

Abstract
Flavodoxins are electron-transfer proteins that contain the prosthetic group flavin mononucleotide. In Escherichia coli, flavodoxin is reduced by the FAD-containing protein NADPH:ferredoxin (flavodoxin) oxidoreductase; flavodoxins serve as electron donors in the reductive activation of anaerobic ribonucleotide reductase, biotin synthase, pyruvate formate lyase, and cobalamin-dependent methionine synthase. In addition, domains homologous to flavodoxin are components of the multidomain flavoproteins cytochrome P450 reductase, nitric oxide synthase, and methionine synthase reductase. Although three-dimensional structures are known for many of these proteins and domains, very little is known about the structural aspects of their interactions. We address this issue by using NMR chemical shift mapping to identify the surfaces on flavodoxin that bind flavodoxin reductase and methionine synthase. We find that these physiological partners bind to unique overlapping sites on flavodoxin, precluding the formation of ternary complexes. We infer that the flavodoxin-like domains of the cytochrome P450 reductase family form mutually exclusive complexes with their electron-donating and -accepting partners, complexes that require conformational changes for interconversion.
Flavodoxins are used by photosynthetic cyanobacteria and anaerobic and aerobic bacteria (1–5). In enteric bacteria, flavodoxin participates in a number of reactions, including the reductive activation of cobalamin-dependent methionine synthase (6), which catalyzes the terminal step in de novo methionine biosynthesis. In this reaction, reduced flavodoxin donates an electron to the cobalamin of methionine synthase and S-adenosylmethionine (AdoMet) serves as the methyl donor in a reductive methylation (6, 7).
Although humans also possess cobalamin-dependent methionine synthase, they do not have flavodoxin and flavodoxin reductase to provide the reducing equivalents for methionine synthase activation. However, an analogous NADPH-dependent electron transfer reaction reactivates methionine synthase in humans through methionine synthase reductase, a protein containing domains with sequence homology to flavodoxin and flavodoxin reductase (8). Thus, features governing the molecular recognition between Escherichia coli flavodoxin, flavodoxin reductase, and methionine synthase are likely to be conserved in their human counterparts.
Interestingly, other proteins, including cytochrome P450 reductase, cytochrome P450BM-3, and nitric oxide synthase, carry domains homologous to flavodoxin and flavodoxin reductase on a single polypeptide chain (Fig. 1). It has been hypothesized that cytochrome P450 reductase arose from a gene fusion of flavodoxin and flavodoxin reductase (9). The structures of the flavin mononucleotide (FMN)- and FAD-containing domains of cytochrome P450 reductase (10) are remarkably similar to flavodoxin (11) and flavodoxin reductase (12, 13). Hence, definition of the molecular interfaces between flavodoxin and its partners should provide insight into the electron transfer interactions in these homologous systems.
Figure 1.

Flavodoxin, flavodoxin reductase, and proteins that contain homologous domains. Domains that bind FMN and that are homologous to flavodoxin are shown in gray. In green are the reducing partners of the flavodoxin-like domains, which bind FAD and are homologous to flavodoxin reductase. In red are the oxidizing partners of the flavodoxin-like domains, which bind heme or corrin cofactors.
Methods
Materials.
15NH4Cl and 2H2O were obtained from Isotec, and 13C6-glucose was supplied by Martek (Columbia, MD). All plasmids were prepared by using the Promega Wizard Plus Minipreps DNA purification kit. Restriction enzymes and T4 DNA ligase were supplied by Boehringer Mannheim, Promega, and New England Biolabs. All other reagents were obtained from Sigma.
Preparation of 2H,13C,15N-Labeled Flavodoxin.
For production of 2H,13C,15N-labeled flavodoxin, E. coli K-12 strain PS2209 containing the flavodoxin expression plasmid pDH01 (4) was grown at 37°C on glucose-minimal 3-(N-morpholino)propanesulfonic acid labeling medium (>99% 2H, >99% 13C, >99% 15N, containing 4 g/liter 13C6-glucose and 0.5 g/liter 15N-ammonium chloride in 2H2O) with 100 μg/ml of ampicillin and 35 μM uracil. Flavodoxin was purified by anion exchange chromatography (4). The exchangeable hydrogens were replaced with 1H during protein purification in 1H2O buffers. Flavodoxin concentrations were calculated by using the absorbance of bound FMN (ɛ466 nm = 8,250 M−1 ⋅ cm−1) (14).
Preparation of Proteins.
Purified recombinant wild-type E. coli methionine synthase was prepared as previously described from E. coli K-12 strain XL1-Blue/pKF5a by using anion exchange chromatography (15). The AdoMet-binding fragment was obtained by limited tryptic proteolysis of methionine synthase and purified by anion exchange and size exclusion chromatographies (16). Concentration of this fragment was estimated by using the molar extinction coefficient ɛ280 nm = 68,490 M−1 ⋅ cm−1, an average of the values obtained by using the program peptidesort (Genetics Computer Group, Madison, WI) and the calculation of Pace and colleagues (17). The accuracy of this extinction coefficient was confirmed by thiol titrations with 5,5′-dithiobis- (2-nitrobenzoic acid) under denaturing conditions.
Generation of Histidine-Tagged E. coli Flavodoxin Reductase.
Amino-terminal His6-tagged E. coli flavodoxin reductase (His6flavodoxin reductase) was generated from the Pet11a-FPR plasmid (18) by using restriction digestion and ligation. The resultant plasmid, pCNS5, enables expression of flavodoxin reductase with the addition of amino-terminal residues MetSerTyr2His6. The entire flavodoxin reductase gene was sequenced (University of Michigan DNA Sequencing Core Facility, Ann Arbor, MI) to ensure that no mutations were present. The His6flavodoxin reductase was expressed in cells of E. coli K-12 strain Hms174(DE3) (Novagen) containing the pCNS5 plasmid and purified by nickel affinity chromatography. Protein concentration was calculated by using the absorbance of bound FAD (ɛ456 nm = 7,100 M−1 ⋅ cm−1) (6).
NMR Spectroscopy.
1H,15N-Heteronuclear single-quantum correlation (HSQC) experiments were performed at 25°C on a Varian INOVA 800 MHz spectrometer. Spectra were processed and analyzed with the programs nmrpipe (19) and xeasy (20). Changes in average amide chemical shifts are calculated by using Eq. 1, in which ΔδN represents the change in the amide nitrogen's chemical shift, and ΔδH represents the change in the amide proton's chemical shift (21).
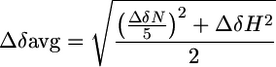 |
1 |
Spectra were recorded for a sample of 0.55 mM oxidized 2H,13C,15N-labeled flavodoxin [Fig. 6 (which is published as supplemental data on the PNAS web site, www.pnas.org)] and for samples of oxidized 2H,13C,15N-labeled flavodoxin titrated with unlabeled oxidized flavodoxin reductase and, separately, with the unlabeled AdoMet-binding domain of methionine. All samples contained 0.1 mM EDTA and 50 mM potassium phosphate, pH 7.0, in 10% 2H2O/90% H2O. Because of dilution, the concentration of 2H,13C,15N-labeled flavodoxin in the titrations ranged from 0.22 mM to 0.55 mM. Titration points were at molar ratios of 0, 0.25, 0.50, 0.75, 2.25, and 11.33 (flavodoxin reductase:flavodoxin and AdoMet-binding domain of methionine synthase:flavodoxin). During the course of titrations, individual residues that span the mapped surfaces underwent uniform changes in the magnitude and direction of their normalized amide chemical shifts, Δδavg/Δδmaximum (data not shown). These factors suggest a homogeneous interface in each titration, rather than multiple binding interactions with different binding affinities.
A third set of experiments was performed in which the AdoMet-binding domain of methionine synthase was added to 15N-labeled flavodoxin in the presence of AdoMet. This experiment was performed at molar ratios of 2.25 and 11.33 (AdoMet-binding domain of methionine synthase:flavodoxin). Methionine synthase has a Kd of 1.2 μM for AdoMet (22). The concentration of AdoMet was ≈4 times that of the methionine synthase domain so that the domain would be saturated with the cofactor.
The interaction between 2H,15N-labeled flavodoxin and unlabeled methionine synthase was also studied with the recently developed bulk saturation transfer technique (23). The results were somewhat ambiguous, because the flavin cofactor of the otherwise deuterated flavodoxin was protonated. The interface determined by this experiment, however, was fully compatible with the NMR chemical shift mapping data (data not shown).
Results and Discussion
The Same Face of Flavodoxin Binds to Methionine Synthase and Flavodoxin Reductase.
To identify the surfaces on flavodoxin that bind its physiological partners, we performed 1H,15N-HSQC NMR experiments in which we titrated 15N,2H-labeled flavodoxin (20 kDa) with flavodoxin reductase (28 kDa) and, separately, with the AdoMet-binding region of methionine synthase (38 kDa). The latter module (methionine synthase residues 897-1227) was used in lieu of the entire 136-kDa methionine synthase protein because it is required for reductive activation (24) and because it was previously shown by crosslinking experiments to contain determinants for binding to flavodoxin (25). Only the peptide backbone and amide-containing side chains of flavodoxin were observed in the HSQC titrations, because flavodoxin reductase and the AdoMet-binding region of methionine synthase were not 15N-labeled. When flavodoxin binds a partner protein, residues at the binding interface experience a change in their chemical environment; consequently, chemical shift positions can be altered. Residues involved in allosteric conformational changes also may experience changes in chemical environment, and their chemical shifts may consequently change.
Flavodoxin is in fast exchange (koff > 102 s−1) with both flavodoxin reductase and the AdoMet-binding domain of methionine synthase, enabling us to track the changes in chemical shifts for individual resonances based on the chemical shift mapping method (21). On the basis of the known resonance assignments of E. coli flavodoxin (26), we were able to map the chemical shift perturbations to defined residues of flavodoxin (Fig. 2). Binding of either flavodoxin reductase or methionine synthase to flavodoxin causes chemical shift changes that are localized to the face of flavodoxin that presents the FMN cofactor (Fig. 3).
Figure 2.

Flavodoxin residues that change in chemical shift on titration with flavodoxin reductase and the AdoMet-binding domain of methionine synthase. Changes in average amide chemical shifts (Δδavg) of flavodoxin on titration with flavodoxin reductase (A) and the AdoMet-binding domain of methionine synthase in the absence and presence of AdoMet (B and C, respectively) are mapped onto the sequence of flavodoxin. The molar ratio of flavodoxin reductase: flavodoxin is 2.25:1 (A). The molar ratio of AdoMet-binding domain of methionine synthase:flavodoxin is 11.33:1 (B and C). The vertical color strips categorize the changes in average amide chemical shifts into high, medium, and low groups for projection onto the three-dimensional structure of flavodoxin in Fig. 3.
Figure 3.

Surfaces of flavodoxin that experience chemical shift changes on binding flavodoxin reductase and the AdoMet-binding domain of methionine synthase. Amide chemical shift perturbations of flavodoxin on titration with flavodoxin reductase (A) and the AdoMet-binding domain of methionine synthase in the absence and presence of AdoMet (B and C, respectively) are projected onto the three-dimensional structure of flavodoxin. Changes in average amide chemical shift are indicated by the vertical color strips in Fig. 2 (i.e., in red are residues that shift the most, in yellow are residues that shift moderately, and in green are residues that shift a lesser amount). Glu-61, which has previously been shown to crosslink with the AdoMet-binding domain of methionine synthase (25), is outlined in B and C. The FMN cofactor is colored black. Two faces of flavodoxin are shown, the FMN-containing surface and the surface after 180° rotation about the horizontal axis. With both partners, chemical shift perturbations are largely confined to the FMN-containing face of flavodoxin.
In the structure of the AdoMet-binding domain of methionine synthase, AdoMet is positioned where it could be involved in a binding interface with flavodoxin (27, 28). Therefore, we compared complex formation between flavodoxin and this domain in the absence and presence of AdoMet. The chemical shift changes were confined to the same regions of flavodoxin in the absence and presence of AdoMet (Fig. 2 B and C and Fig. 3 B and C). The addition of AdoMet significantly increased the magnitude of these changes at a given concentration of methionine synthase, suggesting an enhanced interaction in the presence of AdoMet. This observation supports the physiological relevance of the complex observed, because reductive methylation of the cobalamin of methionine synthase requires a methyl donor, AdoMet, in addition to an electron donor, flavodoxin.
Glutamate 61 of flavodoxin is among the residues that undergo changes in chemical shift on interaction with the AdoMet-binding domain of methionine synthase. This residue has previously been shown to form a covalent crosslink with Lys-959 of this methionine synthase module (25). Glu-61 is located at the periphery of the observed surface (Fig. 3 B and C), where steric accommodation of the succinimidyl ester crosslinking intermediate would be possible without disruption of the complex.
The similarities between the surfaces of flavodoxin that interact with flavodoxin reductase and with the AdoMet-binding domain of methionine synthase are striking (Fig. 3). Each interaction surface is centered on the flavin. Many perturbations are clustered in or near three loops that interact with the FMN cofactor; the region between residues 10 and 15 contains residues that form hydrogen bonds with the phosphate oxygens of the FMN, and regions 55–66 and 90–99 contain residues that are in van der Waals contact with the FMN cofactor (11). The interfaces contain a hydrophobic patch including the exposed dimethyl groups of the flavin and flanking residues Trp-57 and Tyr-58, -59, and -94. This hydrophobic patch is surrounded by charged residues. Similar arrangements have been observed in analyses of other protein–protein interfaces (29, 30). Electrostatic interactions may generally orient the molecules for complex formation and occlude solvent, whereas hydrophobic packing may contribute the majority of the binding energy.
The complexes of flavodoxin with flavodoxin reductase or with the AdoMet-binding domain of methionine synthase both have a stoichiometry of 1:1. This stoichiometry is implied by the observation that the chemical shifts of the different residues in the interface change at the same rate on titration (Figs. 7 and 8, which are published as supplemental data on the PNAS web site, www.pnas.org) and that only one partner molecule can bind to the relatively small interface on flavodoxin at a time (Fig. 3). Examination of the interaction of flavodoxin and flavodoxin reductase by isothermal titration calorimetry reveals a Kd of ≈10 μM but a stoichiometry indicating that only two-thirds of the flavodoxin reductase is in a reactive conformation (D.A.H. and R.G.M., unpublished data). Given this partial reactivity, we obtain from the NMR titration data of flavodoxin and flavodoxin reductase a dissociation constant in the range 1–300 μM (Fig. 7, supplemental data, www.pnas.org). The median value of the order of 10 μM is compatible with the fast chemical exchange observed in the NMR titration, as well as with the isothermal titration calorimetry data. The NMR titration data of the binding of the AdoMet-binding domain of methionine synthase to flavodoxin reveals a dissociation constant of 10 mM. We were not able to observe the interaction between this fragment of methionine synthase and flavodoxin by isothermal titration calorimetry, a finding compatible with a Kd of this magnitude. This weak binding is likely because a subdomain of methionine synthase was used. We do, however, believe this binding is representative of the interaction of the full methionine synthase with flavodoxin. This follows from the fact that all perturbed resonances shift together (Fig. 8, supplemental data, www.pnas.org), excluding the possibility that interactions with multiple nonspecific sites are being monitored. Furthermore, we know that AdoMet strongly affects the tightness of interaction but does not change the binding interface (Fig. 2 B and C). We also know that AdoMet lies in the interface between the activation and B12-binding domains of methionine synthase (M. L. Ludwig and R.G.M., unpublished data). AdoMet would then be in position to interact with flavodoxin as well, as observed. We believe these findings strongly support a physiologically significant, albeit weak, complex.
Modeled Binding Interfaces Between Flavodoxin and Its Partners.
Our experimental findings can be used to evaluate models of the binding interfaces between flavodoxin and its physiological partners. In the structures of flavodoxin (11) and flavodoxin reductase (12, 13), the dimethylbenzene moieties of FMN and FAD are exposed. In a productive complex, these cofactors are thought to be juxtaposed for electron transfer. Structures of flavodoxin reductase reveal a cavity at the FAD-containing face that could accommodate the FMN-containing face of flavodoxin. Our experiments demonstrate involvement of the FMN-containing face of flavodoxin in the binding interface with flavodoxin reductase and thus support this interaction model.
Because cytochrome P450 reductase contains a domain homologous to flavodoxin and a domain homologous to flavodoxin reductase, the structure of this protein (10) suggests an arrangement of these molecules for productive electron transfer. In this structure, the flavodoxin- and flavodoxin reductase-like domains are in proximity for electron transfer from FAD to FMN (Fig. 4A). Van der Waals contacts between the flavodoxin reductase- and the flavodoxin-like domains of cytochrome P450 reductase were mapped by homology onto the E. coli flavodoxin structure. There is good agreement between the surface of flavodoxin predicted to be the interface and the surface that experiences changes in chemical shift positions on titration with flavodoxin reductase (compare Figs. 3A and 4A). Therefore, we demonstrate that the interaction between flavodoxin and flavodoxin reductase indeed resembles that between the domains of cytochrome P450 reductase.
Figure 4.

FMN-reducing and FMN-oxidizing configurations in cytochrome 450 reductase may require a conformational change for interconversion. (A) In cytochrome P450 reductase, electrons are transferred from FAD to FMN in an interdomain reaction. In the structure of cytochrome P450 reductase (10), the FAD and FMN cofactors of the flavodoxin reductase-like (green) and flavodoxin-like (gray) domains are juxtaposed for this electron transfer. Van der Waals contacts between these domains of cytochrome P450 reductase are mapped by homology onto the structure of E. coli flavodoxin, which is shown in the same orientation as in Fig. 3. (B) In cytochrome P450BM-3, electrons are transferred from FMN to the heme in an interdomain reaction. In the structure of the heme/FMN-containing fragment of cytochrome P450BM-3 (34), the FMN-containing domain (gray) is juxtaposed against the heme-containing domain (red) for electron transfer. Van der Waals contacts between these domains of cytochrome P450 BM-3 are mapped by homology onto the structure of E. coli flavodoxin, which is shown in the same orientation as in Fig. 3.
Similarly, our results can address a modeled complex of flavodoxin and methionine synthase (28). Because the x-ray structures of E. coli flavodoxin (11) and the cobalamin- (31) and AdoMet-binding domains of methionine synthase (27) have been determined, these could be positioned for reductive remethylation (Fig. 5). In this model, the FMN of flavodoxin is positioned near the cobalamin to permit facile electron transfer, and AdoMet is positioned for subsequent methyl transfer to cobalamin. The predicted involvement of the FMN-containing face of flavodoxin is compatible with our experimental findings. This model also predicts numerous specific contacts between flavodoxin residues and the AdoMet-binding domain of methionine synthase. Many of these flavodoxin residues change in chemical shift position in our experiments (compare Fig. 5 with Fig. 3 B and C). Several features distinguish complexes with the AdoMet-binding domain of methionine synthase from those with flavodoxin reductase. For example, the loop comprised of the residues 145–150 is identified in complexes with the AdoMet-containing domain but not with flavodoxin reductase. In a docking model (28), this loop is a steric match for methionine synthase. Such overlapping, but not identical, binding sites may tune flavodoxins for specific interactions with a variety of physiological partners.
Figure 5.

Modeled interface of flavodoxin with methionine synthase. The FMN, cobalamin, and AdoMet cofactors of flavodoxin (11) (gray), the cobalamin-binding region (31) (blue), and AdoMet-binding domain (27) (red) of methionine synthase are juxtaposed for electron transfer from FMN to cobalamin and for methyl transfer from AdoMet to cobalamin according to a published docking model (28). Predicted contacts of flavodoxin with the AdoMet-binding domain are mapped onto the surface of flavodoxin, which is shown in the same orientation as in Fig. 3.
Binding Information Predicts Conformational Changes.
Our experimental findings strongly suggest that productive ternary complexes between flavodoxin, flavodoxin reductase, and methionine synthase cannot form. The FMN-containing surface of flavodoxin is buried within complexes between flavodoxin reductase and methionine synthase and is not available for simultaneous interaction with another partner. Indeed, competition between methionine synthase and flavodoxin reductase for binding to flavodoxin has previously been documented in spectrophotometric binding titrations (32). Therefore, we predict sequential interactions of flavodoxin with flavodoxin reductase and then with methionine synthase to enable direct interaction of each partner with the FMN-containing surface of flavodoxin.
A complex of the FAD- and FMN-containing domains of cytochrome P450 reductase with a heme-containing partner (a ternary domain complex) has been proposed (10). Electron transfer from cytochrome P450 reductase to its partners occurs in the following sequence: NADPH→FAD→FMN→heme (33). The structure of cytochrome P450 reductase (10) with its adjacent FAD- and FMN-containing domains represents a FMN-reducing conformation (Fig. 4A). The interface between these domains is similar to that observed in titrations of flavodoxin with flavodoxin reductase (Fig. 3A). For the FMN-oxidizing reaction, however, docking of an additional protein to form a ternary domain complex was suggested (10). In contrast, we find overlapping surfaces on the homologous flavodoxin to be occupied in titrations with its reducing and oxidizing partners and consequently do not concur with this suggestion.
Structural studies of cytochrome P450BM-3 suggested conformational changes as an alternative to ternary domain complex formation for the cytochrome P450 reductase family of proteins (34). Cytochrome P450BM-3 is a fatty acid monooxygenase that possesses domains for binding heme, FMN, and FAD. Expression of a heme/FMN-containing fragment of this protein has enabled biochemical and structural studies of a model FMN-oxidizing complex. In the x-ray structure of this construct, the FMN-containing face is again sequestered (Fig. 4 B). Our titrations of flavodoxin with its FMN-oxidizing partner, methionine synthase, depict a parallel interaction surface (Fig. 3C).
For proteins like cytochrome P450 reductase and cytochrome P450BM-3, which carry the FMN- and FAD-containing domains on a single polypeptide, we predict that large conformational changes are necessary to swing the FAD- and FMN-containing domains apart to permit cycling between FMN-reducing and -oxidizing configurations. An additional domain present between the FAD- and FMN-binding domains of cytochrome P450 reductase and an 11-residue linker between these domains in cytochrome P450 BM-3 could serve as hinges for such conformational changes. We propose similar conformational changes for the other proteins that share sequence homology with cytochrome P450 reductase, like methionine synthase reductase and nitric oxide synthase.
Conclusions
The surfaces on flavodoxin that bind flavodoxin reductase and the AdoMet-binding domain of methionine synthase were mapped by using NMR spectroscopy. The data demonstrate that each protein binds to the FMN-containing face of flavodoxin. Because overlapping surfaces are involved in binding these physiological partners, the formation of a ternary complex of flavodoxin with both species is unlikely. Conformational changes have been proposed as an alternative to ternary domain complex formation for the cytochrome P450 reductase family of proteins, which contains domains homologous to flavodoxin and flavodoxin reductase. Our data depicting involvement of the FMN-containing face in complexes with each redox partner provide evidence in support of this hypothesis.
Supplementary Material
Acknowledgments
We are grateful to Michael Waterman and Christopher Jenkins (Vanderbilt University, Nashville, TN) for the Pet11a-FPR plasmid (18). We also thank Maurizio Pellecchia (Triad Therapeutics, San Diego) for preliminary NMR experiments. This work was supported in part by National Institutes of Health Research Grants GM24908 (R.G.M.), HL58955 (R.G.M.), and GM 52421 (E.R.P.Z.), a Medical Scientist Training Program Fellowship T32 GM07863–20 (D.A.H.), a University of Michigan Rackham Predoctoral Fellowship (D.A.H.), and a University of Michigan Regents Fellowship (C.W.V.K.).
Abbreviations
- AdoMet
S-adenosylmethionine
- FMN
flavin mononucleotide
- HSQC
heteronuclear single-quantum correlation
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Mayhew S G, Tollin G. In: Chemistry and Biochemistry of Flavoenzymes. Müller F, editor. Vol. 3. Boca Raton, FL: CRC; 1992. pp. 389–426. [Google Scholar]
- 2.Smillie R M. Biochem Biophys Res Commun. 1965;20:621–629. doi: 10.1016/0006-291x(65)90445-6. [DOI] [PubMed] [Google Scholar]
- 3.Wong K K, Murray B W, Lewisch S A, Baxter M K, Ridky T W, Ulissi-DeMario L, Kozarich J W. Biochemistry. 1993;32:14102–14110. doi: 10.1021/bi00214a005. [DOI] [PubMed] [Google Scholar]
- 4.Bianchi V, Eliasson R, Fontecave M, Mulliez E, Hoover D M, Matthews R G, Reichard P. Biochem Biophys Res Commun. 1993;197:792–797. doi: 10.1006/bbrc.1993.2548. [DOI] [PubMed] [Google Scholar]
- 5.Ifuku O, Koga N, Haze S, Kishimoto J, Wachi Y. Eur J Biochem. 1994;224:173–178. doi: 10.1111/j.1432-1033.1994.tb20009.x. [DOI] [PubMed] [Google Scholar]
- 6.Fujii K, Huennekens F M. J Biol Chem. 1974;249:6745–6753. [PubMed] [Google Scholar]
- 7.Foster M A, Dilworth M J, Woods D D. Nature (London) 1964;201:39–42. doi: 10.1038/201039a0. [DOI] [PubMed] [Google Scholar]
- 8.Leclerc D, Wilson A, Dumas R, Gafuik C, Song D, Watkins D, Heng H H, Rommens J M, Scherer S W, Rosenblatt D S, Gravel R A. Proc Natl Acad Sci USA. 1998;95:3059–3064. doi: 10.1073/pnas.95.6.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Porter T D, Kasper C B. Biochemistry. 1986;25:1682–1687. doi: 10.1021/bi00355a036. [DOI] [PubMed] [Google Scholar]
- 10.Wang M, Roberts D L, Paschke R, Shea T M, Masters B S, Kim J J. Proc Natl Acad Sci USA. 1997;94:8411–8416. doi: 10.1073/pnas.94.16.8411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoover D M, Ludwig M L. Protein Sci. 1997;6:2525–2537. doi: 10.1002/pro.5560061205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karplus P A, Daniels M J, Herriott J R. Science. 1991;251:60–66. [PubMed] [Google Scholar]
- 13.Ingelman M, Bianchi V, Eklund H. J Mol Biol. 1997;268:147–157. doi: 10.1006/jmbi.1997.0957. [DOI] [PubMed] [Google Scholar]
- 14.Vetter H, Jr, Knappe J. Hoppe-Seyler's Z Physiol Chem. 1971;352:433–446. doi: 10.1515/bchm2.1971.352.1.433. [DOI] [PubMed] [Google Scholar]
- 15.Jarrett J T, Goulding C W, Fluhr K, Huang S, Matthews R G. Methods Enzymol. 1997;281:196–213. doi: 10.1016/s0076-6879(97)81026-9. [DOI] [PubMed] [Google Scholar]
- 16.Drummond J T, Loo R R, Matthews R G. Biochemistry. 1993;32:9282–9289. doi: 10.1021/bi00087a004. [DOI] [PubMed] [Google Scholar]
- 17.Pace C N, Vajdos F, Fee L, Grimsley G, Gray T. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jenkins C M, Waterman M R. Biochemistry. 1998;37:6106–6113. doi: 10.1021/bi973076p. [DOI] [PubMed] [Google Scholar]
- 19.Delaglio F, Grzesiek S, Vuister G W, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 20.Bartels C, Xia T, Billeter M, Güntert P, Wüthrich K. J Biomol NMR. 1995;6:1–10. doi: 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
- 21.Garrett D S, Seok Y J, Peterkofsky A, Clore G M, Gronenborn A M. Biochemistry. 1997;36:4393–4398. doi: 10.1021/bi970221q. [DOI] [PubMed] [Google Scholar]
- 22.Jarrett J T, Huang S, Matthews R G. Biochemistry. 1998;37:5372–5382. doi: 10.1021/bi9730893. [DOI] [PubMed] [Google Scholar]
- 23.Takahashi H, Nakanishi T, Kami K, Arata Y, Shimada I. Nat Struct Biol. 2000;7:220–223. doi: 10.1038/73331. [DOI] [PubMed] [Google Scholar]
- 24.Drummond J T, Huang S, Blumenthal R M, Matthews R G. Biochemistry. 1993;32:9290–9295. doi: 10.1021/bi00087a005. [DOI] [PubMed] [Google Scholar]
- 25.Hall D A, Jordan-Starck T C, Loo R O, Ludwig M L, Matthews R G. Biochemistry. 2000;39:10711–10719. doi: 10.1021/bi001096c. [DOI] [PubMed] [Google Scholar]
- 26.Ponstingl H, Otting G. Eur J Biochem. 1997;244:384–399. doi: 10.1111/j.1432-1033.1997.00384.x. [DOI] [PubMed] [Google Scholar]
- 27.Dixon M M, Huang S, Matthews R G, Ludwig M. Structure (London) 1996;4:1263–1275. doi: 10.1016/s0969-2126(96)00135-9. [DOI] [PubMed] [Google Scholar]
- 28.Drennan C L, Dixon M M, Hoover D M, Jarrett J T, Goulding C W, Matthews R G, Ludwig M L. In: Vitamin B12 and B12-Proteins. Kräutler B, Arigoni D, Golding B T, editors. Weinheim, Germany: Wiley–VCH; 1998. pp. 133–155. [Google Scholar]
- 29.Wells J A. Proc Natl Acad Sci USA. 1996;93:1–6. doi: 10.1073/pnas.93.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bogan A A, Thorn K S. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 31.Drennan C L, Huang S, Drummond J T, Matthews R G, Ludwig M L. Science. 1994;266:1669–1674. doi: 10.1126/science.7992050. [DOI] [PubMed] [Google Scholar]
- 32.Hoover D M, Jarrett J T, Sands R H, Dunham W R, Ludwig M L, Matthews R G. Biochemistry. 1997;36:127–138. doi: 10.1021/bi961693s. [DOI] [PubMed] [Google Scholar]
- 33.Vermilion J L, Ballou D P, Massey V, Coon M J. J Biol Chem. 1981;256:266–277. [PubMed] [Google Scholar]
- 34.Sevrioukova I F, Li H, Zhang H, Peterson J A, Poulos T L. Proc Natl Acad Sci USA. 1999;96:1863–1868. doi: 10.1073/pnas.96.5.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.