

Abstract
Ohdo syndrome–Maat-Kievit-Brunner (OSMKB) type is an X-linked recessive disorder, a subtype of blepharophimosis-intellectual disability syndromes caused by mutations in the mediator complex subunit 12 ( MED12 ) gene. Here we report a familial OSMKB type with two affected siblings and mutation in MED12 gene.
Keywords: Ohdo syndrome–Maat-Kievit-Brunner type, blepharophimosis, intellectual disability, congenital heart disease, high-place winged scapula
Introduction
Blepharophimosis is a birth defect, defined as fixed reduction in the vertical distance between upper and lower eyelids with short palpebral fissures as a subjective finding. 1 Blepharophimosis associated with intellectual disability (ID) and/or malformation syndrome are rare group of disorders with variable clinical presentation. Overlapping features and subtle clinical differences among subtypes of blepharophimosis-ID syndromes make final clinical impression difficult and confusing. Ohdo syndrome (MIM 249620) is one of the syndromes associated with blepharophimosis-ID and was first described in three children of two related families affected by blepharophimosis, ID, congenital heart disease (CHD), and hypoplastic teeth. 2 Clinically related blepharophimosis-ID syndromes (related to Ohdo / Ohdo-like syndrome) were classified into five subgroups. 3 One of the five subtypes is Ohdo syndrome–Maat-Kevit-Brunner (OSMKB) type. The OSMKB subtype is known to be caused by gene MED12 (mediator complex subunit 12) mutations localized to chromosome Xq13, in X-linked recessive inheritance pattern. 4
MED12 gene mutations are known to cause three different distinct syndromes and nonspecific syndromic ID. 4 5 6 The three main distinct syndromes include FG syndrome, Lujan syndrome, and Ohdo syndrome, OSMKB type. In addition, two male siblings are described with ID, feeding difficulties, chronic constipation, severe micrognathia, abnormal semicircular canals, block vertebra, horizontal gaze paresis, and many other overlapping features of three distinct syndromes—thus expanding the spectrum of atypical phenotype—severe micrognathia/ID and MED12 mutations. 7
Here we report a family of two affected male siblings with blepharophimosis-ID (as well as probably affected maternal uncle). The clinical phenotype in this family resembled Ohdo/Ohdo-like syndrome, and the whole exome results revealed known MED12 missense mutation as causative gene, further subclassifying clinical phenotype as OSMKB type of blepharophimosis-ID syndrome. We discuss and compare clinical variability among the two affected male siblings and other reported patients with MED12 mutations/OSMKB phenotype.
Clinical Reports
Patient 1
A 3-year-old male child presented with global developmental delay, blepharophimosis, and CHD. He is a first born child to nonconsanguineous parents. On follow-up, his intelligence quotient at the age of 8 years was 47. There is no clinical suspicion of hearing impairment.
On examination (at the age of 8 years) his facial features were remarkable for triangular face, high forehead, thick arched eyebrows with broad medial end, blepharophimosis, mild left side ptosis, telecanthus, wide nasal ridge (mild), small alae nasi, short smooth philtrum, low-set ears, small mouth, and micrognathia ( Fig. 1A ).
Fig. 1.
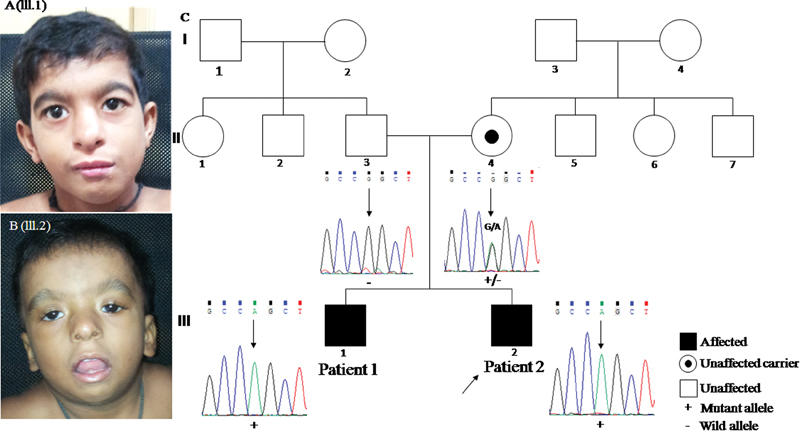
( A ) (III.1) Triangular face, high forehead, thick eyebrows, arched and medial end broad; blepharophimosis, mild left side ptosis, telecanthus, wide nasal ridge (mild), small ala nasi, short smooth philtrum, low-set ears, small mouth, and micrognathia. ( B ) (III.2) Round face, high forehead, thick eyebrows, arched, wide nasal bridge, bilateral ptosis (left > right), blepharophimosis, downslanting eyes, telecanthus, hypertelorism, small nose, anteverted nostrils, somewhat thick ala nasi, long smooth philtrum, open mouth, micrognathia, and low-set ears. ( C ) Pedigree showing inheritance pattern and individual partial Sanger sequences show hemizygous variant, c.887G > A in exon 7 of MED12 in affected individuals (patients 1 and 2). Mother is heterozygous carrier for the variant whereas father has wild-type allele.
In addition, microdontia, bilateral inguinal hernias surgical scars, microphallus, hypospadias with congenital chordee, and right-sided spermatic cord hydrocele were noted. He had friendly disposition.
Anthropometric measurements at the age of 8 years: height 114 cm (at 5th percentile) and head circumference 47.2 cm (between −2 and −3 standard deviation [SD]). Echocardiography showed perimembranous ventricular septal defect (VSD) with posterior upper muscular extension and right coronary cusp (RCC) buckling at the age of 3 years. Follow-up echocardiography study at the age of 8 years was essentially normal as perimembranous VSD was completely covered by RCC (venturi effect) without aortic regurgitation.
Patient 2
A 2-year 5-month-old younger male sibling of patient 1 presented with global developmental delay and bilateral hearing impairment.
On examination facial features were remarkable for round facies, high forehead, thick arched eyebrows, wide nasal bridge, bilateral ptosis (left > right), blepharophimosis, downslanting eyes, telecanthus, hypertelorism, small nose, anteverted nostrils, somewhat thick alae nasi, long smooth philtrum, open mouth, micrognathia, and low-set ears ( Fig. 1B ). In addition, three-dimensional (3D) surface–rendered magnetic resonance images showed relatively underdeveloped tarsal plate of left lower lid ( Supplementary Fig. 1 , available in the online version).
Other features noted included short neck, faint palmar creases, right-sided high-placed winging scapula ( Supplementary Fig. 2 , available in the online version), and single café-au-lait spot on back measuring 2 × 3 cm. Cardiac echocardiography did not show any CHD. Genital examination was unremarkable. Anthropometric measurements at the age of 2 years 5 months: Length 82 cm (between −2 and −3 SD) and head circumference 43.5 cm (< −3 SD).
Maternal uncle is known to have ID. Review of his photographs suggested that he might have had blepharophimosis. However, we were unable to examine him in person.
Investigations and Results
Karyotype at resolution of 500 to 550 bands is normal (46,XY) for both siblings. Thyroid function test were normal for both patients (thyroid-stimulating hormone [TSH] 1.91 uIU/mL [patient 1] and 3.4 uIU/mL [patient 2]) .Genomic DNA was extracted from ethylenediamine tetraacetic acid (EDTA) blood samples of the siblings and the parents using standard phenol-chloroform method. Whole exome sequencing (WES) was performed for patient 1 as described previously. 8 Briefly, DNA capture was achieved with Illumina's Nextera Rapid Capture Exome Kit. NextSEq 500 Sequencer (Illumina, Inc., San Diego, California, United States) along with the NextSEq 500 High Output Kit was used for massive parallel sequencing. bcl2fastq software 2.17.1.14 (Illumina, Inc.) was used to convert raw sequencing reads to standard FASTQ format. Reads were mapped to human reference genome (GRCh37/ hg19) by BWA software. Variant calling was done by GATK HaplotypeCaller, FreeBayes, and SAMtools. Annotation of variants was done by using ANNOVAR. Variant prioritization was performed by a series of filtering steps ( Supplementary Table 1 , available in the online version). These include the exclusion of polymorphisms and variants commonly seen in healthy individuals at a frequency > 0.02. Variants outside the exons, splice site regions, and synonymous variants were excluded. Further, based on the hypothesis of autosomal recessive or an X-linked recessive inheritance pattern, homozygous/hemizygous variants were prioritized. Variants were also filtered against an in-house WES database of approximately 200 families of Indian origin. The above filtering strategy narrowed down the total variants to 32 variants. Of these variants, a known pathogenic variation relevant to the clinical phenotype was observed in MED12 . The other variants were not found to be significant for the given phenotype. The predicted causal variant was then validated by Sanger sequencing in both siblings and unaffected parents to confirm segregation.
Results of Exome Sequencing
Exome sequencing revealed a known hemizygous missense variant, c.887G > A [p.(Arg296Gln)] in the exon seven of MED12 (NM_005120.2) confirmed by Sanger sequencing ( Supplementary Fig. 3 , Fig. 1C , available in the online version). The variant, c.887G > A replaces the amino acid arginine by glutamine at 296th position, causing alteration in the structure of protein. Targeted sequencing for the parents for the aforementioned variant revealed the mother to be a carrier of the variant while the variant was not present in his father ( Fig. 1C ). The same variant was also present in hemizygous state in his affected sibling ( Fig. 1C ).The maternal uncle could not be tested for the mutation identified. The phenotypic features observed in the proband and his sibling is in concordance with clinical features reported for X-linked OSMKB type. The variant c.887G > A [p.(Arg296Gln)] in exon 7 of MED12 gene is previously reported by Caro-Llopis et al (2016) as pathogenic de novo mutation in a 7-year-old affected male child with blepharophimosis-ID syndrome. 9
Discussion
Mediators are complex subunits that play an important role at preinitiation complex, resulting in either transcriptional repression or activation of a gene (through epigenetic mechanism–histone modification), especially during developmental stages. The mediator complex subunit 12 ( MED12 ) is one of the mediators that act as gene repressor through REST-directed G9A-dependent neuronal gene expression (REST—RE1 silencing transcription factors), both in neuronal and non-neuronal cells. 10 Pathogenic mutations in MED12 leads to blunted recruitment of the mediators, resulting in neuronal gene de-repression of certain repressor element 1 silencing transcription (REST) target genes and is suggested as a cause for MED12 -related X-linked ID syndromes. 4 However, the mechanisms underlying different phenotypes and MED12 mutations are not clear.
MED12 hemizygous gene mutations and OSMKB phenotype are well described in 6 families with 10 affected individuals. 4 9 11 12 In four families the mutations reported are familial and in the other two families it is de novo in origin. In the 10 patients with MED12 mutations/OSMKB type, there are similarities/dissimilarities in the clinical findings ( Table 1 ). 4 9 11 12 OSMKB type phenotypes include blepharophimosis, ID (mild-severe), facial phenotype, microdontia, skeletal manifestations, genital abnormalities, hearing loss, and behavioral issues. 3 4 Facial phenotypes include triangular face (obvious at an older age), high anterior hairline/high forehead, sparse eyebrows, blepharophimosis, ptosis (secondary), hypertelorism, telecanthus, epicanthic folds, wide nasal bridge, bulbous nose, long smooth philtrum, thin vermillion, small mouth, low-set ears, and micrognathia (reported in most patients with MED12 mutations) ( Table 1 ). 12 Range of skeletal manifestations and genital abnormalities in 10 affected patients with MED12 mutations/OSMKB phenotype is listed in Table 1 . 4 9 11 12
Table 1. Comparison of clinical features of X-linked Ohdo syndrome—OSMKB subtype with MED12 mutations .
| Physical features | Present patient family | Vulto-van Silfout et al, 2013 | Isidor et al, 2014 | Langley et al, 2015 | Caro-Llopis et al, 2016 |
Total (%) |
|---|---|---|---|---|---|---|
| Face | ||||||
| High forehead | 2/2 | 2/5 | 2/2 | 0/2 | 0/1 | 6/12 (50) |
| Thick and arched eyebrows | 2/2 | 1/1 | 3/12 (25) | |||
| Sparse eyebrows | 5/5 | 2/2 | 2/2 | 9/12 (75) | ||
| Blepharophimosis | 2/2 | 5/5 | 2/2 | 2/2 | 1/1 | 12/12 (100) |
| Telecanthus | 2/2 | 3/5 | 2/2 | 0/2 | 0/1 | 7/12 (58) |
| Hypertelorism | 1/2 | 3/5 | 2/2 | 0/2 | 1/1 | 7/12 (58) |
| Epicanthic folds | 0/2 | 5/5 | 0/2 | 0/2 | 1/1 | 6/12 (50) |
| Downslanting palpebral fissure | 1/2 | 0/5 | 0/2 | 2/2 | 0/1 | 3/12 (25) |
| Long philtrum a | 1/2 | 4/5 | 0/2 | 0/2 | 1/1 | 6/12 (50) |
| Short philtrum | 1/2 | 1/5 | 0/2 | 0/2 | – | 2/12 (17) |
| Ptosis | 2/2 | 5/5 | NA | 2/2 | 0/1 | 9/10 (90) |
| Strabismus | 0/2 | 3/5 | NA | 2/2 | 1/2 | 6/10 (60) |
| Hypermetropia | 0/2 | 2/5 | NA | 0/2 | 1/2 | 3/10 (10) |
| Micro/retrognathia | 2/2 | 5/5 | 0/2 | 0/2 | 1/1 | 8/12 (67) |
| Bulbous nose | 0/2 | 5/5 | 0/2 | 0/2 | 1/1 | 6/12 (50) |
| Wide nasal bridge | 2/2 | 5/5 | 0/2 | 0/2 | 0/1 | 7/12 (58) |
| Anteverted nostrils a | 1/2 | 3/5 | 1/2 | 0/2 | 0/1 | 4/12 (33) |
| Small mouth | 2/2 | 5/5 | 1/2 | 2/2 | 1/1 | 11/12 (92) |
| Thin vermillion of lips a | 0/2 | 3/5 | 1/2 | 2/2 | 0/1 | 6/12 (50) |
| Low-set ears | 2/2 | 2/5 | 2/2 | 2/2 | 1/1 | 12/12 (100) |
| Triangular face Round face |
1/2 1/2 |
5/5 | 1/2 | 2/2 | 0/1 | 9/12 (75) |
| Genitalia | ||||||
| Chordee | 1/2 | 0/5 | 0/2 | 2/2 | 0/1 | 3/12 (25) |
| Microphallus | 1/2 | 2/5 | 0/2 | 1/2 | 0/1 | 4/12 (33) |
| Undescended testis | 0/2 | 3/5 | 1/1 | 0/2 | 1/1 | 5/11 (45) |
| Musculoskeletal system | ||||||
| Overriding toes | 0/5 | 1/5 | 1/2 | 0/2 | 1/1 | 2/12 (17) |
| Clinodactyly | 0/5 | 2/5 | 1/2 | 0/2 | 0/1 | 3/12 (25) |
| Metaphyseal dysplasia | 0/2 | 1/5 | 2/2 | 0/2 | 0/1 | 3/12 (25) |
| Central nervous system | ||||||
| Intellectual disability | 2/2 | 5/5 | NA | 2/2 | 1/1 | 10/10 (100) |
| Microcephaly | 2/2 | 1/5 | 0/2 | 2/2 | 0/1 | 5/12 (42) |
| Hypotonia | 0/2 | 3/5 | 1/1 | 0/2 | 0/1 | 4/11 (36) |
| Hypertonia | 0/2 | 0/5 | 0/1 | 2/2 | 1/1 | 3/11 (27) |
| Behavior issues | 1/2 | 5/5 | NA | 1/2 | 0/1 | 7/10 (70) |
| Other clinical manifestations | ||||||
| Short stature | 1/2 | 3/5 | 0/2 | 1/2 | 0/1 | 5/12 (42) |
| Congenital heart defect | 1/2 | 0/5 | 1/2 | 0/2 | 0/1 | 2/12 (17) |
| Inguinal hernia | 1/2 | 1/5 | 0/1 | 0/2 | 0/1 | 3/11 (27) |
| Hearing loss | 1/2 | 3/5 | NA | 0/2 | 0/1 | 4/10 (40) |
| Microdontia a | 1/2 | 3/5 | NA | 0/2 | 0/1 | 4/10 (40) |
| Narrow auditory canals | 0/2 | 4/5 | 0/2 | 0/2 | 0/1 | 4/12 (33) |
| Constipation | 0/2 | 2/5 | NA | 0/2 | 1/1 | 3/10 (30) |
| Other less frequent clinical findings b | Unilateral high placed winging scapula & underdeveloped tarsal plate of lower eyelid, hypospadias | Nystagmus, microcornea, thick ala nasi, shawl scrotum, hiatus hernia, seizures, other skeletal finidngs c | Hydrops fetalis, Hirschsprung's disease, short humeri, equinovarus feet, brain imaging and renal abnormalities | Hypotelorism, hypertonia, severe GER, wide spaced teeth | Oligohydramnios, Intrauterine growth retardation, cleft palate, hirsutism, astigmatism | |
Abbreviations: GER, gastroesophageal reflux; NA, not applicable (too young for the conclusive evidence of clinical findings); OSMKB, Ohdo syndrome–Maat-Kievit-Brunner.
Neuroimaging findings: Thin and short corpus callosum, bilateral choanal stenosis. Renal abnormalities: Hypoplastic, pyelocaliceal dilation, renal cysts.
Abnormalities not clearly defined as absent or present, some features are marked as present by the author if it is obvious from photographs.
Clinical features described in one patient family.
Narrow thorax, hip dysplasia, scoliosis, high narrow palate.
Some of the infrequent findings reported include short stature, microdontia, wide-spaced teeth, microcephaly, hypotonia, hypertonia, CHD, inguinal hernia, hearing loss, narrow auditory canal, and chronic constipation. There are numerous clinical findings of patients with MED12 mutations/OSMKB phenotype reported only in one patient family ( Table 1 ). Isidor et al (2014) suggested that hydrops fetalis, Hirschsprung's disease, and short humeri with familial MED12 mutation might represent severe forms of OSMKB type. 11
Patients 1 and 2 in our family have facial phenotype similar to OSMKB type described. 4 9 11 Unlike other clinical reports, patients 1 and 2 have thick arched eyebrows except for the patient reported by Caro-Llopis et al. 9 In addition, patient 2 has unilateral high-placed winged scapula and left lower eyelid underdeveloped tarsal plate—both not reported earlier in any of the clinical reports. Hearing impairment and short stature are only seen in patient 2 and are reported earlier in patients with OSMKB/ MED12 mutation. 12 13 Patient 1 has genital abnormalities, normal stature, and CHD reported earlier in patients with OSMKB/ MED12 mutation. 4 9 11 12 These differences in clinical observations of patients 1 and 2 are suggestive of intrafamilial variability.
Caro-Llopis et al reported a de novo MED12 mutation in a child with blepharophimosis-ID. Although Caro-Llopis et al do not clearly discuss the patient as OSMKB phenotype, the patient has features suggestive of OSMKB phenotype. The mutation in this patient reported is de novo c.887G > A and it is the same mutation identified in our patient family. 9 Some of the clinical features do not match between our patients and the patient reported by Caro-Llopis et al suggestive of interfamilial variability. Clinical findings in common with the patient reported by Caro-Llopis et al include microcephaly, facial features such as thick arched eyebrows, hypertelorism, blepharophimosis, micrognathia, long smooth philtrum, and ID. Clinical findings not seen in our patients but seen in the patient reported by Caro-Llopis et al are severity of small mandible (requiring tracheostomy), bulbous nose, cleft palate, undescended testis, strabismus, chronic constipation, and hypertrichosis. 9 These patients together illustrate identical MED12 missense mutation, resulting in variable clinical phenotype (interfamilial and intrafamilial variability). The consistent recognizable clinical findings suggestive of OSMKB type are male child with blepharophimosis, ID, and certain dysmorphic facial features (triangular face, high forehead, hypertelorism/telecanthus, ptosis, epicanthic folds, wide nasal bridge, bulbous nose, long smooth philtrum, thin vermillion, small mouth, low-set ears, and micrognathia). Genital abnormalities and the other systemic clinical manifestations may be important additional clues for the clinical diagnosis of OSMKB phenotype. High-placed winging scapula, underdeveloped tarsal plate of lower eyelid, and hypospadias are the new clinical findings not reported earlier. Further studies are needed to understand the cause of clinical variability among patients with MED12 mutations/OSMKB phenotype and other related variable phenotypes.
Acknowledgement
The authors thank the parents and the patients' family for their participation in this study.
Conflict of Interest None.
Funding
This work was supported and funded by the project of National Institutes of Health titled “Genetic Diagnosis of Heritable Neurodevelopmental Disorders in India: Investigating the Use of Whole Exome Sequencing and Genetic Counseling to Address the High Burden of Neurodevelopmental Disorders” (1R21NS094047–01).
Supplementary Material
Supplementary Figures
Supplementary Figures
References
- 1.Hall B D, Graham J M, Jr, Cassidy S B, Opitz J M. Elements of morphology: standard terminology for the periorbital region. Am J Med Genet A. 2009;149A(01):29–39. doi: 10.1002/ajmg.a.32597. [DOI] [PubMed] [Google Scholar]
- 2.Ohdo S, Madokoro H, Sonoda T, Hayakawa K. Mental retardation associated with congenital heart disease, blepharophimosis, blepharoptosis, and hypoplastic teeth. J Med Genet. 1986;23(03):242–244. doi: 10.1136/jmg.23.3.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verloes A, Bremond-Gignac D, Isidor B et al. Blepharophimosis-mental retardation (BMR) syndromes: a proposed clinical classification of the so-called Ohdo syndrome, and delineation of two new BMR syndromes, one X-linked and one autosomal recessive. Am J Med Genet A. 2006;140(12):1285–1296. doi: 10.1002/ajmg.a.31270. [DOI] [PubMed] [Google Scholar]
- 4.Vulto-van Silfhout A T, de Vries B B, van Bon B W et al. Mutations in MED12 cause X-linked Ohdo syndrome. Am J Hum Genet. 2013;92(03):401–406. doi: 10.1016/j.ajhg.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graham J M, Jr, Schwartz C E. MED12 related disorders. Am J Med Genet A. 2013;161A(11):2734–2740. doi: 10.1002/ajmg.a.36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lesca G, Moizard M P, Bussy G et al. Clinical and neurocognitive characterization of a family with a novel MED12 gene frameshift mutation. Am J Med Genet A. 2013;161A(12):3063–3071. doi: 10.1002/ajmg.a.36162. [DOI] [PubMed] [Google Scholar]
- 7.Prescott T E, Kulseth M A, Heimdal K R et al. Two male sibs with severe micrognathia and a missense variant in MED12. Eur J Med Genet. 2016;59(08):367–372. doi: 10.1016/j.ejmg.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 8.Girisha K M, Shukla A, Trujillano D et al. A homozygous nonsense variant in IFT52 is associated with a human skeletal ciliopathy. Clin Genet. 2016;90(06):536–539. doi: 10.1111/cge.12762. [DOI] [PubMed] [Google Scholar]
- 9.Caro-Llopis A, Rosello M, Orellana C et al. De novo mutations in genes of mediator complex causing syndromic intellectual disability: mediatorpathy or transcriptomopathy? Pediatr Res. 2016;80(06):809–815. doi: 10.1038/pr.2016.162. [DOI] [PubMed] [Google Scholar]
- 10.Ding N, Zhou H, Esteve P O et al. Mediator links epigenetic silencing of neuronal gene expression with X-linked mental retardation. Mol Cell. 2008;31(03):347–359. doi: 10.1016/j.molcel.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Isidor B, Lefebvre T, Le Vaillant C et al. Blepharophimosis, short humeri, developmental delay and Hirschsprung disease: expanding the phenotypic spectrum of MED12 mutations. Am J Med Genet A. 2014;164A(07):1821–1825. doi: 10.1002/ajmg.a.36539. [DOI] [PubMed] [Google Scholar]
- 12.Langley K G, Brown J, Gerber R J et al. Beyond Ohdo syndrome: a familial missense mutation broadens the MED12 spectrum. Am J Med Genet A. 2015;167A(12):3180–3185. doi: 10.1002/ajmg.a.37354. [DOI] [PubMed] [Google Scholar]
- 13.Maat-Kievit A, Brunner H G, Maaswinkel-Mooij P. Two additional cases of the Ohdo blepharophimosis syndrome. Am J Med Genet. 1993;47(06):901–906. doi: 10.1002/ajmg.1320470618. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
Supplementary Figures