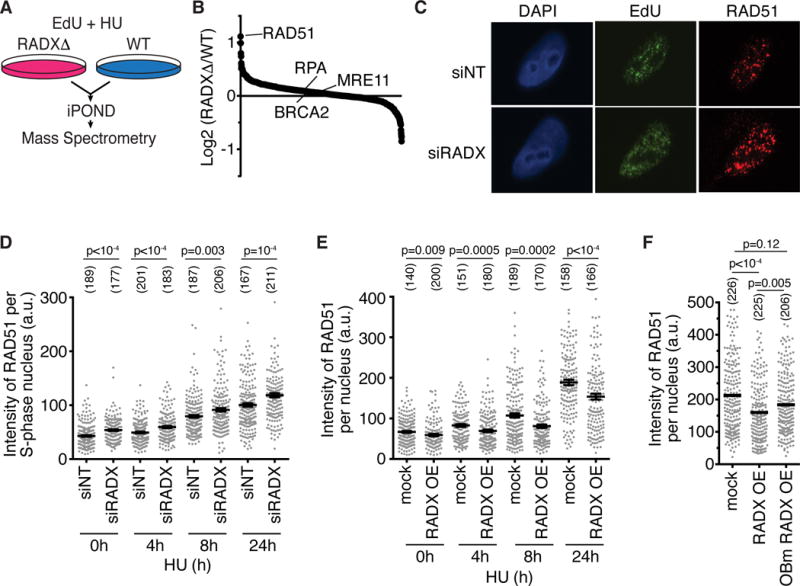
(A) Schematic of iPOND-SILAC-MS experiment. (B) The median of the log2 abundance ratios of the proteins identified in five cell clones is depicted. (C and D) U2OS cells transfected with siRNA were labeled with 10μM EdU for 30 minutes and were treated with 3mM HU. Cells were stained for RAD51 and EdU after detergent extraction. Representative images of RAD51 foci after 24h HU are shown in (C). In (D) the intensity of chromatin bound RAD51 in S phase cells is quantified. The number of nuclei analyzed is indicated and the mean+/−SEM is shown. P-value derived from a Mann-Whitney test. (E) Cells overexpressing GFP-RADX (OE) or parental U2OS cells were treated with 3mM HU. Chromatin-bound RAD51 intensity after detergent extraction with mean+/−SEM is depicted. P-values were derived from a Mann-Whitney test. (F) Cells overexpressing GFP-RADX or GFP-OBm were treated with 3mM HU for 24 hours Chromatin-bound RAD51 intensity with the mean is plotted. P-values were calculated by Kruskal-Wallis ANOVA with Dunn’s posttest. Experiments in C–F are representative of at least two replicates.
Quantitative immunofluorescence imaging of chromatin-bound RAD51 confirmed an increase in RAD51 at replication forks corresponding to an increase in both the number of detectable foci and an increase in their size in RADX-silenced cells, even when cells were not stressed with HU (Figure 4C and 4D). These differences are not due to a difference in the total expression of RAD51 in RADX-silenced and RADXΔ cells (Figure S4A and S4B). In addition, this difference is unlikely to be due to increased end-resection of DSBs at forks since we did not observe an increase in RPA at forks (Figure 4B), an accumulation of more ssDNA in RADX-deficient cells as measured by native BrdU staining (Figure S4C), more RPA on chromatin (Figure S4D), or an increase in RPA S4/S8 phosphorylation which is often used as a marker for end resection (Figure S4D). Thus, even though the amount of ssDNA is not appreciably different than in wild-type cells, inactivating RADX allows more RAD51 to accumulate at forks.
These data collectively suggest that RADX might antagonize RAD51 recruitment. If so, then overexpression of RADX should reduce RAD51 accumulation at forks. As predicted, cells overexpressing GFP-RADX have decreased RAD51 accumulation in detergent-resistant foci, both in the absence of added exogenous stress, and in response to replication stress over a time course in HU (Figure 4E). The decreased RAD51 is not due to a decrease in the percentage of cells in S phase in the GFP-RADX cells (Figure S4E and S4F), but is dependent on the ability of RADX to bind DNA since it is less apparent in cells overexpressing OBm RADX (Figure 4F). As noted previously, RADX overexpression causes DNA damage as measured by both an increase in DSBs (Figure 3E) and increased γH2AX (Figure S4G), indicating that the differences in RAD51 accumulation at forks in RADXΔ and RADX overexpressing cells cannot simply be explained by changes in the amount of breaks and replication stress. Thus, we conclude that RADX inhibits RAD51 accumulation at forks, a function that is dependent on its ability to bind DNA.
Excessive RAD51 activity causes fork collapse in RADX-deficient cells
Since RADX causes decreased RAD51 at replication forks, we reasoned that the replication defects such as decreased replication elongation and increased fork collapse in RADX-deficient cells might be caused by too much RAD51 activity. This hypothesis predicts that RAD51 silencing should rescue the replication defects caused by RADX deficiency. Strikingly, reducing RAD51 expression with RNA interference rescues both the fork elongation defect and the increased DSBs observed in RADX-deficient cells (Figure 5A–C, and S4A–B). Thus, despite RAD51 being a DSB repair protein, RAD51 silencing decreases the DSBs caused by loss of RADX.
Figure 5. Excessive RAD51 activity causes fork collapse in RADX-deficient cells. See also Figure S4 and S5.
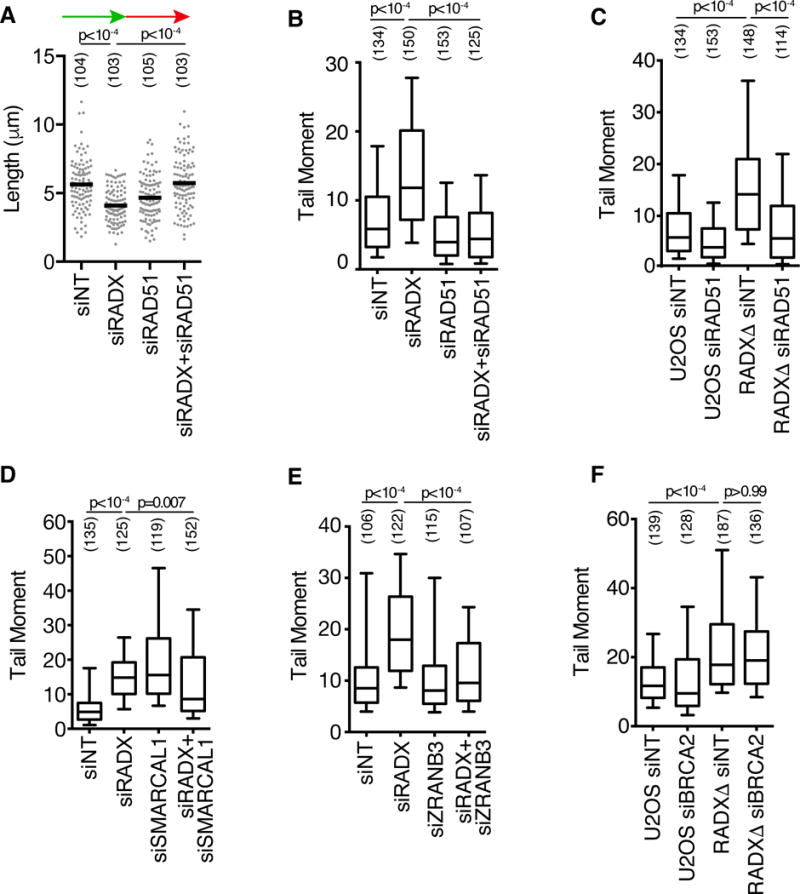
(A) U2OS cells transfected with siRNAs were labeled with IdU (15 min) followed by CldU (15 min). CldU fiber lengths are plotted. (B–F) DSBs were measured by neutral comet assay in parental or RADXΔ U2OS cells transfected with siRNAs. The number of DNA fibers or comet tails examined is indicated in parentheses. P-values were calculated with a Mann Whitney test in A or a Kruskal-Wallis ANOVA and Dunn’s post-test in all other panels. Experiments are representative of at least two replicates.
Since a DSB repair activity of RAD51 is unlikely to explain why we observe less fork breakage after silencing RAD51 in RADX-deficient cells, we considered the other known activities of RAD51 at replication forks. RAD51 is required for fork reversal (Zellweger et al., 2015) and replication fork stabilization (Schlacher et al., 2011). Unregulated fork reversal has previously been shown to lead to fork collapse (Bansbach et al., 2009; Couch et al., 2013; Sogo et al., 2002), so we considered the possibility that excessive fork reversal underlies the increase in DSBs observed in RADX-deficient cells. To test this idea, we asked whether depletion of the fork reversal enzymes SMARCAL1 and ZRANB3 could also rescue the fork collapse seen in RADX-deficient cells. As previously noted, SMARCAL1 depletion in wild-type cells increases DSB frequency (Bansbach et al., 2009; Ciccia et al., 2009; Yuan et al., 2009; Yusufzai et al., 2009). Nonetheless, depleting SMARCAL1 or ZRANB3 in RADX-deficient cells reduces the DSBs observed (Figure 5D, 5E, S5A, and S5B). Thus, these data suggest that RADX is needed to prevent inappropriate RAD51-, ZRANB3- and SMARCAL1-dependent fork remodeling that results in slow forks and cleavage by MUS81, and support the idea that the proper equilibrium between fork reversal and fork restoration is essential to prevent aberrant fork processing (Couch and Cortez, 2014). Interestingly, unlike RAD51, ZRANB3 and SMARCAL1, silencing BRCA2 did not decrease the DSBs seen in RADX-deficient cells (Figure 5F and S5C), consistent with previous observations that some RAD51 functions at forks are independent of BRCA2 (Chaudhuri et al., 2016; Tarsounas et al., 2003).
Deleting RADX restores fork protection to BRCA2-deficient cells without restoring HDR
BRCA2 is needed to stabilize RAD51 filaments and prevent fork degradation (Schlacher et al. 2011). Since our data indicates that RADX antagonizes RAD51 at replication forks, we tested whether removing this negative regulator might be sufficient to rebalance RAD51 functions and reverse the nascent strand degradation observed in BRCA2-deficient cells. Indeed, RADX silencing rescued the degradation phenotype that is observed upon BRCA2-silencing (Figure 6A, S5D and S5E). RADX depletion also restored fork protection to the BRCA2-mutant CAPAN-1 pancreatic cell line (Figure 6B, S5E). Thus, RADX inactivation is capable of compensating for the decreased stability of RAD51 at forks in BRCA2-deficient cells. In addition, overexpression of RADX by itself causes an increase in nascent strand degradation, further arguing that RADX antagonizes RAD51 at forks (Figure 6C, S5E).
Figure 6. RADX depletion in BRCA2-deficient cells prevents nascent strand degradation. See also Figure S5.
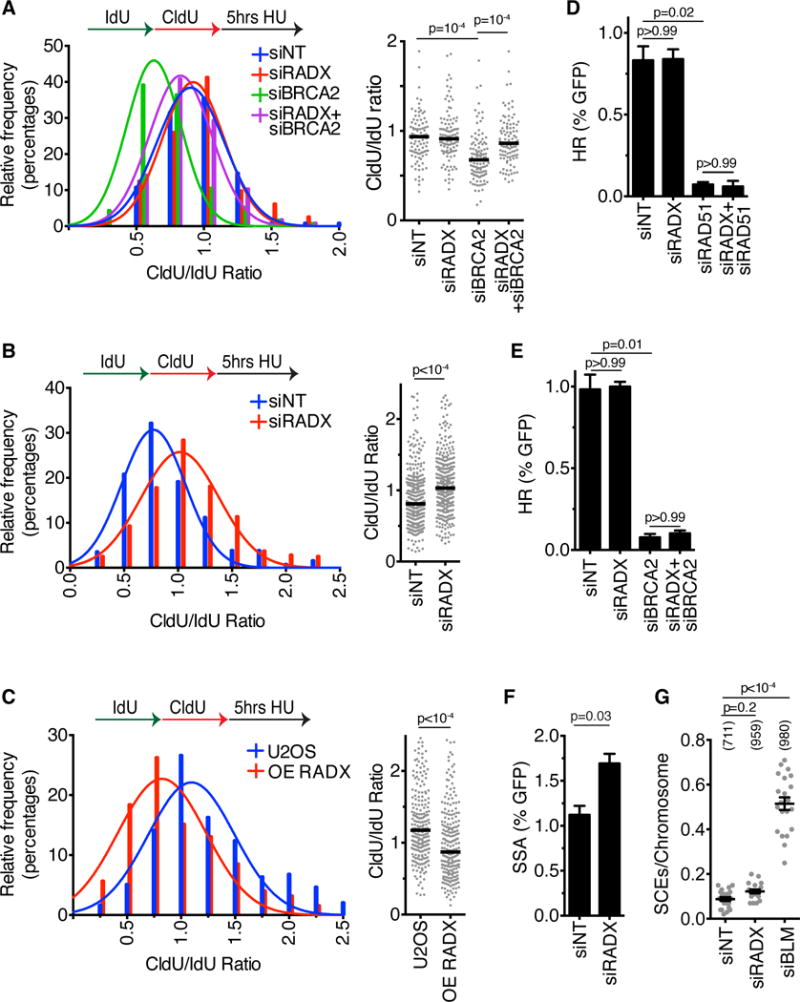
(A) U2OS cells or (B) CAPAN-1 cells transfected with siRNA, or (C) U2OS cells overexpressing RADX (OE) were labeled with IdU and CldU prior to treatment with 3mM HU for five hours. The lengths of DNA fibers were scored and plotted as histograms and dot plots. P-values were derived using Mann Whitney test. Experiments are representative of at least two repeats. Figure S5E lists the numbers of fibers analyzed. (D and E) The percentage of DR-GFP-positive U2OS cells after transfection with siRNAs and I-SceI expression vector. (Mean+/−SD, n=3 in which 25,000 cells were scored per experiment). (F) The percentage of SA-GFP-positive U2OS cells after transfection with siRNAs and I-SceI expression vector. (Mean+/−SD, n=3 in which 25,000 cells were scored per experiment). (G) The number of sister chromatid exchanges per chromosome were scored in U2OS cells transfected with siRNAs. The experiment is representative of three repeats. The number of chromosomes analyzed is indicated in parentheses.
We next asked whether RADX-silencing would affect the DSB repair activities of RAD51 and BRCA2. RADX silencing by itself has no effect on the rate of HDR (Figure 6D), as would be expected for a RAD51 antagonist since RAD51 overexpression does not change the rate of HDR (Stark et al., 2004). RADX silencing also does not restore the ability of RAD51- or BRCA2-deficient cells to perform HDR at a site-specific DSB (Figure 6D, 6E, S4A, S5D). This failure to restore HDR persists even when we titrated the amount of RAD51 siRNA to yield only a partial HDR-defect (Figure S5F). However, overexpression of RADX can interfere with RAD51 recruitment to DSBs and decrease the efficiency of HDR (Figure S5G, S5H), indicating that if RADX is too highly expressed it can interfere with DSB repair.
If RADX is an antagonist of RAD51, then we would expect that silencing RADX might increase the frequency of single-strand annealing repair of DSBs since overexpression of RAD51 has this effect (Stark et al., 2004). As predicted, we did observe a modest increase in the frequency of single-strand annealing as measured by an SA-GFP assay in RADX-deficient cells (Fig. 6F).
Finally, break repair at replication forks may be different than at site-specific DSBs (Willis et al., 2014). Therefore, we examined the rate of sister chromatid exchanges (SCEs) in RADX deficient cells to monitor HDR at forks. The BLM helicase, which functions as an anti-recombinase, prevents excessive SCEs (Sarbajna and West, 2014); however, we did not observe a large induction of SCEs in RADX deficient cells (Figure 6G). A small SCE increase (that was not statistically significant) was observed after RADX silencing in multiple experiments. This small increase may result from the increased fork breakage in these cells. However, these data suggest that RADX is not a general anti-recombinase.
RADX deletion causes chemo- and PARP-inhibitor resistance in BRCA2/RAD51-compromised cells
Although the ability of RADX to antagonize RAD51 function in the absence of replication stress may be important to prevent unwanted fork processing, RAD51 also has critical functions to protect and repair damaged replication forks (Hartlerode and Scully, 2009; Schlacher et al., 2011; Zellweger et al., 2015). Thus, reducing RAD51 expression with siRNA causes hypersensitivity to replication stress agents including HU, cisplatin, and camptothecin (Figure 7A–7C). Since RADX antagonizes RAD51 activity at forks, and restores fork protection to BRCA2-depleted cells, we tested whether removing RADX might also be sufficient to suppress the hypersensitivity of cells with reduced BRCA2/RAD51 function. Indeed, siRNA depletion of RADX suppressed the HU, cisplatin, and camptothecin hypersensitivity of cells with reduced RAD51 expression (Figure 7A–7C).
Figure 7. RADX depletion rescues the chemotherapy hypersensitivity of RAD51- and BRCA2-depleted cells. See also Figure S5 and S6.
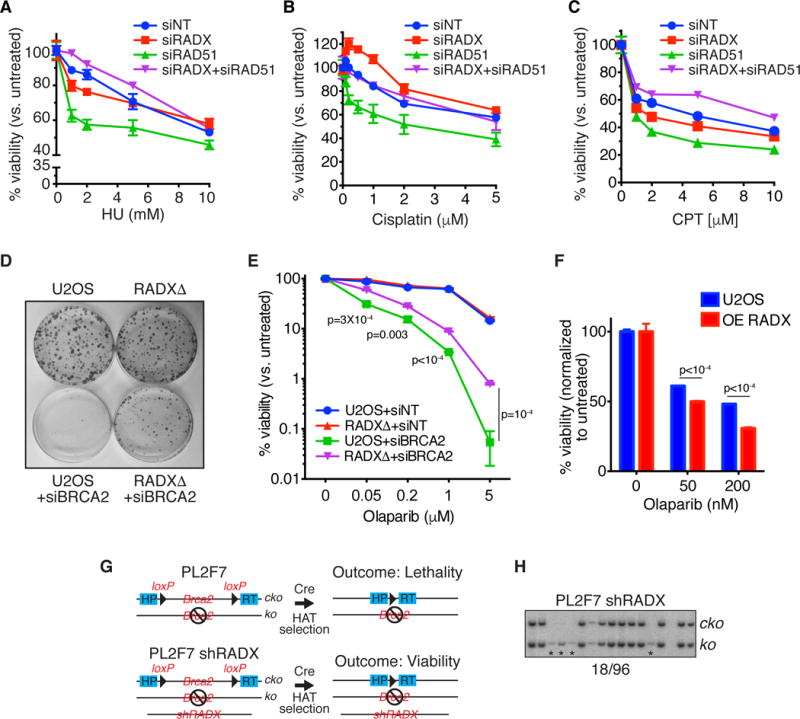
(A–C) Cells transfected with siRNAs were treated with HU, camptothecin or cisplatin and viability was measured 72 hours later. (mean+/−SEM, n=3). (D) Parental or RADXΔ U2OS cells transfected with non-targeting or BRCA2 siRNAs to achieve extensive knockdown were plated in the absence of drug and surviving colonies examined by methylene blue staining. (E) siRNA transfected cells were treated with the indicated concentrations of Olaparib, and surviving colonies were quantified. (mean+/−SD, n=3). P-values derived from unpaired t-tests corrected for a 1% FDR are shown for differences between siBRCA2 knockdown in U2OS and RADXΔ cells. The overall ANOVA-derived p value for these two curves is <10−4. (F) Parental and RADX overexpressing U2OS cells were treated with Olaparib and viability of surviving colonies quantified (mean +/− SEM, n=3). P-values were derived from FDR-corrected, unpaired t-tests. (G) Schematic of the PL2F7 mESC cell model (cko, Brca2 conditional knockout allele). (H) Representative southern blot from clones showing the rescue of Brca2 null mESC cell lethality with RADX knocked-down by shRNA. Asterisks depict the rescued null cells, which grew slower yielding less genomic DNA. All viability experiments are representative of at least two replicates.
RADX deletion also increases the viability of BRCA2-silenced cells in the absence of damage (Figure 7D), and partially rescues the hypersensitivity of the BRCA2-silenced cells to low doses of PARP inhibitor (Figure 7E). However, RADX deficiency does not rescue the hypersensitivity of BRCA2-silenced cells to ionizing radiation, consistent with its function being largely confined to replication forks (Figure S5I). Thus, RADX silencing causes resistance to cancer therapies in BRCA2-compromised cells and this resistance correlates with restoration of replication fork stability without changes in HDR. Finally, overexpression of RADX increases cellular sensitivity to Olaparib consistent with overexpression causing fork degradation and inhibiting HDR (Figure 7F).
Brca2 is essential for the viability of mouse embryonic stem cells (mESC) (Kuznetsov et al., 2008). To test if RADX knockdown can rescue the lethality of Brca2 null mESC, we knocked down RADX in PL2F7 mESC (Kuznetsov et al., 2008), which have one null and one conditional knockout allele of Brca2 (Figure 7G). We then expressed Cre recombinase in these cells to delete the Brca2 conditional allele and checked for the presence of viable Brca2 null cells as described previously (Ding et al., 2016). Usually deletion of Brca2 in mESC results in no viable clones (Chaudhuri et al., 2016; Ding et al., 2016; Kuznetsov et al., 2008); however, 19% (18 out of 96) of the cell clones recovered from the RADX knockdown population were Brca2 null (Figure 7H). Thus, RADX silencing can suppress the lethality caused by Brca2 deletion, although these cells did grow more slowly than those that did not delete Brca2.
Intriguingly, breast and lung cancer patients with high levels of RAD51 tend to do poorly as compared to patients with lower levels (Figure S6). Conversely, patients with higher levels of RADX tend to survive better. These patients are often treated with chemotherapeutic agents that cause replication stress, suggesting that modulation of RAD51 function by RADX could impact cancer patient responses to treatments targeting DNA replication and DNA repair.
DISCUSSION
Here, we report the identification of RADX as a new ssDNA binding protein with similarity to RPA, but with a specific activity in maintaining genome stability by regulating RAD51 function at replication forks. Strikingly, excessive RAD51 activity at forks slows elongation and leads to fork collapse when RADX is absent. In addition, loss of RADX confers chemotherapy resistance and restores fork protection in BRCA2-defcient cancer cells even though it cannot rescue HDR. We propose that RADX antagonizes RAD51 activities at replication forks to ensure the proper balance of fork remodeling and protection without interfering with the capacity of cells to complete HDR of DSBs. These results highlight that achieving the right balance of RAD51-dependent fork remodeling is critical for genome stability.
RADX prevents fork collapse
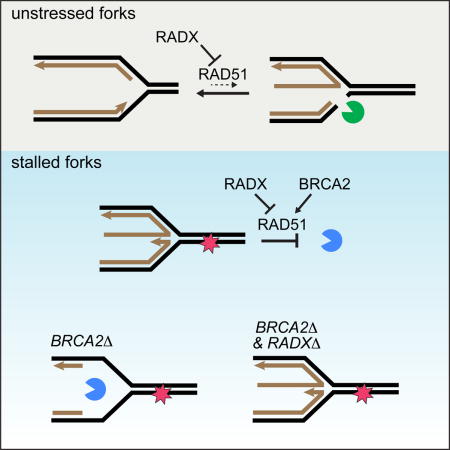
Our data support the following model: RAD51, SMARCAL1, and ZRANB3 promote fork reversal reactions to stabilize stalled replication forks (Bétous et al., 2012, 2013; Ciccia et al., 2012; Zellweger et al., 2015). However, fork reversal can be dangerous since inappropriate fork reversal would slow fork elongation and can result in fork cleavage by structure specific nucleases (Bansbach et al., 2009; Couch and Cortez, 2014; Couch et al., 2013; Sogo et al., 2002). RADX is an essential regulator of these processes. It binds ssDNA and prevents inappropriate accumulation of RAD51 at replication forks. This regulation ensures the right balance of active fork elongation and RAD51/SMARCAL1/ZRANB3-mediated fork reversal (Figure S7A). Too much or too little fork reversal causes DNA damage and fork collapse. Thus, RADX works in parallel to other regulatory mechanisms of fork regression including phosphorylation of SMARCAL1 by ATR (Couch et al., 2013), and regulation of SMARCAL1 substrate preference by RPA (Bétous et al., 2013; Bhat et al., 2015) to prevent fork collapse. Interestingly, while RAD51-, SMARCAL1- and ZRANB3- silencing rescues the DSBs seen in RADX-deficient cells, BRCA2-loss does not. Mechanistically, this result indicates that RAD51 has BRCA2-independent functions, and can be explained if BRCA2 is not required for RAD51-dependent fork reversal. This hypothesis will require direct testing with electron microscopy analyses of replication intermediates, but it is consistent with other reports which have found that BRCA2 is dispensable for some RAD51 functions at replication forks (Chaudhuri et al., 2016; Tarsounas et al., 2003).
BRCA2 stabilizes RAD51 at forks to protect them from MRE11-dependent degradation (Ciccia and Elledge, 2011; Schlacher et al., 2011) (Figure S7B). Lack of fork protection in BRCA2-mutant cells causes sensitivity to DNA damaging agents and PARP inhibitors (Schlacher et al. 2011; Chaudhuri et al. 2016). RADX deletion stabilizes the nascent DNA strands in BRCA2-deficient cells and causes chemoresistance. The simplest explanation is that it does so by rebalancing the functions of RAD51. In other words, removal of the negative RAD51 regulator (RADX) makes the positive regulator (BRCA2) dispensable at forks, but not at resected DSBs. This proposal is consistent with the ability of RAD51 overexpression to suppress the fork instability of BRCA2-deficient cells (Schlacher et al., 2012).
Importantly, HDR silencing RADX in BRCA2- or RAD51-deficient cells did not restore HDR. The amount of ssDNA at a stalled fork versus at resected DSBs may be key to explaining why RADX deletion suppresses RAD51 defects at forks, but not at DSBs. Moreover, this idea clarifies the need for a protein like RADX to regulate the fork protection activities of RAD51. Successful HDR involves extensive DSB resection and RAD51 filament formation; whereas electron microscopy analyses indicates that there are only short stretches of ssDNA at normal and stalled forks (Hashimoto et al., 2010; Ray Chaudhuri et al., 2012; Zellweger et al., 2015). Therefore, deletion of RADX may be sufficient to restore enough RAD51 function to maintain fork protection in BRCA2-deficient cells. However, it cannot suppress defects associated with DSBs because of a need for more RAD51 protein for HDR. In this model, proper balancing of RAD51 functions at forks cannot happen through regulation of RAD51 protein levels since RAD51 must be highly expressed in anticipation of DSB formation. Thus, modulation of RAD51 fork binding by RADX provides a finely tuned regulatory mechanism to yield the right amount of fork reversal and nascent strand stabilization while maintaining a high capacity for repair of DSBs by HDR.
Alternative models include the possibility that RADX directly recruits proteins such as MRE11 that degrade the nascent DNA in BRCA2- or RAD51-deficient cells, or that RADX binds to RAD51 directly to regulate its fork reversal and protection functions. Although these models cannot be excluded, we note that there was no RADX-dependent change in MRE11 levels at stalled forks and we have not been able to detect an interaction between RADX and MRE11 or RAD51 in preliminary screens for RADX-binding proteins.
In addition to RADX, cells have additional ways of fine-tuning RAD51 function through negative regulation. For example, the BLM helicase counteracts recombination during replication to prevent excessive sister chromatid exchanges (Larsen and Hickson, 2013). In addition, cyclin-dependent kinase phosphorylation of the C-terminus of BRCA2 disrupts its ability to stabilize RAD51 filaments and regulates fork protection (Davies and Pellegrini, 2007; Esashi et al., 2005; Schlacher et al., 2011). This multitude of both positive and negative regulatory mechanisms emphasizes the importance of appropriately balancing RAD51 activities.
Functional similarity to RecX
RAD51 is the eukaryotic homolog of bacterial RecA. Like RAD51, RecA also acts at stalled replication forks to promote fork reversal and block extensive nuclease degradation of DNA (Horii and Suzuki, 1968; Robu et al., 2001; Satta et al., 1979). RecA is aided in its fork protection function by the RecFOR proteins that mediate the loading and stabilization of RecA onto ssDNA similar to BRCA2 (Chow and Courcelle, 2004; Courcelle et al., 1997). The recX gene is often linked to recA in bacterial genomes. For example, E. coli recX is expressed from the recA promoter but at reduced transcript levels (Pagès et al., 2003). RecX suppresses the growth inhibitory consequences of RecA overexpression and acts to inhibit RecA by binding RecA-DNA filaments (Cox, 2007). Thus, RecX helps establish the proper balance of RecA activities. We propose that RADX serves a similar function in vertebrate cells although the details of the mechanism differ.
Clinical implications
Many cancers are caused by inactivation of BRCA2, a property that makes the tumor cells hypersensitive to chemotherapies and PARP inhibitors. However, drug resistance is a problem. Most described resistance mechanisms involve the acquisition of secondary BRCA2 mutations that result in a functional protein, or the re-establishment of HDR (such as by loss of 53BP1, a key antagonist of the HDR pathway) (Lord et al., 2015). However, resistance can be generated without restoring HDR (Guillemette et al., 2015), and recent studies described two mechanisms that contribute to resistance by restoring fork protection to BRCA2-deficient cells (Chaudhuri et al., 2016; Ding et al., 2016). These studies also indicated that there are as yet undiscovered mechanisms of fork protection that are relevant to tumor chemotherapy resistance. RADX inactivation should be considered as a candidate mechanism contributing to resistance.
The amount of functional RAD51 is important for both the etiology and treatment of cancer. Low activity can yield genome instability through defects in fork protection and HDR, but it also improves the cell killing by chemotherapeutics and PARP inhibitors (Budke et al., 2016; Quiros et al., 2011). On the other hand, RAD51 overexpression causes genome instability (Klein, 2008; Richardson et al., 2004), and RAD51 is frequently overexpressed in cancers (Klein, 2008). Thus, excessive RAD51 activity may drive tumorigenesis and generate drug resistance (Klein, 2008; Mason et al., 2014; Tennstedt et al., 2013). Since RADX loss mimics RAD51 overexpression, and reduces the PARP inhibitor sensitivity of BRCA2-deficient cells, it will be important to determine if RADX functionality is a determinant of how tumors respond to therapy.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David Cortez (david.cortez@vanderbilt.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
U2OS and HEK293T cells were cultured in DMEM with 7.5% fetal bovine serum (FBS). RPE-hTERT cells were cultured in DMEM F12, 7.5% FBS, and 7.5% sodium bicarbonate. A549 and BT549 cells were cultured in RPMI with 10% FBS. CAPAN-1 cells were cultured in RPMI with 20% FBS, and 1mM sodium pyruvate. All cell lines were purchased from ATCC, tested for mycoplasma, and authentication verified using short tandem repeat profiling. All cells were cultured at 37 °C and 5% CO2. U2OS, HEK293T, RPE-hTERT and A549 are female cell lines, while CAPAN-1 and BT549 cells are male.
METHOD DETAILS
CRISPR/CAS Editing
HEK293T RADXΔ and U2OS RADXΔ cells were generated using CRISPR/Cas9. Briefly, cells were transfected with pSpCas9(BB)-2A-Puro 4 (Addgene plasmid no. 48139) containing guide RNAs that target the intron-exon junction of the second exon of RADX (3′-CACCGAATCAAAACTGCGATACTA-5′ and 3′-CACCGTTACCATTACATGTTAAAC-5′), selected with 2 μg/ml puromycin for two days prior to plating for individual clones. Homozygous editing of the RADX locus was confirmed by genomic DNA PCR. The RADXΔ cell lines were also validated for loss of RADX expression by immunoblotting and qRT-PCR of mRNA. Complementation of RADXΔ cells with cDNA expression vectors was completed by lentiviral infection and selection for the linked G418 resistance cassette. Generation of stable cell lines overexpressing RADX was performed by lentiviral infection of a cDNA expression vector and G418 selection. Since the cells attenuate the level of RADX overexpression over time, overexpression experiments were completed within a few passages of cell line generation.
Transfection reagents
Plasmid transfections were performed with polyethylenimine. FUGENE HD was used for transfection of pCBASceI plasmid in the DR-GFP assays. siRNA transfections were performed with Dharmafect1 (Dharmacon) for U2OS, A549 and BT549 cells, Dharmafect4 (Dharmacon) for CAPAN-1 cells and RNAiMax (Thermo Fisher) for RPE-hTERT cells.
Plasmids
The RADX cDNA was obtained from the ThermoScientific Open Biosystems Human ORFeome collection (Catalog number OHS-1770). The mutant RADX was constructed using gene blocks synthesized by IDT and assembled using the Gibson Assembly method (NEB). The mutations made were R240E, R248E, K252E, K255E, K256E, W279A, K304E, R310E and E327A.
Viability Assays
Short-term viability assays were completed with Alamar Blue. PARP inhibitors and cisplatin were left in the growth media for the duration of the assay, while HU was removed after 24 hours. All viability measurements are presented as percent of the untreated control. Each assay was completed in triplicate. For clonogenic survival assays, cells were plated and treated with Olaparib for approximately two weeks. Colonies were scored by methylene blue staining (48% methanol, 2% methylene blue, 50% water). All clonogenic survival assays were completed in triplicate.
Flow Cytometry
Cells were collected 72 hours after siRNA transfection, fixed in 70% ethanol, treated with RNaseA, stained with propidium iodide and analyzed on a BD Biosciences FACSCalibur. For BrdU flow cytometry, cells were labeled with 10 μM BrdU for 30 minutes prior to harvest, fixed with 70% ethanol, denatured with 2N HCl for 30 minutes before staining with anti-BrdU antibody.
Immunofluorescence
For immunofluorescence experiments, U2OS cells were detergent extracted with 0.1% Triton X100 prior to fixing with 3% paraformaldehyde/2% sucrose. Slides were blocked with 5% BSA in PBS and incubated with antibody. EdU was detected using click chemistry with an Alexa Fluor 594-conjugated azide. Immunofluorescent images were obtained with a Nikon microscope with fixed camera exposure times. Staining intensities were determined using Cell Profiler software.
To detect changes in ssDNA, U2OS cells were labeled with BrdU for 22 hours, incubated in fresh media for 2 hours and then treated with 3 mM HU for different times prior to detergent extraction with 0.1% Triton. Following fixation and blocking, coverslips were stained with a mouse anti-BrdU antibody without DNA denaturation and analyzed by microscopy. Mean BrdU intensity per nucleus was scored for each sample using Cell Profiler.
GFP-repair assays
DR-GFP U2OS reporter cells were used as described previously (Xia et al., 2006). SA-GFP U2OS reporter cells were obtained from Jeremy Stark and used as described previously (Gunn and Stark, 2012). Briefly, 2.5×105 cells were transfected with the respective siRNAs in a 6-well dish. 24 hours post transfection, media was refreshed and cells were transfected with 2 μg of I-SceI expressing plasmid pCBASceI using FUGENE HD. After 72 hours, cells were analyzed by flow cytometry. P-values for all GFP-based assays were calculated using two-tailed unpaired t-tests.
iPOND-SILAC Mass Spectrometry
Changes in abundance of replication fork proteins in RADXΔ cells were determined using iPOND-SILAC MS as described previously(Dungrawala et al., 2015). Briefly, labelled cells were harvested following cross-linking with 1% formaldehyde for 10 minutes and quenched with glycine. Cells were permeabilized in 0.25% TritonX-100/PBS for 30 minutes. RADXΔ and wild type cells were combined 1:1 prior to performing the click reaction for 2 hours. Streptavidin coupled C1 magnabeads were utilized to capture DNA-protein complexes, washed and boiled in sample buffer to reverse the cross-links. The samples were separated by SDS-PAGE gel and digested with trypsin. MudPIT analysis was performed on the extracted peptides using an eight-step gradient. MaxQuant was utilized for peptide and protein identification. For iPOND analysis in RADXΔ293T cells, 4×108 cells were utilized. For iPOND analysis in RADXΔ U2OS cells, 5×108 cells were utilized. To examine changes in response to HU, cells were incubated for 10 minutes with 10 μM EdU followed by 24 hours of 3 mM HU leaving EdU in the media.
Neutral Comet Assay
Trevigen comet assay kit was utilized to detect changes in DSBs. Tail moments were scored using the CometScore software (TriTek). All comet assay data is presented with box and whisker plots where the box depicts 25–75%, whiskers are 10–90%, and median value is indicated.
DNA Fiber Analysis
DNA fiber analysis of DNA replication was carried out essentially as described previously (Couch et al., 2013). Briefly, cells were labeled with 20 μM IdU for 15 minutes, washed twice with HBSS and labeled with 100 μM CldU for 15 minutes. Cells were washed twice again with HBSS and then treated with or without HU prior to collection. Following stretching and fixation on glass slides, DNA was denatured in 2.5M HCl for 80 minutes, washed three times with PBS and blocked in 10% goat serum/PBS with 0.1% triton X-100 for 1 hour. The DNA was stained with antibodies recognizing IdU and CldU for 1 hour and probed subsequently with secondary antibodies for 30 minutes. Images were obtained using a 40X oil objective (Nikon Eclipse Ti) and fiber lengths analyzed using NI-elements software. The percentage of asymmetric forks was calculated as those with greater than 33.3% length difference between sister forks.
siRNAs, Antibodies and Chemicals
Four different ON-TARGETplus CXorf57 siRNA (Dharmacon) target sequences were utilized for knockdown of RADX: J-014634-21 (5′-CAUAGAGGCCAGCCGUAUA-3′), J-014634-20 (5′-GCUUGAACUCUCUCGUAUA-3′), J-014634-19 (5′-CUUCAGAAAUAGAGCGCAC-3′), J-014634-17 (5′-GAACACAACUUUAGCGAUA-3′). J-014634-21 siRNA was utilized for all RADX knockdown experiments unless otherwise noted. Other siRNAs used were as follows: Qiagen Flexitube Hs_RAD51_7 (5′-AAGGGAATTAGTGAAGCCAAA-3′), Qiagen Flexitube Hs_RAD51_9 (5′-CAGGTGGTAGCTCAAGTGGAT-3′), Qiagen Flexitube Hs_BRCA2_6 (5′-CAGGACACAATTACAACTAAA-3′),siGENOME Dharmacon MUS81 (5′-GGGUAUACCUGGUGGAAGA-3′), ON-TARGETplus Dharmacon SMARCAL1 (5′-GCUUUGACCUUCUUAGCAA-3′), Ambion silencer select siZRANB3 (5′-CAAGAGAUAUCAUCGAUUAtt-3′), siGENOME Dharmacon siBLM D-007287-01 (5′-GAGCACAUCUGUAAAUUAA-3′), siGENOME Dharmacon siBLM D-007287-03 (5′-GAGAAACUCACUUCAAUAA-3′), siGENOME Dharmacon siBLM D-007287-04 (5′-CAGGAUGGCUGUCAGGUUA-3′) and siGENOME Dharmacon siBLM D-007287-05 (5′-CUAAAUCUGUGGAGGGUUA-3′. siRAD51_7 and siRAD51_9 were pooled and used for all RAD51 knockdown experiments. A pool of all four siRNAs were used for BLM knockdown experiments. The following antibodies were used: rabbit anti-RAD51 (1:1000, ab63801, Abcam), rabbit anti-RAD51 (1:200 for IF, SantaCruz,sc8349), mouse anti-BRCA2 (1:250, OP95, Calbiochem),mouse anti-MUS81 (1:1000, ab14387, Abcam), rabbit anti-SMARCAL1 (1:1000), rabbit anti-ZRANB3 (1:1000, Bethyl, A303-033A), mouse anti-RPA32 (1:1000, abcam, ab2175, rabbit anti-RPA32 S4/S8 (1:1000, Bethyl, A300-245A) and rabbit anti-histone H3 (1:10,000, Abcam, ab46765). Primary antibody to RADX was made by Bethyl Laboratories using a RADX peptide containing residues 846–855 of human RADX. The antibody was validated by examining overexpressed RADX and cell lysates from RADXΔ cells. It was utilized at 1:500 dilution for immunoblotting. Mouse anti-BrdU (1:100, 347580, BD) and rat anti-BrdU (1:100, ab6326, Abcam) were used for DNA fiber analyses to recognize IdU and CldU respectively.
Protein Purifications
Flag-RADX from baculovirus-infected insect cells was purified using the same methodology as previously described for Flag-SMARCAL1 purification (Bétous et al., 2013; Bhat et al., 2015). Briefly, cells were lysed in buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 0.1 mM EDTA, 1 mM DTT, 0.2 mM PMSF, 1 mg/mL leupeptin, 1 mg/mL aprotinin, and 0.1% Triton X-100. After high-speed centrifugation, the cleared lysates were incubated with Flag-M2 beads (Sigma) for 4 h at 4°C. The beads were washed three times in LiCl buffer (lysis buffer containing 0.3 M LiCl) and twice in KCl buffer (20 mM HEPES at pH 7.6, 20% glycerol, 0.1 M KCl, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM DTT, 0.2 mM PMSF, 0.01% IGEPAL CA-630). The bound proteins were eluted in KCl buffer containing 0.25 mg/mL Flag peptide on ice, flash-frozen, and stored at −80°C.
Biotin-DNA pull-down assays
Dynabeads T1 (Life Technologies) were washed twice in TE buffer and bound to biotinylated DNA substrates at room temperature for 30min. Beads were washed twice again in TE buffer followed with two washes in binding buffer (80mM Tris, pH 7.5, 100mM KCl, 5mM MgCl2, 2mM DTT and 100μg/ml BSA in RNase/DNase free water). 1ul of beads with 4 picomoles of bound DNA was resuspended in binding buffer. Approximately 500 femtomoles of purified protein was added to the mix and rotated at room temperature for 30min. The supernatant was discarded and the beads were boiled in 2X sample buffer for 5min. Captures were analyzed by immunoblotting. The ssDNA oligo utilized for pulldown assays was a poly dT50. The dsDNA was created by annealing5′-/Biosg/GGATGATGACTCTTCTGGTCCGGATGGTAGTTAAGTGTTGAG-3′ (IDT) with its complimentary oligo.
Electrophoretic Mobility Shift Assays for DNA binding
32P-labeled oligo-dT50 ssDNA was incubated with the indicated concentrations of RADX in binding buffer containing 40 mM Tris (pH 7.5), 100 mM KCl, 5 mM MgCl2, 100 g/ml BSA, and 2 mM DTT for 30 minutes at room temperature. After adding 15% Ficoll dye to a final concentration of 2.5%, the reactions were separated by electrophoresis on an 8% gel (19:1 polyacrylamide, 1× TBE) at 80 V for 80 min. Gels were dried and quantified using a Molecular Imager FX system (Bio-Rad). The dsDNA substrate was created by annealing the following oligonucleotide purchased from IDT with its complimentary oligo. 5′-TCGATAGTCGGATCCTCTAGACAGCTCCATGTAGCAAGGCACTGGTAGAATTCGGCAGCGT. A non-linear regression fit in was completed in Prism. The R2 goodness of fit is 0.98 for ssDNA, and 0.96 for dsDNA.
mESC viability assay
Generation of the Brca2cko/ko PL2F7 mESC was described previously(Kuznetsov et al., 2008). Lentiviral shRNA against mouse RADX was purchased from Sigma (TRCN0000128185). Generation of stable KD clones in PL2F7 cells was performed as described(Ding et al., 2016). qPCR was performed to verify knockdown using iTaq Universal SYBR Green Supermix (Bio-rad) and by using the following primers to detect mouse RadX mRNA: Forward: 5′ TTGGAGCACCCCGAAAGGGATCAGG; Reverse: 5′ ATCTAGGGACTCCCCACAGTGGACC. The southern blot to examine Brca2 deletion after the addition of Cre with selection for the HPRT gene product performed as described (Ding et al., 2016).
Structural homology modeling
Modeling of the 3D structure of putative human RADX domains was performed using the template-based protein structure modeling program RaptorX (Källberg et al., 2012) (webserver version) using the full length sequence (amino acids 1-855). Comparative modeling of OB-2 domain complexed with DNA was performed using the program MODELLER v9.16(Eswar et al., 2006) using the complex of RPA70A bound to ssDNA, extracted from the X-ray crystal structure of the RPA70AB-dC8 complex (PDB code 1JMC), as a template. The objective function score was used for evaluating the quality of each of the 100 models. The best model was selected for designing mutations in the putative DNA binding site of RADX. Representations of the structural models for figures were generated using Pymol (Schrodinger LLC).
Sister Chromatid Exchange Assay
24hrs post transfection with the indicated siRNAs, cells were labelled with 10μM BrdU for approximately two cycles (48hrs). Colcemid was added to a final concentration of 150ng/ml to enrich for mitotic cells. Cells were trypsinized and spread for metaphases as described previously (German and Alhadeff, 2001). Cells were stained with 0.1mg/ml acridine orange and slides were mounted in Sorenson buffer (0.1 M Na2HPO4, 0.1 M NaH2PO4, pH 6.8).
Chromatin Fractionation
Chromatin fractionation experiments were performed as described previously(Lee et al., 2013). Briefly, cells were harvested and resuspended in Buffer A (100mM NaCl, 300mM Sucrose, 3mM MgCl2, 10mM Pipes pH 6.8, 1mM EGTA, 0.2% TX-100, 1mM DTT, 1mM NaF, 1mM Na2VO3 and protease inhibitors) for 5 minutes on ice to collect the soluble fraction. The pellet was then washed once with Buffer A and then resuspended in Buffer B (50mM Tris-HCl pH 7.5, 150mM NaCl, 5mM EDTA, 1% TritonX-100, 0.1% SDS, 1mM DTT. 1mM NaF, 1mM Na2VO3 and protease inhibitors) for 10 minutes on ice. The insoluble fraction was collected following sonication and centrifugation.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were completed using Prism. If the data conformed to a normal distribution, a two tailed t-test was used. In most cases a normal distribution was not assumed and so a Mann-Whitney test was used. An ANOVA was used when comparing more than two samples, with a Dunn’s post-test to correct for multiple comparisons. The authors were blinded to all sample identities. No statistical methods or criteria were used to estimate sample size or to include/exclude samples. All experiments were performed at least twice unless otherwise stated, and representative experiments are depicted. Individual statistical details for each experiment including sample size, significance values and tests performed are indicated in the figure legends, figures and method details.
DATA AND SOFTWARE AVAILABILITY
All data is available by request to the corresponding author. The iPOND data presented in Figure 1B was extracted from previously published datasets and are available within that manuscript (Dungrawala et al., 2015). The iPOND data presented in Figure 4B is available as a supplemental table (Table S1). All the raw microscope files and western blots have been deposited in Mendeley and are available at http://dx.doi.org/10.17632/rfhrr9nb9h.1.
Supplementary Material
SUPPLEMENTAL ITEM
Table S1. iPOND data comparing proteins enriched at stalled forks in RADXΔ to RADX wild-type cells (related to Figure 4). The values listed are Log2 of the ratios obtained from SILAC mass spectrometry. Note that BRCA2 was only observed in three of the five experiments but is included for comparison.
Highlights.
RADX (CXorf57) is a single-strand DNA binding protein related to RPA
RADX prevents MUS81-dependent replication fork collapse
RADX antagonizes RAD51 to prevent excessive replication fork remodeling
RADX deletion restores fork protection and chemoresistance to BRCA2-deficient cells
Acknowledgments
This research was supported by NIH grant R01GM116616 to DC with additional funding from the Breast Cancer Research Foundation (BCRF 16-029), and R35GM118089 to WJC. Purification of RADX was supported by P01CA092584 and assistance from Miaw-Sheue Tsai. KB is supported by F99CA212435. SA-GFP cells were a gift from Jeremy Stark. We thank James Berger, Ilya Finkelstein, James Dewar, and Andre Nussensweig for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
H.D. and K.B. conducted most of the experiments with assistance from A.S. S.W. and R.Z and direction from D.C.; X.D. conducted the BRCA2 ES experiment with direction from S.S.; R.L.M. generated the homology structure model with direction from W.C.; H.D., K.B., and D.C. conceived of the project and wrote the manuscript.
References
- Ball HL, Myers JS, Cortez D. ATRIP binding to replication protein A-single-stranded DNA promotes ATR-ATRIP localization but is dispensable for Chk1 phosphorylation. Mol Biol Cell. 2005;16:2372–2381. doi: 10.1091/mbc.E04-11-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansbach CE, Bétous R, Lovejoy CA, Glick GG, Cortez D. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 2009;23:2405–2414. doi: 10.1101/gad.1839909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass TE, Luzwick JW, Kavanaugh G, Carroll C, Dungrawala H, Glick GG, Feldkamp MD, Putney R, Chazin WJ, Cortez D. ETAA1 acts at stalled replication forks to maintain genome integrity. Nat Cell Biol. 2016;18:1185–1195. doi: 10.1038/ncb3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bétous R, Mason AC, Rambo RP, Bansbach CE, Badu-Nkansah A, Sirbu BM, Eichman BF, Cortez D. SMARCAL1 catalyzes fork regression and holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012;26:151–162. doi: 10.1101/gad.178459.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bétous R, Couch FB, Mason AC, Eichman BF, Manosas M, Cortez D. Substrate-selective repair and restart of replication forks by DNA translocases. Cell Rep. 2013;3:1958–1969. doi: 10.1016/j.celrep.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KP, Bétous R, Cortez D. High-affinity DNA-binding domains of replication protein A (RPA) direct SMARCAL1-dependent replication fork remodeling. J Biol Chem. 2015;290:4110–4117. doi: 10.1074/jbc.M114.627083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budke B, Lv W, Kozikowski AP, Connell PP. Recent Developments Using Small Molecules to Target RAD51: How to Best Modulate RAD51 for Anticancer Therapy? ChemMedChem. 2016;11:2468–2473. doi: 10.1002/cmdc.201600426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri AR, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016;535:382–387. doi: 10.1038/nature18325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow KH, Courcelle J. RecO Acts with RecF and RecR to Protect and Maintain Replication Forks Blocked by UV-induced DNA Damage in Escherichia coli. J Biol Chem. 2004;279:3492–3496. doi: 10.1074/jbc.M311012200. [DOI] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. The DNA Damage Response: Making It Safe to Play with Knives. 2011 doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Bredemeyer AL, Sowa ME, Terret ME, Jallepalli PV, Harper JW, Elledge SJ. The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev. 2009;23:2415–2425. doi: 10.1101/gad.1832309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Nimonkar AV, Hu Y, Hajdu I, Achar J, Izhar L, Petit SA, Adamson B, Yoon JC, Kowalczykowski SC, et al. The ZRANB3 translocase associates with poly-ubiquitinated PCNA to promote fork restart and limit recombination after replication stress. Mol Cell. 2012;47:396–409. doi: 10.1016/j.molcel.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch FB, Cortez D. Fork reversal, too much of a good thing. Cell Cycle. 2014;13:1049–1050. doi: 10.4161/cc.28212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Bétous R, Carroll CM, Jung SY, Qin J, Cimprich KA, et al. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 2013;27:1610–1623. doi: 10.1101/gad.214080.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courcelle J, Carswell-Crumpton C, Hanawalt PC. recF and recR are required for the resumption of replication at DNA replication forks in Escherichia coli. Proc Natl Acad Sci U S A. 1997;94:3714–3719. doi: 10.1073/pnas.94.8.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox MM. Regulation of Bacterial RecA Protein Function. Crit Rev Biochem Mol Biol. 2007;42:41–63. doi: 10.1080/10409230701260258. [DOI] [PubMed] [Google Scholar]
- Davies OR, Pellegrini L. Interaction with the BRCA2 C terminus protects RAD51-DNA filaments from disassembly by BRC repeats. Nat Struct Mol Biol. 2007;14:475–483. doi: 10.1038/nsmb1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X, Chaudhuri AR, Callen E, Pang Y, Biswas K, Klarmann KD, Martin BK, Burkett S, Cleveland L, Stauffer S, et al. Synthetic viability by BRCA2 and ARP1/ARTD1 deficiencies. Nat Commun. 2016;7:12425. doi: 10.1038/ncomms12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drees JC, Lusetti SL, Chitteni-Pattu S, Inman RB, Cox MM. A RecA filament capping mechanism for RecX protein. Mol Cell. 2004;15:789–798. doi: 10.1016/j.molcel.2004.08.026. [DOI] [PubMed] [Google Scholar]
- Dungrawala H, Rose KL, Bhat KP, Mohni KN, Glick GG, Couch FB, Cortez D. The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Mol Cell. 2015;59:998–1010. doi: 10.1016/j.molcel.2015.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duursma AM, Driscoll R, Elias JE, Cimprich KA. A Role for the MRN Complex in ATR Activation via TOPBP1 Recruitment. Mol Cell. 2013;50:116–122. doi: 10.1016/j.molcel.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–1115. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- Esashi F, Christ N, Gannon J, Liu Y, Hunt T, Jasin M, West SC. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature. 2005;434:598–604. doi: 10.1038/nature03404. [DOI] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. Comparative protein structure modeling using Modeller. 2006 doi: 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn RL, Zou L. Oligonucleotide/oligosaccharide-binding fold proteins: a growing family of genome guardians. Crit Rev Biochem Mol Biol. 2010;45:266–275. doi: 10.3109/10409238.2010.488216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German J, Alhadeff B. Analysis of sister-chromatid exchanges. Curr Protoc Hum Genet. 2001 doi: 10.1002/0471142905.hg0806s02. Chapter 8, Unit 8.6. [DOI] [PubMed] [Google Scholar]
- Guillemette S, Serra RW, Peng M, Hayes JA, Konstantinopoulos PA, Green MR, Green MR, Cantor SB. Resistance to therapy in BRCA2 mutant cells due to loss of the nucleosome remodeling factor CHD4. Genes Dev. 2015;29:489–494. doi: 10.1101/gad.256214.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn A, Stark JM. I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol Biol. 2012;920:379–391. doi: 10.1007/978-1-61779-998-3_27. [DOI] [PubMed] [Google Scholar]
- Haahr P, Hoffmann S, Tollenaere MAX, Ho T, Toledo LI, Mann M, Bekker-Jensen S, Räschle M, Mailand N. Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat Cell Biol. 2016;18:1196–1207. doi: 10.1038/ncb3422. [DOI] [PubMed] [Google Scholar]
- Hartlerode AJ, Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J. 2009;423:157–168. doi: 10.1042/BJ20090942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Chaudhuri AR, Lopes M, Costanzo V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol. 2010;17:1305–1311. doi: 10.1038/nsmb.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horii ZI, Suzuki K. Degradation of the DNA of Escherichia coli K12 rec- (jc1569b) after irradiation with ultraviolet light. Photochem Photobiol. 1968;8:93–105. doi: 10.1111/j.1751-1097.1970.tb05976.x. [DOI] [PubMed] [Google Scholar]
- Källberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, Xu J. Template-based protein structure modeling using the RaptorX web server. Nat Protoc. 2012;7:1511–1522. doi: 10.1038/nprot.2012.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass EM, Moynahan ME, Jasin M. When Genome Maintenance Goes Badly Awry. Mol Cell. 2016;62:777–787. doi: 10.1016/j.molcel.2016.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein HL. The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair (Amst) 2008;7:686–693. doi: 10.1016/j.dnarep.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, de Antoni A, Técher H, Baldi G, Costanzo V. Moonlighting at replication forks: a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett. 2017 doi: 10.1002/1873-3468.12556. [DOI] [PubMed] [Google Scholar]
- Kowalczykowski SC. An Overview of the Molecular Mechanisms of Recombinational DNA Repair. Cold Spring Harb Perspect Biol. 2015;7 doi: 10.1101/cshperspect.a016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov SG, Liu P, Sharan SK. Mouse embryonic stem cell-based functional assay to evaluate mutations in BRCA2. Nat Med. 2008;14:875–881. doi: 10.1038/nm.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen NB, Hickson ID. RecQ helicases: Conserved guardians of genomic integrity. Adv Exp Med Biol. 2013;767:161–184. doi: 10.1007/978-1-4614-5037-5_8. [DOI] [PubMed] [Google Scholar]
- Lee KY, Fu H, Aladjem MI, Myung K. ATAD5 regulates the lifespan of DNA replication factories by modulating PCNA level on the chromatin. J Cell Biol. 2013;200:31–44. doi: 10.1083/jcb.201206084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–294. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- Lord CJ, Tutt ANJ, Ashworth A. Synthetic Lethality and Cancer Therapy: Lessons Learned from the Development of PARP Inhibitors. Annu Rev Med. 2015;66:455–470. doi: 10.1146/annurev-med-050913-022545. [DOI] [PubMed] [Google Scholar]
- Mason JM, Logan HL, Budke B, Wu M, Pawlowski M, Weichselbaum RR, Kozikowski AP, Bishop DK, Connell PP. The rad51-stimulatory compound rs-1 can exploit the rad51 overexpression that exists in cancer cells and tumors. Cancer Res. 2014;74:3546–3555. doi: 10.1158/0008-5472.CAN-13-3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, Karlan BY, Taniguchi T, Swisher EM. Secondary Somatic Mutations Restoring BRCA1/2 Predict Chemotherapy Resistance in Hereditary Ovarian Carcinomas. J Clin Oncol. 2011;29:3008–3015. doi: 10.1200/JCO.2010.34.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor M. Targeting the DNA damage response in cancer. Mol Cell. 2015;60:547–560. doi: 10.1016/j.molcel.2015.10.040. [DOI] [PubMed] [Google Scholar]
- Oakley GG, Patrick SM. Replication protein A: directing traffic at the intersection of replication and repair. Front Biosci (Landmark Ed) 2010;15:883–900. doi: 10.2741/3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagès V, Koffel-Schwartz N, Fuchs RPP. recX, a new SOS gene that is co-transcribed with the recA gene in Escherichia coli. DNA Repair (Amst) 2003;2:273–284. doi: 10.1016/s1568-7864(02)00217-3. [DOI] [PubMed] [Google Scholar]
- Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiros S, Roos WP, Kaina B. Rad51 and BRCA2–New molecular targets for sensitizing glioma cells to alkylating anticancer drugs. PLoS One. 2011;6:e27183. doi: 10.1371/journal.pone.0027183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, Lopes M. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol. 2012;19:417–423. doi: 10.1038/nsmb.2258. [DOI] [PubMed] [Google Scholar]
- Richard DJ, Bolderson E, Khanna KK. Multiple human single-stranded DNA binding proteins function in genome maintenance: structural, biochemical and functional analysis. Crit Rev Biochem Mol Biol. 2009;44:98–116. doi: 10.1080/10409230902849180. [DOI] [PubMed] [Google Scholar]
- Richardson C, Moynahan ME, Jasin M. Double-strand break repair by interchromosomal recombination: Suppression of chromosomal translocations. Genes Dev. 1998;12:3831–3842. doi: 10.1101/gad.12.24.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson C, Stark JM, Ommundsen M, Jasin M. Rad51 overexpression promotes alternative double-strand break repair pathways and genome instability. Oncogene. 2004;23:546–553. doi: 10.1038/sj.onc.1207098. [DOI] [PubMed] [Google Scholar]
- Robu ME, Inman RB, Cox MM. RecA protein promotes the regression of stalled replication forks in vitro. Proc Natl Acad Sci U S A. 2001;98:8211–8218. doi: 10.1073/pnas.131022698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–1120. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbajna S, West SC. Holliday junction processing enzymes as guardians of genome stability. Trends Biochem Sci. 2014;39:409–419. doi: 10.1016/j.tibs.2014.07.003. [DOI] [PubMed] [Google Scholar]
- Satta G, Gudas LJ, Pardee AB. Degradation of Escherichia coli DNA: Evidence for limitation in vivo by protein X, the recA gene product. MGG Mol Gen Genet. 1979;168:69–80. doi: 10.1007/BF00267935. [DOI] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Wu H, Jasin M. A Distinct Replication Fork Protection Pathway Connects Fanconi Anemia Tumor Suppressors to RAD51-BRCA1/2. Cancer Cell. 2012;22:106–116. doi: 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodinger LLC The PyMOL Molecular Graphics System, Version 0.99rc6
- Sogo JM, Lopes M, Foiani M. Fork Reversal and ssDNA Accumulation at Stalled Replication Forks Owing to Checkpoint Defects. Sci. 2002;297:599–602. doi: 10.1126/science.1074023. [DOI] [PubMed] [Google Scholar]
- Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M. Genetic Steps of Mammalian Homologous Repair with Distinct Mutagenic Consequences. Mol Cell Biol. 2004;24:9305–9316. doi: 10.1128/MCB.24.21.9305-9316.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohl EA, Brockman JP, Burkle KL, Morimatsu K, Kowalczykowski SC, Seifert HS. Escherichia coli RecX inhibits RecA recombinase and coprotease activities in vitro and in vivo. J Biol Chem. 2003;278:2278–2285. doi: 10.1074/jbc.M210496200. [DOI] [PubMed] [Google Scholar]
- Symington LS. End resection at double-strand breaks: Mechanism and regulation. Cold Spring Harb Perspect Biol. 2014;6 doi: 10.1101/cshperspect.a016436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarsounas M, Davies D, West SC. BRCA2-dependent and independent formation of RAD51 nuclear foci. Oncogene. 2003;22:1115–1123. doi: 10.1038/sj.onc.1206263. [DOI] [PubMed] [Google Scholar]
- Tennstedt P, Fresow R, Simon R, Marx A, Terracciano L, Petersen C, Sauter G, Dikomey E, Borgmann K. RAD51 overexpression is a negative prognostic marker for colorectal adenocarcinoma. Int J Cancer. 2013;132:2118–2126. doi: 10.1002/ijc.27907. [DOI] [PubMed] [Google Scholar]
- Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, Lukas J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell. 2013;155:1088–1103. doi: 10.1016/j.cell.2013.10.043. [DOI] [PubMed] [Google Scholar]
- Venkatesh R, Ganesh N, Guhan N, Reddy MS, Chandrasekhar T, Muniyappa K. RecX protein abrogates ATP hydrolysis and strand exchange promoted by RecA: Insights into negative regulation of homologous recombination. Proc Natl Acad Sci U S A. 2002;99:12091–12096. doi: 10.1073/pnas.192178999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierling S, Weber T, Wohlleben W, Muth G. Transcriptional and mutational analyses of the Streptomyces lividans recX gene and its interference with recA activity. J Bacteriol. 2000;182:4005–4011. doi: 10.1128/jb.182.14.4005-4011.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis NA, Chandramouly G, Huang B, Kwok A, Follonier C, Deng C, Scully R. BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature. 2014;510:556–559. doi: 10.1038/nature13295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ, Livingston DM. Control of BRCA2 Cellular and Clinical Functions by a Nuclear Partner, PALB2. Mol Cell. 2006;22:719–729. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Xu X, Vaithiyalingam S, Glick GG, Mordes DA, Chazin WJ, Cortez D. The basic cleft of RPA70N binds multiple checkpoint proteins, including RAD9, to regulate ATR signaling. Mol Cell Biol. 2008;28:7345–7353. doi: 10.1128/MCB.01079-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Ghosal G, Chen J. The annealing helicase HARP protects stalled replication forks. Genes Dev. 2009;23:2394–2399. doi: 10.1101/gad.1836409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusufzai T, Xiangduo K, Kyoko Y, Kadonaga JT. The annealing helicase HARP is recruited to DNA repair sites via an interaction with RPA. Genes Dev. 2009;23:2400–2404. doi: 10.1101/gad.1831509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, Lopes M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol. 2015;208:563–579. doi: 10.1083/jcb.201406099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L. DNA Replication Checkpoint: New ATR Activator Identified. Curr Biol. 2017;27:R33–R35. doi: 10.1016/j.cub.2016.11.025. [DOI] [PubMed] [Google Scholar]
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTAL ITEM
Table S1. iPOND data comparing proteins enriched at stalled forks in RADXΔ to RADX wild-type cells (related to Figure 4). The values listed are Log2 of the ratios obtained from SILAC mass spectrometry. Note that BRCA2 was only observed in three of the five experiments but is included for comparison.