

Abstract
Sensorineural hearing loss (SNHL) is a common defect with a multifactorial etiology. Congenital cytomegalovirus infection (cCMV) is the most common infectious cause, and its early detection allows a prompt pharmacological treatment that can improve hearing prognosis. In a consistent percentage of profound SNHL, genetic causes and/or inner ear malformations are involved; their prompt diagnosis might change therapeutic options. This study reports a case of a 3- year-old female patient with symptomatic cCMV infection who also exhibits developmental delay, dysmorphic facial features, bilateral hearing loss, and cochlear incomplete partition, type 2, in 7q21.3 deletion. This deletion includes the genes DLX5 and DLX6 , which could be the candidate genes for the ear malformation named incomplete partition, type 2.
Keywords: cochlea, malformation, intellectual disability, syndrome, 7q21 deletion, DLX5, DLX6
Introduction
Sensorineural hearing loss (SNHL) is the most common sensory deficit in developed societies, with an estimated prevalence of approximately 1.5 children out of 1,000. 1 Updated results derived from newborn hearing screening programs suggest that this rate could be higher: approximately 2 to 4 children out of 1,000. 2
Etiological causes of SNHL remain largely unknown. Among the cases with an identified cause, half of them are supposed to have an environmental origin, including congenital or acquired infections, ototoxic drugs, and prematurity with hypoxic–ischemic complications, whereas the other half is ascribable to genetic causes and/or malformations. 3 4
Congenital cytomegalovirus infection (cCMV) infection is the leading cause of neurodevelopmental delay and SNHL. Overall, the cCMV estimated prevalence in newborns is 0.64%, and the prevalence of permanent sensorineural sequelae is 12.7% of the affected, but it increases to 40 to 58% among children with a symptomatic onset. The fraction of SNHL attributed to cCMV ranges between 17 and 23%. 5 6 Prompt treatment with ganciclovir or valganciclovir in newborns affected by cCMV can decrease the incidence and slow the progression of SNHL and neurodevelopmental delay, 7 although some authors suggest that the treatment could have modest benefit in the long term. 8 Prenatal and newborn testing for cCMV infection is increasing over time, and a screening program has been proposed by several authors. 9
Approximately 70% of genetic SNHL cases are considered nonsyndromic; the mode of inheritance is usually autosomal recessive. Autosomal dominant, X-linked, and mitochondrial transmission have also been reported. On the other hand, deafness is a feature of several monogenic syndromes 4 and is associated with some microdeletion syndromes 10 ; genetic SNHL can be associated with inner ear malformations.
Cranial computed tomography and/or magnetic resonance imaging (MRI) are considered the gold standard for the detection of inner ear malformations related to SNHL. Incomplete partition type 2 (IP2) is a cochlear, usually bilateral, malformation characterized by a cystic expansion of the medial and apical cochlear turns that are fused together and associated with the dilatation of the vestibule and of vestibular aqueduct. The modiolus and the interscalar septa are defective. It usually coincides with profound SNHL. 11
SLC26A4 , responsible for Pendred's syndrome, is the only gene associated with isolated inner ear malformations, including IP2; however, only a minority of the cases are associated with mutations in this gene, 12 13 and other loci are supposed to be involved.
SNHL has been described in patients with 7q21.3 deletion syndrome, 14 15 16 associated with or without split hand/foot malformations. 17 18 19
Case Report
A preterm baby was delivered by emergency cesarean section at 31 weeks of gestation. Apgar scores were 6 and 9 at 1 minute and 5 minutes, respectively; weight was 1,160 kg (5–10th centile), length was 39 cm (10–25th centile), and head circumference was 25 cm (<5th centile). Maternal preeclampsia, oligohydramnios, and intrauterine growth restriction were recorded on gestation charts. Mechanical ventilation was applied for 5 days (nasal continuous positive airway pressure, FiO 2 max 28%).
Physical examination revealed hypotonia, hepatosplenomegaly, and petechial rash. Laboratory tests displayed moderate anemia, thrombocytopenia, direct hyperbilirubinemia, and increased glutamic–pyruvic transaminase levels. Cranial ultrasound scanning revealed ventricular dilation and bilateral frontal lobe hypoplasia. Transiently evoked otoacoustic emissions showed bilateral deafness. Clinical suspicion of cCMV was confirmed by the laboratory on the third day of life, with positive CMV–DNA studies in urine and plasma and negative bacterial cultures; therefore, the baby received a prompt 6-week treatment with ganciclovir. Clinical and laboratory remission was obtained after 5 days of treatment.
Standard multidisciplinary diagnostic workload, including careful clinical, expert ophthalmological, audiological, and neuroradiological examinations, started at the age of 4 months, when the infant was admitted to the Regional Centre for Perinatal Infection of Federico II Medical School of Naples. Auditory brainstem response confirmed a bilateral, profound SNHL (threshold over 90 dB). No auditory skill improvements were detected after middle ear prostheses. At electroencephalogram, a mild dysfunction in the temporoparietal regions was reported. Ophthalmological examinations revealed microphthalmia and corneal leukoma. Cranial MRI displayed mild cerebellar hypoplasia and a malformation of the inner ears. It was characterized by symmetrical, mild dilatation of the basal turn of the cochlea with cystic enlargement of the medial and apical turns, which also appeared fused. Enlargement of the internal acoustic canals and agenesis of the osseous fundus and of the modiolus were detected. Both of the cochlear nerves were severely hypoplastic. A marked dilation of vestibules and vestibular aqueducts and a slight enlargement of part of the lateral and of the superior semicircular canals were noted ( Fig. 1 ). Neuroimaging was consistent with the diagnosis of bilateral incomplete IP2. Expert dysmorphological examination pointed out microcephaly, left microphthalmia, epicanthus, low-set ears, and pilonidal cyst. Familiar anamnesis excluded the occurrence of other cases of congenital malformations. An array comparative genomic hybridization (CGH) was requested, detecting a 17-Mb deletion in the region 7q21.11-q21.3 (80,001,166–97,260,436; hg18).
Fig. 1.
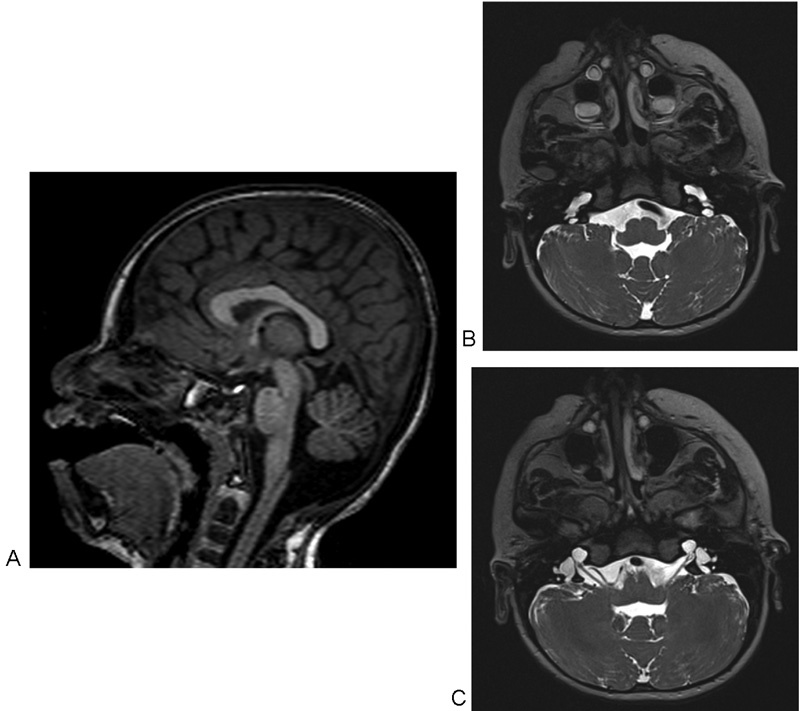
Brain magnetic resonance imaging at 4 months of life. ( A ) Cerebellar vermis hypoplasia. ( B ) Bilateral dilatation of the endolymphatic sac, and dilatation of the cochlear basal turn. ( C ) Internal acoustic canals enlargement and agenesis of their osseous fundus, agenesis of the modiolus, dilation of vestibules and vestibular aqueducts, and cystic enlargement of the medial and apical turns, which appear fused.
At the last clinical evaluation (age 3 years 9 months) the patient displayed a stature of 89.3 cm, weight of 9.4 kg, and head circumference of 42 cm, all <3rd centile. Verbal language was absent, nonverbal language was poor, disposition appeared aggressive, and gait was clumsy. Denver test score was 6 to 12 months. Several dysmorphic features were recognizable: microcephaly with brachycephaly; epicanthus; low-set, prominent, and dysmorphic ears with hypoplasia of the helix; wide nasal bridge; prominent columella; and micrognathia ( Fig. 2 ; informed consent has been signed).
Fig. 2.
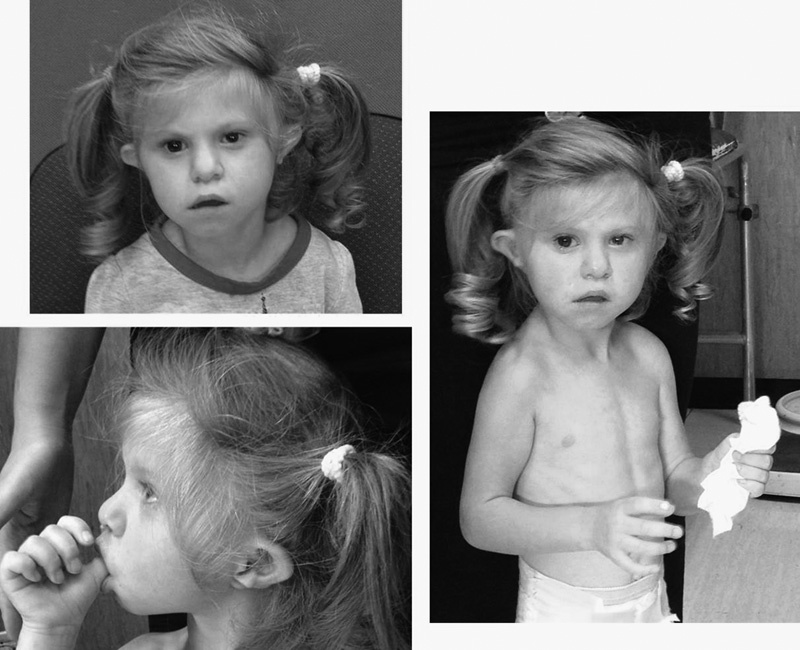
Patient at the age of 3 years and 9 months. Notice left microphthalmia; epicanthus; low-set, prominent, and dysmorphic ears with hypoplasia of the helix; wide nasal bridge; prominent columella; Cupid's bow upper lip; and microretrognatia.
A second brain MRI was performed at 3 years of life. Some white matter T2 hyperintensity, consistent with gliosis, was noted peripheral to the trigones, probably not appreciable at the first MRI because of the immature myelination but related to the infection as well. Other small hyperintense areas were also present at the subcortical white matter of the cerebral hemispheres, consistent with delayed myelination. Brainstem implant is still in progress.
Discussion
SNHL can occur due to several environmental causes, including infections, ototoxic drugs, prematurity, and hypoxic–ischemic damage. In developed countries, cCMV infection is the most frequent cause. At birth, infected infants might show severe or moderate signs of disease (anemia, thrombocytopenia, hyperbilirubinemia), as in our case, thus leading to the diagnosis, or might appear asymptomatic. Neurodevelopmental delay and SNHL can complicate the disease in the short or long term with different grades of severity, whereas exterior malformations do not usually occur. According to the high prevalence of the cCMV and level B evidence of the efficacy of short- and long-term antiviral tertiary prophylaxis (6 weeks and 6 months treatment with ganciclovir and valganciclovir, respectively), the inclusion of CMV viruria on the standard diagnostic panel for SNHL has been widely suggested. 20 In our case, clinical suspicion allowed an early diagnosis and thus pharmacological treatment in a hospital with a neonatal intensive care unit. Unfortunately, EOTA pointed out hearing loss (HL), whereas cranial ultrasonography did not detect internal ear anomalies. Dysmorphic features, including epicanthus and low-set ears, although not typical for a cCMV, were underestimated at birth because of their mildness and overlap with the common facial appearance of preterm infants. Only after admission at a University Hospital and following a careful dysmoprhological evaluation, the suspicion of an associated genetic defect arose, due to the presence of the cochlear malformation and dysmorphic features, and for these reasons genetic tests were performed. Early detection of a malformation can dramatically influence prognosis and practices. In this case, the marked hypoplasia of both the cochlear nerves discouraged a cochlear implantation.
7q21 deletion syndrome is a rare contiguous gene syndrome characterized by growth impairment, microcephaly, craniofacial abnormalities, HL, split hand/foot, and intellectual disability. 21 Split hand/foot and other limbs anomalies have a reduced penetrance (54–80% in previous reports). 19 22 While the diagnosis is accessible for patients with a full phenotype, it could be insidious in patients displaying only some of the clinical features, such as the present case. The phenotypic variability is probably a consequence of the variability of the breakpoints of the deletions and of the genes involved.
The strongest candidate genes for HL are DLX5 and DLX6 , which are involved in the deletion described here. These are target genes of the retinoic acid dependent activation of FGF3 and FGF10, and their expression is critical for the development of the inner ear. 23 24 The disruption of these genes has been showed to cause HL, craniofacial anomalies, and mental disability in mice. 25
In humans, there are several descriptions of patients with HL and 7q21 deletions, but in only two of the cases a cochlear malformation has been reported, probably because brain MRI has not been performed in most of the cases. To our knowledge, this case is the third description of a cochlear malformation associated with 7q21 deletion. 16 21 Thus, DLX5 and DLX6 could be considered, along with SLC26A4 , as loci to study in the presence of such type of ear malformations.
The identification of comorbid conditions in a patient with cCMV is crucial to determine critical decisions, such as the duration of a pharmacological treatment (which must be carried on until the further investigations are performed) and the eligibility of prosthetic treatment, and to avoid the adverse effects of a prolonged antiviral treatment. When the clinical signs are not consistent with a specific condition, array CGH is the most suitable analysis because HL is a sign shared by several microdeletion and microduplication syndromes.
Conflict of Interest None.
Note
Informed consent has been obtained from the patients.
References
- 1.Parving A, Hauch A M, Christensen B. Hearing loss in children--epidemiology, age at identification and causes through 30 years [in Danish] Ugeskr Laeger. 2003;165(06):574–579. [PubMed] [Google Scholar]
- 2.Wessex Universal Neonatal Hearing Screening Trial Group.Controlled trial of universal neonatal screening for early identification of permanent childhood hearing impairment Lancet 1998352(9145):1957–1964. [PubMed] [Google Scholar]
- 3.Smith R J, Bale J F, Jr, White K R.Sensorineural hearing loss in children Lancet 2005365(9462):879–890. [DOI] [PubMed] [Google Scholar]
- 4.Morzaria S, Westerberg B D, Kozak F K. Systematic review of the etiology of bilateral sensorineural hearing loss in children. Int J Pediatr Otorhinolaryngol. 2004;68(09):1193–1198. doi: 10.1016/j.ijporl.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 5.Dollard S C, Grosse S D, Ross D S. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev Med Virol. 2007;17(05):355–363. doi: 10.1002/rmv.544. [DOI] [PubMed] [Google Scholar]
- 6.Barbi M, Binda S, Caroppo S, Ambrosetti U, Corbetta C, Sergi P. A wider role for congenital cytomegalovirus infection in sensorineural hearing loss. Pediatr Infect Dis J. 2003;22(01):39–42. doi: 10.1097/00006454-200301000-00012. [DOI] [PubMed] [Google Scholar]
- 7.Kimberlin D W, Lin C Y, Sánchez P J et al. Effect of ganciclovir therapy on hearing in symptomatic congenital cytomegalovirus disease involving the central nervous system: a randomized, controlled trial. J Pediatr. 2003;143(01):16–25. doi: 10.1016/s0022-3476(03)00192-6. [DOI] [PubMed] [Google Scholar]
- 8.Kimberlin D W, Jester P M, Sánchez P J et al. Valganciclovir for symptomatic congenital cytomegalovirus disease. N Engl J Med. 2015;372(10):933–943. doi: 10.1056/NEJMoa1404599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cannon M J, Griffiths P D, Aston V, Rawlinson W D. Universal newborn screening for congenital CMV infection: what is the evidence of potential benefit? Rev Med Virol. 2014;24(05):291–307. doi: 10.1002/rmv.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zarchi O, Attias J, Raveh E, Basel-Vanagaite L, Saporta L, Gothelf D. A comparative study of hearing loss in two microdeletion syndromes: velocardiofacial (22q11.2 deletion) and Williams (7q11.23 deletion) syndromes. J Pediatr. 2011;158(02):301–306. doi: 10.1016/j.jpeds.2010.07.056. [DOI] [PubMed] [Google Scholar]
- 11.Zheng Y, Schachern P A, Cureoglu S, Mutlu C, Dijalilian H, Paparella M M. The shortened cochlea: its classification and histopathologic features. Int J Pediatr Otorhinolaryngol. 2002;63(01):29–39. doi: 10.1016/s0165-5876(01)00642-5. [DOI] [PubMed] [Google Scholar]
- 12.Huang S, Han D, Yuan Yet al. Extremely discrepant mutation spectrum of SLC26A4 between Chinese patients with isolated Mondini deformity and enlarged vestibular aqueduct J Transl Med 20119167. Doi: 10.1186/1479-5876-9-167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee H J, Jung J, Shin J W et al. Correlation between genotype and phenotype in patients with bi-allelic SLC26A4 mutations. Clin Genet. 2014;86(03):270–275. doi: 10.1111/cge.12273. [DOI] [PubMed] [Google Scholar]
- 14.Saitsu H, Kurosawa K, Kawara H et al. Characterization of the complex 7q21.3 rearrangement in a patient with bilateral split-foot malformation and hearing loss. Am J Med Genet A. 2009;149A(06):1224–1230. doi: 10.1002/ajmg.a.32877. [DOI] [PubMed] [Google Scholar]
- 15.Bernardini L, Palka C, Ceccarini C et al. Complex rearrangement of chromosomes 7q21.13-q22.1 confirms the ectrodactyly-deafness locus and suggests new candidate genes. Am J Med Genet A. 2008;146A(02):238–244. doi: 10.1002/ajmg.a.32093. [DOI] [PubMed] [Google Scholar]
- 16.Haberlandt E, Löffler J, Hirst-Stadlmann A et al. Split hand/split foot malformation associated with sensorineural deafness, inner and middle ear malformation, hypodontia, congenital vertical talus, and deletion of eight microsatellite markers in 7q21.1-q21.3. J Med Genet. 2001;38(06):405–409. doi: 10.1136/jmg.38.6.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown K K, Reiss J A, Crow K et al. Deletion of an enhancer near DLX5 and DLX6 in a family with hearing loss, craniofacial defects, and an inv(7)(q21.3q35) Hum Genet. 2010;127(01):19–31. doi: 10.1007/s00439-009-0736-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tzschach A, Menzel C, Erdogan F et al. Characterization of a 16 Mb interstitial chromosome 7q21 deletion by tiling path array CGH. Am J Med Genet A. 2007;143(04):333–337. doi: 10.1002/ajmg.a.31601. [DOI] [PubMed] [Google Scholar]
- 19.Tackels-Horne D, Toburen A, Sangiorgi E et al. Split hand/split foot malformation with hearing loss: first report of families linked to the SHFM1 locus in 7q21. Clin Genet. 2001;59(01):28–36. doi: 10.1034/j.1399-0004.2001.590105.x. [DOI] [PubMed] [Google Scholar]
- 20.De Leenheer S, Janssens E, Loose D, Leroy B P, Dhooge I J, Padalko,Etiological diagnosis in the hearing impaired newborn: proposal of a flow chart Eur Ann Otorhinolaryngol Head Neck Dis 2015729615153–162. [Google Scholar]
- 21.Wieland I, Muschke P, Jakubiczka S, Volleth M, Freigang B, Wieacker P F.Refinement of the deletion in 7q21.3 associated with split hand/foot malformation type 1 and Mondini dysplasia J Med Genet 20044105e54. Doi: 10.1136/jmg.2003.010587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rattanasopha S, Tongkobpetch S, Srichomthong C, Kitidumrongsook P, Suphapeetiporn K, Shotelersuk V. Absent expression of the osteoblast-specific maternally imprinted genes, DLX5 and DLX6, causes split hand/split foot malformation type I. J Med Genet. 2014;51(12):817–823. doi: 10.1136/jmedgenet-2014-102576. [DOI] [PubMed] [Google Scholar]
- 23.Frenz D A, Liu W, Cvekl A et al. Retinoid signaling in inner ear development: a “Goldilocks” phenomenon. Am J Med Genet A. 2010;152A(12):2947–2961. doi: 10.1002/ajmg.a.33670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chatterjee S, Kraus P, Lufkin T.A symphony of inner ear developmental control genes BMC Genet 20101168. Doi: 10.1186/1471-2156-11-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Birnbaum R Y, Everman D B, Murphy K K, Gurrieri F, Schwartz C E, Ahituv N. Functional characterization of tissue-specific enhancers in the DLX5/6 locus. Hum Mol Genet. 2012;21(22):4930–4938. doi: 10.1093/hmg/dds336. [DOI] [PMC free article] [PubMed] [Google Scholar]