

Abstract
Wolf-Hirschhorn syndrome (WHS) is a multiple congenital anomaly-intellectual disability syndrome caused by a deletion involving chromosome 4p16.3. We report clinical and genetic findings of the first WHS patient diagnosed in central Africa. This boy who presented with cleft palate, microcephaly, severe growth delay, and intellectual disability was 12 years old. Typical craniofacial features were present, though the characteristic “Greek helmet” appearance of the nose was less evident, probably reflecting a variable expression related to the genetic background. The clinical diagnosis of WHS was confirmed by array CGH, which revealed a terminal 4p16.3 deletion of 3.47 Mb, typically associated with a milder phenotype, contributing to the long survival of this child in a developing country.
Keywords: Wolf-Hirschhorn syndrome, cleft palate, severe growth delay, intellectual disability, sacral dimples
Introduction
Wolf-Hirschhorn syndrome (WHS) is a contiguous gene deletion syndrome due to a deletion of distal region of chromosome 4p, characterized by distinct craniofacial features, growth impairment, intellectual disability, and seizures. 1 Its incidence in newborns is estimated between 1/50,000 and 1/100,000. 2 3 Genotype-phenotype correlation studies have not been able to unambiguously correlate single genes to different phenotypic features, but it is clear that haploinsufficiency of two genes are major contributing factors; the Wolf-Hirschhorn syndrome candidate 1 ( WHSC1) is associated with the craniofacial features and growth delay, whereas Homo Sapiens leucine zipper-EF-hand containing transmembrane protein 1 ( LETM1) contributes to seizures. 4 5 6 However, the observation of atypical deletions 7 8 and careful genotype-phenotype correlations 9 suggest that other genes may contribute to the phenotype as well. Many cases have been published, but only few among these are Africans. As far as we know, there has not been a single case described from central Africa. This case report aims to describe the clinical and genetic findings in the first case of WHS in a Congolese individual, clinically diagnosed and confirmed by molecular karyotyping.
Case Report
The index is a 12-year-old Congolese boy, who was initially referred to the surgery department of the University Hospital of the University of Lubumbashi because of cleft palate. He has one elder sister and four younger brothers, who are alive and do not present malformations or developmental delay.
He is the second child of healthy, young, and unrelated parents. At the time of birth, his mother was 20 years old and his father 23 years old. During the first trimester of gestation, his mother had an episode of malaria, treated with quinine, and urinary infections treated with amoxicillin. She consumed clay (Pemba) during the entire pregnancy and did not receive any vaccinations or folic acid supplementation. The first consultation was at the sixth month of pregnancy. The boy was referred at age 11 years for a cleft soft palate, which was further surgically corrected. Severe growth delay was evident, with weight 18.5 kg (standard deviation [SD]: −5, CDC [Centers for Disease Control and Prevention] growth charts), height 118 cm (SD: −3.8). Body mass index was 13.2 (SD: −2.9). His development was moderately delayed. He walked by the age of 4 years; he did not speak but could dress by himself. In addition, he presented with microcephaly (head circumference 48 cm, SD: −3.8), high forehead and prominent glabella, hypertelorism with prominent eyes, and divergent strabismus. The ears were normally placed with a preauricular pit on the right ear. His neck was long. There were sacral dimples ( Fig. 1 ).
Fig. 1.
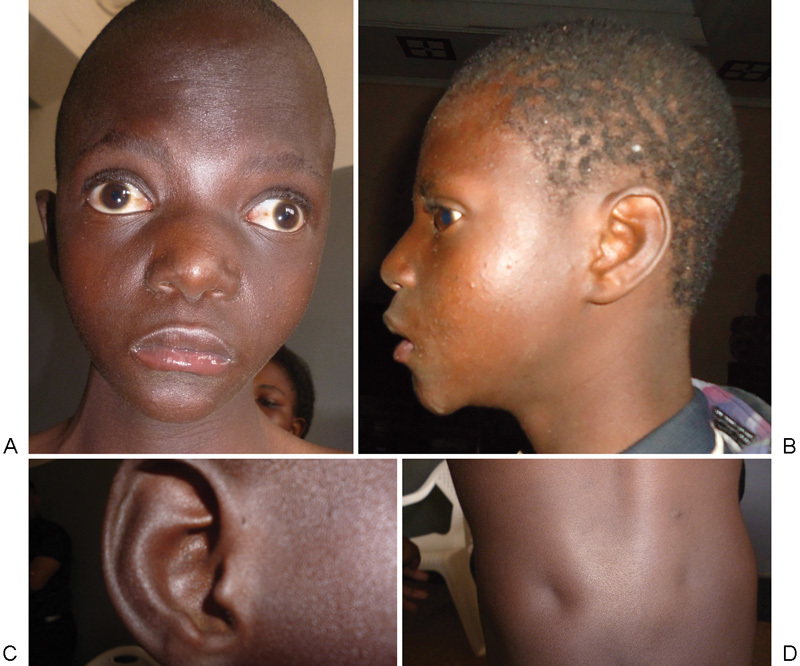
Abnormalities observed in the patient: ( A ) frontal view—high forehead, hypertelorism, divergent strabismus, arched eyebrows. (B) ) Profile view—absence of typical “Greek helmet” feature. (C) Preauricular pit. (D) Sacral dimples.
Genomic DNA was isolated from peripheral blood leukocytes using standard protocols, and screened for copy number alterations using the Oxford Gene Technology 8x60 k Array platform CytoSure ISCA v2 design (catalog number 020040, Oxford, United Kingdom). This slide includes nearly 60,000 oligonucleotides covering the whole genome, with special enrichment providing high coverage at ISCA-defined regions. The backbone resolution is one oligo every 70 kb whereas the resolution at targeted regions is one oligo every 48 kb. The WHS region is one of the targeted regions for this platform.
Array CGH results were interpreted using Oxford Gene technology CytoSure Interpret Software_ v.3.3.2 (OGT CytoSure, OGT Oxford, United Kingdom). A chromosomal copy number change is called when it includes at least five consecutive probes. All genome coordinates were according to NCBI human genome build 19 (hg19 Feb 2009). Analysis in our patient showed a terminal 3.47 Mb deletion arr[hg19] 4p16.3(59,233–3,527,184) × 1 ( Fig. 2 ). No additional copy number variations were detected.
Fig. 2.
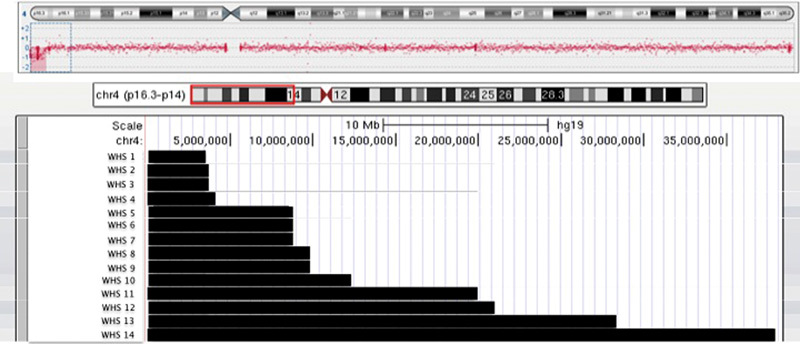
Summary of molecular-cytogenetic findings of 13 reported Wolf-Hirschhorn syndrome (WHS) patients with cleft palate. WHS1 Current index WHS2 Van Buggenhout et al, 2004; patient 5 22 WHS3 Maas et al, 2008; patient 11 23 WHS4 Van Buggenhout et al, 2004; patient 1 22 WHS5 Shimizu et al, 2014; patient 10 1 WHS6 Shimizu et al, 2014; patient 11 1 WHS7 Shimizu et al, 2014; patient 12 1 WHS8 Ji et al, 2010; patient 1 24 WHS9 Ji et al, 2010; patient 5 24 WHS10 Shimizu et al, 2014; patient 16 1 WHS11 Maas et al, 2008; patient 20 23 WHS12 Shimizu et al, 2014; patient 20 1 WHS13 Shimizu et al, 2014; patient 21 1 WHS14 Maas et al, 2008; patient 21 23
Discussion
We describe the clinical and genetic features of a young Central-African boy with WHS. Previously, minimal diagnostic criteria for WHS were proposed, including the characteristic craniofacial appearance with the “Greek warrior helmet,” intellectual disability, growth delay, and seizures or electroencephalographic (EEG) anomalies. 10 11 We diagnosed this patient clinically with WHS, who presented three of the four major criteria including most of the craniofacial features although the “Greek warrior helmet” appearance was less evident. The patient did not manifest seizures and we did not perform an EEG. The possibility of no seizures or EEG anomalies in WHS patients has been reported previously, 12 13 it is often correlated with interstitial deletions spaning the terminal 751 kb of chromosome 4p. 14 The telomeric segment of 0.6 to 0.9 Mb is hypothesized to function synergistically with the seizure candidate region to promote seizures in WHS patients. 15 Our patient also presented the characteristic sacral dimples, located at the cranial part of the sacroiliac joints, a feature we find helpful in reaching this diagnosis.
The severity of WHS correlates with the size of the deletion, and three different categories of the WHS phenotype have been proposed. 1 10 The deletion found in our patient belongs to the group of deletions smaller than 3.5 Mb. In this group, the symptoms are mild, and, in most cases, the patients do not manifest major malformations. We think that the clinical presentation of our patient is consistent with the finding of a deletion of this category, even though he has one major malformation, that is, a cleft palate. In contrast to the mild phenotype of our patient, more severe manifestations, including malformations, are observed in case of larger chromosome 4p deletions between 5 and 18 Mb. These cases are more frequent probably because clinically severe cases are more easily recognized and diagnosed. We expect that the introduction of molecular karyotyping, which permits genome-wide screening for imbalances, will reduce this ascertainment bias in the future.
As in many other cases with WHS, our patient had severe growth retardation with height and weight far below the 3rd percentile. WHSCR1 and WHSCR2 may contribute to the short stature and low weight. 16 17 Facial clefting is commonly observed in WHS. Some cases present cleft lip with or without cleft palate (CL +/− CP), but also cleft palate only (CPO) is observed. CL +/− CP and CPO have distinct etiopathogenesis. 18 Therefore, we explored the possibility that in WHS too distinct genes may be involved in causing these two different malformations ( Fig. 2 ). Occasionally, authors do not differentiate between these two malformations and lump them into one entity. 10 19 20 However, we identified a minimal region of more than 3.5 Mb shared by some WHS patients with CL +/− CP. It is reported that the percentage of cleft lip/palate in WHS patient is different according to the size of the deletion—8% in patients with deletions less than 3.5 Mb compared with 25% and 44% observed in patients with 5 to 18 Mb and greater than 22 to 25 Mb deletions, respectively. 10
Almost all the patients with cleft palate associated with WHS present a terminal deletion of more than 3.5 Mb, whereas our patient has one of the smallest deletions associated with cleft palate. This indicates that candidate genes for cleft palate are located in the distal 3.5 Mb of chromosome 4p. Clinical recognition of WHS strongly depends on the characteristic facial features. The characteristic facial phenotype appears to be less pronounced in patients with smaller deletions, suggesting that although WHSC1 may be essential for the craniofacial features, deletion of additional genes may contribute to the severity of the core phenotype. 17 21 22 23 In the literature, most reports concern Caucasian patients, even though, increasingly, patients from other ethnicities are being reported. 1 24 In the present case, the WHS facial features are recognizable. However, the typical “Greek helmet” appearance was less evident. This feature consists of a broadening of the nasal bridge but mostly the nose bridge continuing in a straight line onto the prominent glabella. In the present patient, the nasal bridge was wider, resulting in the characteristic hypertelorism but not clearly more prominent. The nasal bridge is more depressed in Central African individuals, indicating that ethnic background may influence the expression of certain genetic syndromes. Of interest, when we evaluated the facial dysmorphism applying the FDNA Face2Gene solution, WHS syndrome did not rank within the first 30 matches. 25 However, when adding the detailed clinical description including the dysmorphism as outlined in this report, WHS ranked first. This further illustrates that the ethnic background of the patient influences the phenotypic manifestations of certain syndromes. 25
In Central Africa, an estimated one in seven children dies before the age of 5 years. 26 27 For children with a congenital malformation, life expectancy is even lower. 28 The present child with WHS is still alive at the age of 12 years. He belongs to the milder spectrum of the disorder that probably contributed to his survival. The present report illustrates that knowledge of genetic syndromes, which mostly originates from industrialized countries, is also applicable to Central Africa. However, there might be some local variation in expression. This underlines the urgent need for training in medical genetics and syndromology in Central Africa because currently little awareness and knowledge exists in this field. 29
Acknowledgments
The authors thank the family of the patient for their kind cooperation. The authors thank the staff members of the Center for Human Genetics, KU Leuven, for support. S.M. was supported by a scholarship from interfaculty Council for Development Co-operation (IRO), KU Leuven, and GROS, Holsbeek (Belgium).
Footnotes
Author Contributions Sébastien Mbuyi-Musanzayi: clinical examination, treatment of the patient, and redaction of article. Aimé Lumaka: array testing and manuscript correction. Erick Kasamba Ilunga: first-step DNA extraction. Toni Lubala Kasole: clinical examination. Bienvenu Yogolelo Asani: ophthalmologic examination. Prosper Lukusa Tshilobo: manuscript correction. Prosper Kalenga Muenze: manuscript correction. François Tshilombo Katombe: clinical examination. Hervé Reychler: manuscript corrections. Koenraad Devriendt: clinical examination, diagnosis, and manuscript corrections. All coauthors have read, contributed, and approved the manuscript. Conflict of Interest None.
References
- 1.Shimizu K, Wakui K, Kosho T et al. Microarray and FISH-based genotype-phenotype analysis of 22 Japanese patients with Wolf-Hirschhorn syndrome. Am J Med Genet A. 2014;164A(03):597–609. doi: 10.1002/ajmg.a.36308. [DOI] [PubMed] [Google Scholar]
- 2.Lurie I W, Lazjuk G I, Ussova Y I, Presman E B, Gurevich D B. The Wolf-Hirschhorn syndrome. I. Genetics. Clin Genet. 1980;17(06):375–384. doi: 10.1111/j.1399-0004.1980.tb00167.x. [DOI] [PubMed] [Google Scholar]
- 3.Shannon N L, Maltby E L, Rigby A S, Quarrell O W. An epidemiological study of Wolf-Hirschhorn syndrome: life expectancy and cause of mortality. J Med Genet. 2001;38(10):674–679. doi: 10.1136/jmg.38.10.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zollino M, Neri G.Genotype-phenotype correlations in Wolf-Hirschhorn syndrome Eur J Hum Genet 2001902150. Doi: 10.1038/sj.ejhg.5200611 [DOI] [PubMed] [Google Scholar]
- 5.Zollino M, Lecce R, Fischetto R et al. Mapping the Wolf-Hirschhorn syndrome phenotype outside the currently accepted WHS critical region and defining a new critical region, WHSCR-2. Am J Hum Genet. 2003;72(03):590–597. doi: 10.1086/367925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodríguez L, Zollino M, Climent S et al. The new Wolf-Hirschhorn syndrome critical region (WHSCR-2): a description of a second case. Am J Med Genet A. 2005;136(02):175–178. doi: 10.1002/ajmg.a.30775. [DOI] [PubMed] [Google Scholar]
- 7.Hannes F, Hammond P, Quarrell O, Fryns J P, Devriendt K, Vermeesch J R. A microdeletion proximal of the critical deletion region is associated with mild Wolf-Hirschhorn syndrome. Am J Med Genet A. 2012;158A(05):996–1004. doi: 10.1002/ajmg.a.35299. [DOI] [PubMed] [Google Scholar]
- 8.Zollino M, Orteschi D, Ruiter M et al. Unusual 4p16.3 deletions suggest an additional chromosome region for the Wolf-Hirschhorn syndrome-associated seizures disorder. Epilepsia. 2014;55(06):849–857. doi: 10.1111/epi.12617. [DOI] [PubMed] [Google Scholar]
- 9.Andersen E F, Carey J C, Earl D L et al. Deletions involving genes WHSC1 and LETM1 may be necessary, but are not sufficient to cause Wolf-Hirschhorn syndrome. Eur J Hum Genet. 2014;22(04):464–470. doi: 10.1038/ejhg.2013.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zollino M, Murdolo M, Marangi G et al. On the nosology and pathogenesis of Wolf-Hirschhorn syndrome: genotype-phenotype correlation analysis of 80 patients and literature review. Am J Med Genet C Semin Med Genet. 2008;148C(04):257–269. doi: 10.1002/ajmg.c.30190. [DOI] [PubMed] [Google Scholar]
- 11.Battaglia A, Filippi T, Carey J C. Update on the clinical features and natural history of Wolf-Hirschhorn (4p-) syndrome: experience with 87 patients and recommendations for routine health supervision. Am J Med Genet C Semin Med Genet. 2008;148C(04):246–251. doi: 10.1002/ajmg.c.30187. [DOI] [PubMed] [Google Scholar]
- 12.Yang W X, Pan H, Li L et al. Analyses of genotypes and phenotypes of ten Chinese patients with Wolf-Hirschhorn syndrome by multiplex ligation-dependent probe amplification and array comparative genomic hybridization. Chin Med J (Engl) 2016;129(06):672–678. doi: 10.4103/0366-6999.177996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iype T, Alakbarzade V, Iype Met al. A large Indian family with rearrangement of chromosome 4p16 and 3p26.3 and divergent clinical presentations BMC Med Genet 201516104. Doi: 10.1186/s12881-015-0251-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ho K S, South S T, Lortz A et al. Chromosomal microarray testing identifies a 4p terminal region associated with seizures in Wolf-Hirschhorn syndrome. J Med Genet. 2016;53(04):256–263. doi: 10.1136/jmedgenet-2015-103626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bi W, Cheung S W, Breman A M, Bacino C A. 4p16.3 microdeletions and microduplications detected by chromosomal microarray analysis: new insights into mechanisms and critical regions. Am J Med Genet A. 2016;170(10):2540–2550. doi: 10.1002/ajmg.a.37796. [DOI] [PubMed] [Google Scholar]
- 16.Rauch A, Schellmoser S, Kraus C et al. First known microdeletion within the Wolf-Hirschhorn syndrome critical region refines genotype-phenotype correlation. Am J Med Genet. 2001;99(04):338–342. doi: 10.1002/ajmg.1203. [DOI] [PubMed] [Google Scholar]
- 17.Wright T J, Costa J L, Naranjo C, Francis-West P, Altherr M R. Comparative analysis of a novel gene from the Wolf-Hirschhorn/Pitt-Rogers-Danks syndrome critical region. Genomics. 1999;59(02):203–212. doi: 10.1006/geno.1999.5871. [DOI] [PubMed] [Google Scholar]
- 18.Jugessur A, Shi M, Gjessing H K et al. Fetal genetic risk of isolated cleft lip only versus isolated cleft lip and palate: a subphenotype analysis using two population-based studies of orofacial clefts in Scandinavia. Birth Defects Res A Clin Mol Teratol. 2011;91(02):85–92. doi: 10.1002/bdra.20747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Battaglia A, Carey J C. Wolf-Hirschhorn syndrome and the 4p-related syndromes. Am J Med Genet C Semin Med Genet. 2008;148C(04):241–243. doi: 10.1002/ajmg.c.30189. [DOI] [PubMed] [Google Scholar]
- 20.South S T, Whitby H, Battaglia A, Carey J C, Brothman A R. Comprehensive analysis of Wolf-Hirschhorn syndrome using array CGH indicates a high prevalence of translocations. Eur J Hum Genet. 2008;16(01):45–52. doi: 10.1038/sj.ejhg.5201915. [DOI] [PubMed] [Google Scholar]
- 21.Zollino M, Di Stefano C, Zampino G et al. Genotype-phenotype correlations and clinical diagnostic criteria in Wolf-Hirschhorn syndrome. Am J Med Genet. 2000;94(03):254–261. doi: 10.1002/1096-8628(20000918)94:3<254::aid-ajmg13>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 22.Van Buggenhout G, Melotte C, Dutta B et al. Mild Wolf-Hirschhorn syndrome: micro-array CGH analysis of atypical 4p16.3 deletions enables refinement of the genotype-phenotype map. J Med Genet. 2004;41(09):691–698. doi: 10.1136/jmg.2003.016865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maas N M, Van Buggenhout G, Hannes F et al. Genotype-phenotype correlation in 21 patients with Wolf-Hirschhorn syndrome using high resolution array comparative genome hybridisation (CGH) J Med Genet. 2008;45(02):71–80. doi: 10.1136/jmg.2007.052910. [DOI] [PubMed] [Google Scholar]
- 24.Ji T Y, Chia D, Wang J M et al. Diagnosis and fine localization of deletion region in Wolf-Hirschhorn syndrome patients. Chin Med J (Engl) 2010;123(13):1663–1667. [PubMed] [Google Scholar]
- 25.Lumaka A, Cosemans N, Lulebo Mampasi A et al. Facial dysmorphism is influenced by ethnic background of the patient and of the evaluator. Clin Genet. 2016 doi: 10.1111/cge.12948. [DOI] [PubMed] [Google Scholar]
- 26.Forae G D, Uchendu O J, Igbe A P; GD.An audit of paediatric mortality patterns in a Nigerian teaching hospital Niger Med J 20145502130–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fotso J C, Ezeh A C, Madise N J, Ciera J.Progress towards the child mortality millennium development goal in urban sub-Saharan Africa: the dynamics of population growth, immunization, and access to clean water BMC Public Health 20077218. Doi: 10.1186/1471-2458-7-218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Modell B, Berry R J, Boyle C Aet al. Global regional and national causes of child mortality Lancet 2012380(9853):1556, author reply 1556–1557 [DOI] [PubMed] [Google Scholar]
- 29.Lumaka A, Mubungu G, Nsibu C, Tady B P, Lukusa T, Devriendt K. X-linked adrenal hypoplasia congenita: a novel DAX1 missense mutation and challenges for clinical diagnosis in Africa. Eur J Pediatr. 2012;171(02):267–270. doi: 10.1007/s00431-011-1523-5. [DOI] [PubMed] [Google Scholar]