

Abstract
B1a cells, particularly the PD-L2+ B1a cell subset, are enriched with autoantigen-specific receptors. However, the underlying molecular mechanism responsible for the skewed selection of autoreactive B1a cells remains unclear. Here we find that B1 cells express only RasGRP1, whereas B2 cells express mostly RasGRP3 and little RasGRP1. RasGRP1 is indispensable for transduction of weak signals. RasGRP1-deficiency markedly impairs B1a cell development and reduces serum natural IgM production; in particular, B1a cells that express autoantigen receptors, such as anti-PtC B1a cells, are virtually eliminated. Thus, unlike Btk and other signalosome components, RasGRP1 deficiency selectively affects only the B1a cell population with autoantigen receptors rather than the entire pool of B1a cells.
Keywords: RasGRP1, B1a cells, PD-L2, Signal transduction, Autoreactive antigen receptor
INTRODUCTION
B1 cells constitute a distinct B lymphocyte lineage that derives from a specific progenitor (1, 2) and bears a unique phenotype. B1 cells were originally recognized on the basis of CD5 expression and can be further divided into B1a and B1b subsets. B1 cells differ from follicular (FO) B2 cells and marginal zone (MZ) B cells in many ways that include surface marker expression, anatomical location, and functional activity (3). Notably, B1 and B2 cells differ in signaling parameters: B1 cells show evidence of constitutive activation in baseline expression of phosphorylated ERK and STAT3 while B2 cells do not (4–6), and B1 cells respond differently to various stimuli as compared to B2 cells (7).
B1 cells are the major source of serum natural IgM (8), which is both anti-microbial and autoreactive. Recently, we found that B1 cells are heterogeneous in antigen receptor specificities, and that PD-L2+ B1a cells are enriched with autoreactive natural IgM (9), such as PtC-specific natural IgM (10). Natural antibody tends to be germline-like due to minimal N-addition and somatic mutation, and is essential for infection prevention and organismal homeostasis. For example, B1a cell-derived natural IgM is critical for protection against bacterial (11) and viral (12) infections. Furthermore, autoreactive natural IgM, such as antibody that recognizes phosphatidylcholine (PtC), plays a critical role in the clearance of senescent cells and apoptotic debris (13, 14) and in so doing fulfills a key physiological function.
Ras guanyl nucleotide releasing proteins (RasGRPs) constitute a family comprising 4 members (RasGRP1 through RasGRP4). They have a catalytic domain, a DAG-binding C1 domain, and two EF hands (15). Upon antigen stimulation, RasGRPs are recruited to the membrane through DAG binding, where they play an important role in Ras activation (16, 17). RasGRPs are prominently expressed in blood cells (15). Numerous types of cells including T, B2, iNK T, and mast cells express RasGRP1 (18–21). In lymphocytes, T cells preferentially express RasGRP1, which is essential for T cell activation and positive selection (19), and T cells expressing low affinity receptors rely more on RasGRP1 than those with high affinity receptors (22). B2 cells express mostly RasGRP3 and little RasGRP1. RasGRP1 deficiency has minimal effect on B2 cell activation and development (18). In the present study, we find that B1 cells express only RasGRP1, which transduces a weak signal. RasGRP1 deficiency selectively disrupts development of B1a cells with autoantigen receptors and natural IgM production.
MATERIALS AND METHODS
Mice
C57BL/6 mice, B6.129S7-Rag1tm1Mom/J, and B6.SJL-Ptprca Pepcb/BoyJ mice at 8–14 weeks of age were obtained from the Jackson Laboratory. RasGRP1-deficient mice kindly provided by Dr. James C. Stone have been described (19) and were backcrossed at least 9 generations onto the C57BL/6 background. 6-week-old RasGRP1-deficient mice and littermate control mice were used in this study. All mice were cared for and handled in accordance with National Institutes of Health and institutional guidelines, and studies with these mice were approved by the Institutional Animal Care and Use Committee.
Antibodies and reagents
Affinity-purified F(ab′)2 fragments of polyclonal goat anti-mouse IgM (anti-Ig) and polyclonal goat anti-rabbit Ab-APC were obtained from Jackson ImmunoResearch Laboratories. Anti-mouse mAbs against CD19-APC, B220-PE, B220-FITC, CD5-PE-Cy7, CD43-PE, PD-L2-PE, and PD-L2-APC, CD86-PE, CD44-PE, CD9-PE, CD80-PE, Thy-1-PE, CD45.1-FITC, phospho-p38, and phospho-ERK-APC were obtained from BD pharmingen. Polyclonal anti-pERK, anti-NFATc1, anti-RasGRP3, and anti-phospho-JNK antibodies were obtained from Cell Signaling Technology. Anti-RasGRP1 antibody was obtained from Santa Cruz Biotechnology. Phorbol ester myristate (PMA), RIPA buffer, phenylmethylsulfonyl fluoride (PMSF), and protein inhibitor cocktails were obtained from Sigma Aldrich.
B cell isolation
FO B2 cells (B220+CD23+CD21+) were sorted from spleen cell suspensions. B1 cells (CD19+CD43+) were sorted from peritoneal cavity washouts. All B cell subsets were sorted on a BD Influx 4 (BD Pharmingen).
B cell activation
For ERK activation, B1 cells were stimulated with PMA (25 ng/ml) or anti-Ig (15 μg/ml) for 2 minutes. For p38 activation, peritoneal lymphocytes were stimulated with anti-Ig (15 μg/ml) for 1 min. For JNK activation, B1 cells were stimulated with anti-Ig (15 μg/ml) for 5 and 10 min. For NFATc1 expression, sorted B1 cells and B2 cells were stimulated with anti-Ig (15 μg/ml) for 0, 1, and 2 days.
Qualitative real-time PCR
Total RNA was obtained from sorted follicular B2 cells (B220+CD23+CD21+), MZ B cells (B220+CD23low-negCD21hi), or B1 cells (CD19+CD5+CD43+). cDNA was prepared using AMV reverse transcriptase (Roche Applied Sciences). RasGRP1-specific primers (forward: GGCTTTCCA CACAACTTTC; reverse: TCATCCCGCAGTCTTTAC), RasGRP3-specific primers (forward: CACTGGTGTTGGAGCCTAGA; reverse: TTCTGAAGCCCACTCCAGAG), and beta2-microglobulin-specific primers (forward: CCCGCCTCACATTGAAATCC; reverse: GCGTATGTAT CAGTCTCAGTGG.) were used for real time PCR. RasGRP1 and RasGRP3 expression was assessed and normalized to beta2-microglobulin.
Immunoblot analysis
B1 cells, MZ B cells, and B2 cells were sorted from wild type (WT) mice or RasGRP1-deficient mice and were lysed in RIPA buffer. For NFATc1 expression, unstimulated and anti-Ig-stimulated B1 and B2 cells were lysed in RIPA buffer. Equal amounts of protein for each condition (15–30 μg) were subjected to SDS-PAGE separation followed by immunoblot. Immunoreactive proteins were detected by ECL (Amersham Biosciences). Immunoblots were stripped and reprobed with control Ab to verify that equal amounts of protein were loaded in each lane.
Flow cytometry
For B cell subset separation, single cell suspensions of peritoneal washout cells were incubated with varying combinations of the following antibodies or reagents: anti-FcγII (Fc-blocking antibody), anti-B220, anti-CD5, anti-CD43, anti-PD-L2 and fluorochrome-encapsulating, liposomes (23), a kind gift of Dr. S. H. Clarke (University of North Carolina, Chapel Hill, NC). For analysis of B1 cells in spleen, single cell suspensions of spleen cells were incubated with varying combinations of the following antibodies: anti-FcγII (Fc-blocking antibody), anti-B220, anti-CD5, anti-CD19. For intracellular phospho-ERK, single cell suspensions of peritoneal washout cells were stimulated with PMA or anti-Ig for 2 min followed by fixation and permeabilization. The resultant cells were stained with anti-CD19, anti-CD5, anti-CD43, and anti-phospho-ERK antibodies.
ELISA
Sera were collected from RasGRP1-deficient mice and littermate wild type control mice at 6 weeks and 2–3 months of age. Immunoglobulin titers were measured by standard ELISA methods. For total IgM, plates were coated with anti-immunoglobulin (5 μg/ml). For PtC specific IgM, pre-coated plates with a single high-quality lipid species were obtained from Avanti Polar Lipids. Sera were applied as serial dilutions of 1:2 in PBS with 1% BSA and incubated at room temperature for 2 hours. The plates were then incubated with alkaline-phosphatase-conjugated goat anti-mouse IgM antibody.
Bone marrow or B1a cell adoptive transfer
Lin−B220−CD19− bone marrow cells (5 × 105) were sorted from RasGRP1-deficient mice (CD45.1−), littermate wild type control mice (CD45.1−), and C57BL/6 congenic mice (CD45.1+). A mixture (at a ratio of 1:1) of bone marrow sub-populations from RasGRP1-deficient mice (CD45.1−) and C57BL/6 congenic mice (CD45.1+), or from littermate wild type control mice (CD45.1−) and C57BL/6 congenic mice (CD45.1+) mice, was injected intravenously into lethally irradiated (500 Rad) Rag1−/− mice (C57BL/6J background). After two months, recipient mice were euthanized and the proportion of B cell subsets derived from different donors was analyzed by flow cytometry.
Statistical analysis
Student’s t-test was used for statistical analysis; results with p values below 0.05 were considered significant.
RESULTS
B1 cells express only RasGRP1 protein
B1 and B2 cells exhibit different BCR signaling outcomes. We examined the expression levels of RasGRP1 and RasGRP3 in B1 and B2 cells. B1 cells express a high level of RasGRP1 messenger RNA (mRNA) and a very low level of RasGRP3 mRNA, whereas B2 cells predominantly express RasGRP3 mRNA (Fig. 1A). Consistent with transcription, B1 cells express a high level of RasGRP1 protein, whereas RasGRP3 protein expression is undetectable. In keeping with a previous report (18), B2 cells express mostly RasGRP3 and little RasGRP1 protein (Fig. 1B). These results indicate that B1 and B2 cells express different members of the RasGRP family.
Figure 1. B1 cells express only RasGRP1.
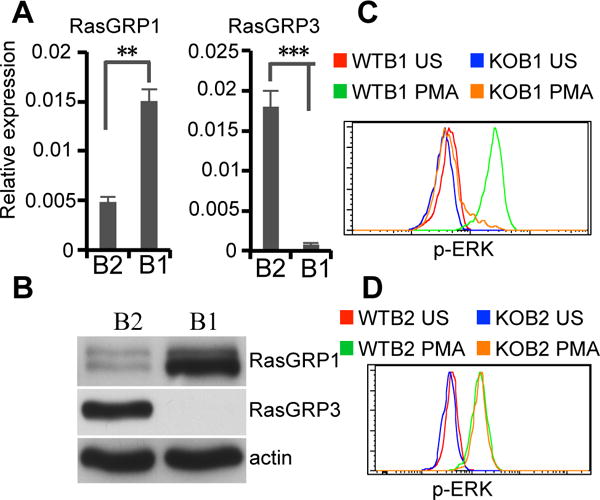
(A) RasGRP1 and RasGRP3 mRNA expression in peritoneal B1 (B1) and splenic B2 cells (B2) was determined by real time PCR analysis. Results are means ± SD of five experiments. ns, no significance. **, P<0.01. ***, P<0.005. (B) RasGRP1 and RasGRP3 protein expression in peritoneal B1 (B1) and splenic B2 cells (B2) was determined by immunoblot analysis. Actin was determined as a loading control. (C) RasGRP1 is required for connecting PLC-γ2 to Ras-ERK in B1 cells. WT and RasGRP1-deficient peritoneal lymphocytes were unstimulated or stimulated with PMA (25 ng/ml) for 2 min. ERK activation in B1 cells was analyzed by intracellular immunofluorence staining for p-ERK and flow cytometry. (D) RasGRP1 is redundant for connecting PLC-γ2 to Ras-ERK in B2 cells. WT and RasGRP1-deficient spleen cells were unstimulated or stimulated with PMA (25 ng/ml) for 2 min. ERK activation in FO B2 cells (B220+CD23+CD21+) was analyzed by intracellular immunofluorence staining for p-ERK and flow cytometry. Green lines represent PMA-stimulated (PMA) WT B1 (C) or FO B2 cells (D). Orange lines represent PMA-stimulated (PMA) RasGRP1-deficient B1 (C) or FO B2 cells (D). Red lines represent unstimulated (US) WT B1 (C) or FO B2 cells (D). Blue lines represent unstimulated (US) KO B1 (C) or FO B2 cells (D). Results represent one of three comparable experiments.
RasGRP1 connects PLC-γ2 to Ras-ERK in B1 cells
Since B1 cells express only RasGRP1, loss of RasGRP1 might impair signal transduction from PLC-γ2 to Ras-ERK. To test this, peritoneal lymphocytes were stimulated with PMA, an analog of diacylglycerol (DAG) that is produced by BCR-initiated activation of PLC-γ2. ERK phosphorylation (p-ERK) in B1 cells (CD19+CD43+) was analyzed. PMA stimulates substantial p-ERK in wild type (WT) B1 cells and RasGRP1 deficiency completely abrogates PMA-stimulated p-ERK (Fig. 1C). Because B2 cells express mostly RasGRP3 and little RasGRP1, RasGRP1 deficiency does not affect PMA-stimulated p-ERK in B2 cells (Fig. 1D). These results indicate that RasGRP1 is the principal molecule linking PLC-γ2 to Ras-ERK in B1 cells.
The RasGRP1-dependent BCR signaling pathway exhibits a low activation threshold in B1 cells
Since B1 cells express only RasGRP1, loss of RasGRP1 might interfere with overall BCR signal transduction. In contrast to B2 cells, B1 cells exhibit constitutive p-ERK, which is initiated by sustained signals (4, 5). RasGRP1 mediates sustained p-ERK in T cells (22), suggesting that RasGRP1 might also transduce sustained signals for constitutive p-ERK in B1 cells. To address this issue, WT B1 cells, WT B2 cells, and RasGRP1-deficient B1 cells were sorted and constitutive p-ERK was examined. As expected, WT B1 cells exhibit constitutive p-ERK whereas B2 cells do not, and RasGRP1 deficiency completely abrogates constitutive p-ERK (Fig. 2A). These results indicate that RasGRP1 is required for mediating sustained signals for constitutive p-ERK in B1 cells.
Figure 2. The RasGRP1-dependent BCR signaling pathway exhibits a low activation threshold in B1 cells.
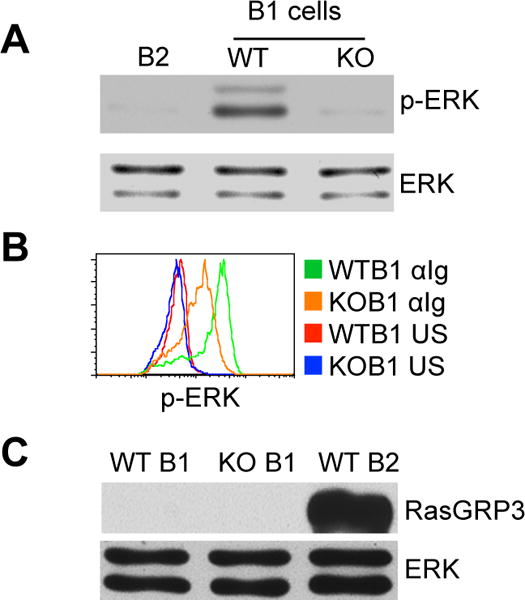
(A) RasGRP1 mediates constitutive p-ERK in B1 cells. B1 cells were sorted from wild type (WT) and RasGRP1-deficient mice (KO). ERK phosphorylation was evaluated by immunoblot and compared with that in B2 cells. Membranes were stripped and reprobed with ERK-specific antibody as a loading control. (B) BCR-engagement stimulates ERK activation through RasGRP1-dependent and -independent pathways. Wild type and RasGRP1-deficient peritoneal lymphocytes were unstimulated or stimulated with anti-Ig (15 μg/ml) for 2 min. ERK phosphorylation in B1 cells (CD19+CD43+) was analyzed by flow cytometric assay. Green lines represent anti-Ig-stimulated (αIg) WT B1 cells. Orange lines represent anti-Ig-stimulated (αIg) RasGRP1-deficient B1 cells. Red lines represent unstimulated (US) WT B1 cells. Blue lines represent unstimulated (US) KO B1 cells. Results represent one of three comparable experiments. (C) RasGRP1-deficent B1 cells do not express compensatory RasGRP3. Wild-type B1 cells (WT B1), RasGRP1-deficient B1 cells (KO B1), and Wild-type B2 cells (WT B2) were sorted, and RasGRP3 expression in these cells was evaluated by immunoblot. Membranes were stripped and reprobed with ERK-specific antibody as a loading control. Results represent one of three comparable experiments.
We then questioned whether disruption of RasGRP1-dependent BCR signaling would affect BCR-stimulated ERK activation. To address this issue, peritoneal lymphocytes were stimulated with anti-Ig, and p-ERK in B1 cells was examined. As expected, anti-Ig stimulates a marked increase of p-ERK in WT B1 cells; surprisingly, unlike constitutive p-ERK that is completely dependent on RasGRP1, BCR-stimulated p-ERK is only partially abrogated by RasGRP1 deficiency (Fig. 2B). This incomplete blockage of p-ERK is not due to compensatory RasGRP3 expression, which is undetectable in RasGRP1-deficient B1 cells (Fig. 2C). Further, the connection from PLC-γ2 to Ras-ERK is completely blocked in RasGRP1-deficient B1 cells, as PMA fails to stimulate p-ERK (Fig. 1C). These results indicate that ERK activation is mediated by both RasGRP1-dependent and RasGRP1-independent signaling pathways in B1 cells. The RasGRP1-dependent signaling pathway exhibits a low activation threshold and is indispensable for transducing weak signals, such as sustained signaling that produces suboptimal p-ERK. In contrast, strong signals, such as anti-Ig-stimulated BCR signals that produce optimal p-ERK, activate both RasGRP1-dependent and RasGRP1-independent signaling pathways.
RasGRP1 is dispensable for other BCR signaling outcomes in B1 cells
As noted above, RasGRP1 transduces signaling for MAP kinase ERK activation. To test whether RasGRP1 is also required for BCR-stimulated activation of other MAP kinase families, sorted B1 cells or peritoneal lymphocytes were stimulated with anti-Ig, and JNK phosphorylation (p-JNK) and p38 phosphorylation (p-p38) in B1 cells were examined. We find that BCR-crosslinking stimulates substantial p-JNK and p-p38 in WT B1 cells, and these phosphorylation events are completely RasGRP1-independent (Fig. 3A, 3B). These results indicate that, although RasGRP1 is critical for BCR-stimulated p-ERK in B1 cells, it is dispensable for BCR-initiated JNK and p38 activation.
Figure 3. RasGRP1 is dispensable for BCR signaling outcomes beyond p-ERK in B1 cells.
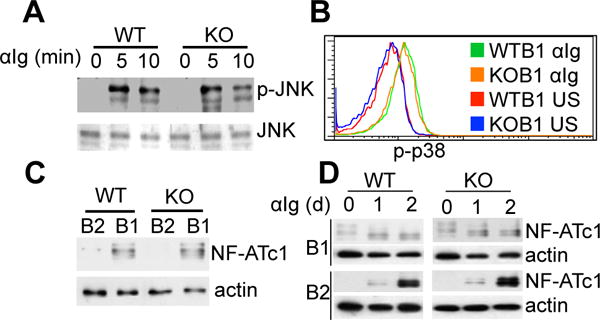
(A) RasGRP1 is dispensable for BCR-stimulated p-JNK. B1a cells were sorted from wild type (WT) and RasGRP1-deficient (KO) mice and were stimulated with anti-Ig (15 μg/ml) (αIg) for 5 and 10 min. p-JNK was evaluated by immunoblot. Membranes were stripped and reprobed with JNK-specific antibody as a loading control. (B) RasGRP1 is dispensable for BCR-stimulated p-p38. Peritoneal lymphocytes from wild type and RasGRP1-deficient mice were unstimulated (US) (WT: red line; KO: blue line) or stimulated (WT: green line; KO: orange line) with anti-Ig (αIg) (15 μg/ml) for 1 min. p-p38 in B1 cells (CD19+CD43+) was evaluated by intracellular immunofluorence staining and flow cytometry. (C) Constitutive expression of NFATc1 is intact in RasGRP1-deficient B1 cells. NFATc1 protein expression was evaluated in WT B1 cells, WT B2 cells, KO B1 cells, and KO B2 cells by immunoblot. Membranes were stripped and reprobed with actin-specific antibody as a loading control. (D) BCR-stimulated NFATc1 protein expression is intact in RasGRP1-deficient B1 cells. WT B1 cells (left upper panels), WT B2 cells (left lower panels), KO B1 cells (right upper panels), and KO B2 cells (right lower panels) were stimulated with anti-Ig (15 μg/ml) (αIg) for 0, 1, or 2 days. NFATc1 protein expression was evaluated by immunoblot. Membranes were stripped and reprobed with actin-specific antibody as a loading control. Results represent one of three comparable experiments.
B1 cells constitutively express a high level of NFATc1 protein, which is critical for B1 cell development (24). Naïve B2 cells express little NFATc1, which is significantly elevated upon BCR crosslinking (25), suggesting that constitutive NFATc1 expression in B1 cells may be due to sustained signaling. RasGRP1 transduces sustained signals for constitutive p-ERK. To examine whether RasGRP1 is required for constitutive NFATc1 expression, WT B1 cells, WT B2 cells, RasGRP1-deficient B1 cells, and RasGRP1-deficient B2 cells were sorted and NFATc1 protein expression was evaluated. Both WT and RasGRP1-deficient B1 cells constitutively express similar levels of NFATc1 protein (Fig. 3C). To test whether BCR-stimulated NFATc1 expression is dependent on RasGRP1, WT B1 cells, WT B2 cells, RasGRP1-deficient B1 cells, and RasGRP1-deficient B2 cells were stimulated with anti-Ig for 0, 1, or 2 days, and NFATc1 expression was evaluated. In agreement with a previous report (25), BCR crosslinking substantially elevates NFATc1 expression in B2 cells (lower panels) and, to a lesser extent, in B1 cells (upper panels). NFATc1 expression at day 0, day 1, and day 2 in WT B1 cells is similar with that in RasGRP1-deficient B1 cells (Fig. 3D). These results indicate that both constitutive and BCR-stimulated NF-ATc1 expression are RasGRP1-independent in B1 cells.
RasGRP1-dependent signaling contributes to constitutive B1a cell activation
Unlike B2 cells, B1a cells continually exhibit activation phenotypes, which results from sustained signaling (5). RasGRP1-dependent BCR signaling transduces sustained signaling for p-ERK suggesting that RasGRP1 may be critical for constitutive B1 cell activation. To test this, surface activation markers including CD86, CD80, and CD44 were examined. Consistent with previous reports (26, 27), WT B1 cells express substantial CD86, CD80, and CD44. RasGRP1 deficiency partially reduces expression of each of these surface antigens (Fig. 4A–4C). These results indicate that RasGRP1-dependent sustained signaling contributes to constitutive B1 cell activation.
Figure 4. RasGRP1-dependent signaling contributes to constitutive surface antigen expression in B1a cells.
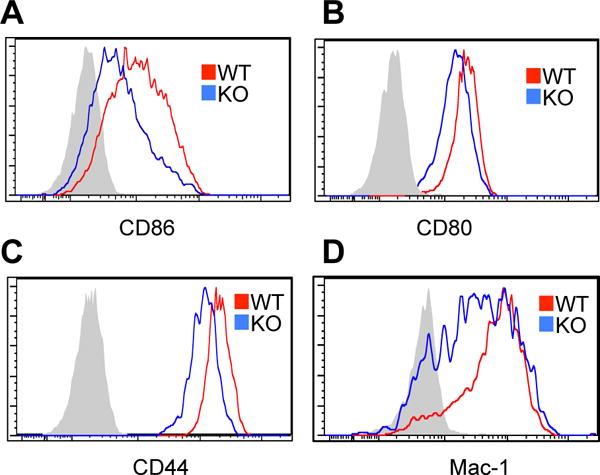
(A–C) RasGRP1-dependent signaling contributes to constitutive B1 cell activation. Peritoneal B1a cells (CD19+CD5+B220low-neg) from WT control mice (red lines) and RasGRP1-deficient mice (blue lines) were stained with antibodies against CD86 (A), CD80 (B), or CD44 (C) followed by flow cytometric analysis. Shaded areas represent isotype controls. (D) RasGRP1-dependent signaling contributes to Mac-1 expression. Peritoneal B1a cells (CD19+CD5+B220low-neg) from WT control mice (red lines) and RasGRP1-deficient mice (blue lines) were stained with antibodies against Mac-1 followed by flow cytometric analysis. Shaded areas represent isotype control. Results represent one of three comparable experiments.
In addition, B1 cells also express a high level of Mac-1 that is involved in B1 cell trafficking. To test whether expression of this surface marker depends on RasGRP1, surface Mac-1 expression was examined. WT B1 cells express substantial Mac-1. RasGRP1-deficiency substantially but incompletely reduces expression of this surface antigen (Fig. 4D). These results indicate that RasGRP1-dependent sustained signaling contributes to Mac-1 expression in B1 cells.
RasGRP1-deficiency impairs B1a cell development and reduces serum natural IgM production
MAP kinase ERK expression is critical for B cell development (28). Constitutive and BCR-stimulated p-ERK is impaired in RasGRP1-deficient B1 cells suggesting a potential role for RasGRP1 in B1 cell development. To explore this issue, we analyzed B1 cells in the spleen and peritoneal cavity. Because adult RasGRP1-deficient mice have already developed lupus-like autoimmune phenotypes (18), all mice used in these experiments were 6 weeks old. We find a 3-fold reduction in percentage and absolute number of splenic B1 cells (CD19+CD5+B220low-neg) in RasGRP1-deficient mice compared with those in WT control mice (Fig. 5A). Moreover, the percentage of B1a cells (CD19+CD5+CD43+) in total peritoneal B cells drops from 47% in WT mice to 15% in RasGRP1-deficient mice. The absolute number of peritoneal B1a cells is proportionally decreased (Fig. 5B). Interestingly, RasGRP1 deficiency does not significantly affect development of B1b cells (CD19+CD5−CD43+) nor B2 cells (CD19+CD5−CD43−) in the peritoneal cavity. In fact, the percentage and absolute number of B2 cells are increased in RasGRP1-deficient mice (Fig. 5B). These results indicate that RasGRP1 deficiency results in impaired B1a cell development.
Figure 5. RasGRP1 deficiency results in impaired B1a cell development and reduced natural IgM in serum.
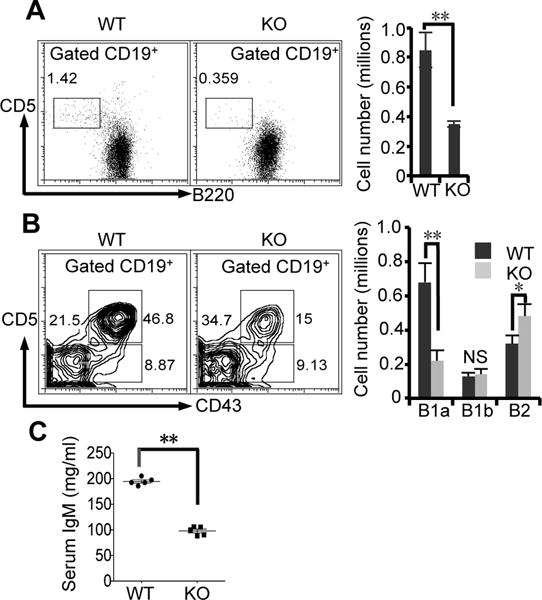
(A) The number of B1 cells in spleen is reduced in RasGRP1-deficient mice. Flow cytometric analysis of B1 cells (CD19+CD5+B220lo-neg) in spleens from RasGRP1-deficient mice (KO) and WT control mice (WT) is shown. (B) Flow cytometric analysis of B1a cells (CD19+CD5+CD43+), B1b cells (CD19+CD5−CD43+), and B2 cells (CD19+CD5−CD43−) in peritoneal cavities from RasGRP1-deficient mice (KO) and WT control mice (WT) is shown. The numbers adjacent to the boxed areas (see panels) are percentages of B cells representing one of five comparable experiments. The absolute numbers of individual B cell subsets are shown in the right panel. The results are means ± SD of five experiments. (C) RasGRP1 deficiency results in reduced natural IgM secretion in serum. Sera were collected from 6-week old wild type mice (WT) and RasGRP1-deficient mice (KO). Serum IgM was analyzed by ELISA. Each spot represents one mouse. The lines represent means ± SD of five mice. NS, not significant. *, p<0.05. **, p<0.01.
B1a cells are the major source of natural IgM antibody in serum. Impaired B1a cell development may affect natural IgM production in RasGRP1-deficient mice. To address this issue, we collected sera from young mice and examined the content of natural IgM. We find that young RasGRP1-deficient mice produce less serum IgM (~2 fold decrease) than littermate WT control mice (Fig. 5C). The decreased natural IgM content correlates with the reduced number of B1a cells. These results indicate that RasGRP1 deficiency results in impaired B1a cell development and, accordingly, a reduced production of natural IgM. Compared with young RasGRP1-deficient mice, adult RasGRP1-deficient mice produce a similar amount of serum IgM as WT mice (18). The different serum IgM production between young and adult RasGRP1-deficient mice may be due to the existence of excessive numbers of plasma cells in adult RasGRP1-deficient mice (18) that contribute to serum IgM production.
Impaired B1a cell development in RasGRP1-deficient mice is B cell autonomous
Because several types of cells express RasGRP1, impaired B1a cell development could be a secondary effect. To exclude this possibility, we prepared mixed-allotype chimeras. Different progenitors in bone marrow give rise to B2 cells, B1a cells and B1b cells (1, 29). Lin−CD19−B220− bone marrow (BM) cells were sorted from RasGRP1-deficient mice (CD45.2+), littermate control mice (CD45.2+), and WT C57BL/6 congenic mice (CD45.1+). A mixture of BM subpopulations from RasGRP1-deficient mice and WT C57BL/6 congenic mice or from littermate control mice and WT C57BL/6 congenic mice (as a control) was injected in a 50:50 mixture into lethally irradiated Rag1−/− mice. Two months later, B cells in the recipient mice were analyzed. RasGRP1 deficiency does not affect B2 cell development (18), because 30–36% B2 cells either in spleen or peritoneal cavity of recipient mice are CD45.1− (Fig. 6A, 6B). Further, RasGRP1 deficiency does not affect B1b cell development, as 34% B1b cells in the peritoneal cavity are CD45.1− (Fig. 6B). In marked contrast, we found that only 15% B1 cells both in spleen and peritoneal cavity are CD45.1− (1:5.7 ratio of RasGRP1−/− to WT) (Fig. 6A, 6B). In the recipient mice that received littermate control BM mixed with WT C57BL/6 BM, again 35% B cells, including B1a cells, B1b and B2 cells, either in spleen or peritoneal cavity are CD45.1− and thus derived from littermate control mice. These results indicate that defective B1a cell development in RasGRP1-deficient mice is cell autonomous. The ratio of CD45.1− B cells versus CD45.1+B cells in recipient mice may reflect variable numbers of B cell progenitors in BM from donor mice with different backgrounds, as the original RasGRP1-deficient mice are on mixed C57BL/6 and 129 background even though the mice used in this study were backcrossed into C57BL/6 mice for 9 generations. Consistent with this prediction, 50% B cells are CD45− when recipient mice received a mixture of BM sub-populations at a ratio of 1:1 from C57BL/6 mice (CD45.2+) and C57BL/6 congenic mice (CD45.1+) (data not shown).
Figure 6. Impaired B1a cell development is cell autonomous in RasGRP1-deficient mice.
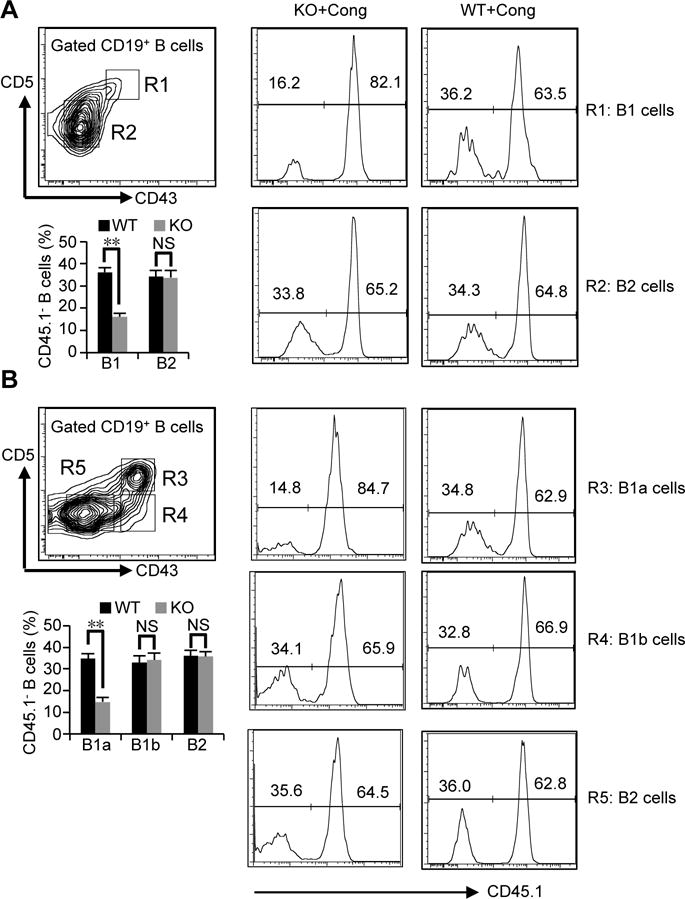
Lin−CD19−B220− bone marrow (BM) cells were sorted from RasGRP1-deficient mice (CD45.2+) (KO), littermate wild type control mice (WT), or wild type C57BL/6 congenic mice (CD45.1+) (Cong). Chimeras were generated by injection of these subpopulations from RasGRP1-deficient mice (CD45.2+) (KO) and wild type C57BL/6 congenic mice (CD45.1+) or from littermate wild type control mice and wild type C57BL/6 congenic mice (CD45.1+) at a ratio of 1:1 into lethally irradiated Rag1−/− mice. B cell subsets from spleen (A) or peritoneal cavity (B) in recipients were analyzed 2 months after injection. Results in (A) and (B) represent one mouse in one of three comparable experiments. Bar graphs represent the mean±SD of combined results of three experiments with 6 recipient mice. NS, not significant. **, p<0.01.
B1a cells expressing autoantigen receptors critically depend on RasGRP1
Autoreactive natural IgM exhibits low affinity in binding interactions with self-antigen (30, 31). The RasGRP1-dependent BCR signaling pathway (PLC-γ2-RasGRP1-Ras) exhibits a low activation threshold and transduces weak BCR signals for ERK activation, which is critical for B cell development (28). We then hypothesized that development of autoreactive B1a cells may especially rely on RasGRP1. To test this, development of anti-phosphatidylcholine (PtC) B1a cells, which secrete a prototypical natural autoantibody that binds to a cryptic determinant exposed on senescent red blood cells, was examined. We find that anti-PtC B1a cells are severely impaired and virtually eliminated in RasGRP1-deficient mice (Fig. 7A, 7B). In line with reduced PtC-specific B1a cells, PtC-specific serum IgM is also significantly reduced in RasGRP1-deficient mice (Fig. 7C). We previously showed that B1a cell receptors are heterogeneous in autoreactive antigen specificities, and PD-L2+ B1a cells are enriched with autoreactivity (9). Thus, we find that, although RasGRP1 deficiency reduces the total number of B1a cells (Fig. 4B), PD-L2+ B1a cells are severely and disproportionately affected and experience marked reduction whereas PD-L2− B1a cells are less affected (Fig. 7D–7F) in RasGRP1-deficient mice.
Figure 7. RasGRP1 deficiency selectively impairs B1a cells with autoantigen receptors.
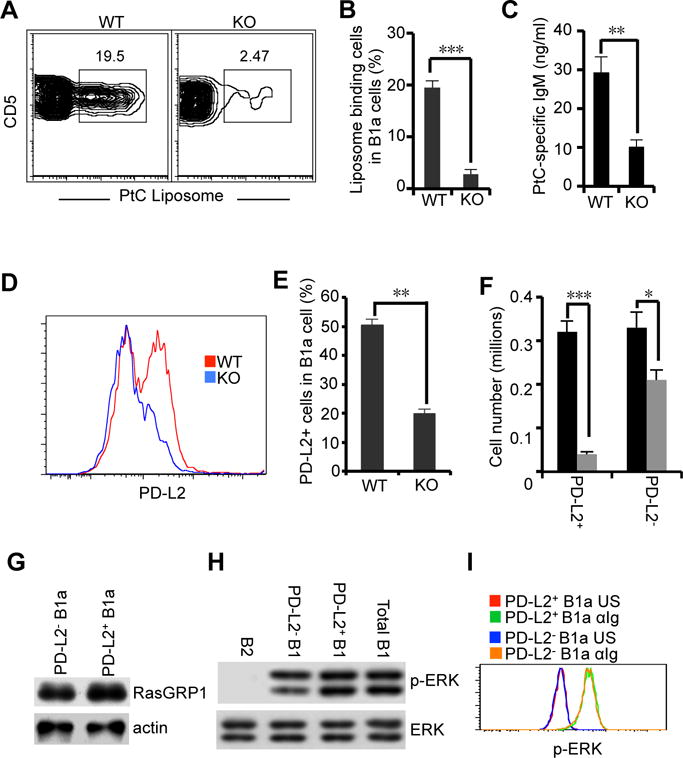
(A–C) PtC-binding B1a cell development is defective in RasGRP1-deficient mice. PtC binding B1a cells (CD19+CD5+CD43+) were analyzed by flow cytometry (A and B). Serum PtC-specific IgM in wild type (WT) and RasGRP1-deficient (KO) mice was examined by ELISA (C). (D–F) PD-L2+ B1a cell development is severely impaired in RasGRP1-deficient mice. Peritoneal PD-L2+ B1a cells (CD19+CD5+CD43+PD-L2+) were analyzed in wild type (WT) and RasGRP1-deficient (KO) mice by flow cytometric analysis (D). The percentage and absolute numbers of PD-L2+ B1 cells and PD-L2− B1 cells in wild type (WT) and RasGRP1-deficient (KO) mice were analyzed (E and F) (G) PD-L2+ B1a cells and PD-L2− B1a cells express an equal amount of RasGRP1 protein. PD-L2+ B1a cells and PD-L2− B1a cells were sorted from C57BL/6 mice, and RasGRP1 protein expression was determined by immunoblot analysis. Actin was determined as a loading control. (H) PD-L2+ B1a cells and PD-L2− B1a cells exhibit similar levels of constitutive p-ERK. PD-L2+ B1a cells and PD-L2− B1a cells were sorted from the peritoneal lymphocytes in C57BL/6 mice. ERK phosphorylation was evaluated by immunoblot. Membranes were stripped and reprobed with ERK-specific antibody as a loading control. (I) PD-L2+ B1a cells and PD-L2− B1a cells exhibit similar levels of p-ERK in response to anti-Ig stimulation. Peritoneal lymphocytes from C57BL/6 mice were unstimulated or stimulated with anti-Ig (15 μg/ml) for 2 min. ERK phosphorylation in PD-L2+ B1a cells and PD-L2− B1a cells was analyzed by flow cytometric assay. Green line represents anti-Ig-stimulated (αIg) PD-L2+ B1a cells. Orange line represents anti-Ig-stimulated (αIg) PD-L2− B1a cells. Red line represents unstimulated (US) PD-L2+ B1a cells. Blue line represents unstimulated (US) PD-L2− B1a cells. Bar graphs in (B), (C), (E), and (F) represent means ± SD of five experiments. Results in (A), (D), (G), (H), and (I) represent one of five comparable experiments. *, p<0.05. **, p<0.01. ***, p<0.005.
Loss of RasGRP1 preferentially affects PD-L2+ B1a cells suggesting the possibility that PD-L2+ B1a cells and PD-L2− B1a cells might differentially express RasGRP1 protein, loss of which may have a different impact on these two B1a cell subsets. To test this, PD-L2+ B1a cells and PD-L2− B1a cells were sorted and RasGRP1 protein expression was examined by immunoblot. Although RasGRP1-deficiency principally affects PD-L2+ B1a cells, RasGRP1 protein expression is similar between PD-L2+ B1a cells and PD-L2− B1a cells (Fig. 7G). Another potential explanation for preferential impairment of PD-L2+ B1a cells in RasGRP1-deficient mice is that PD-L2+ B1a cells and PD-L2− B1a cells have different BCR signaling capabilities. To test this, PD-L2+ B1a cells and PD-L2− B1a cells were sorted, and constitutive p-ERK was examined by immunoblot. In addition, peritoneal lymphocytes were stimulated with anti-Ig, and ERK phosphorylation in PD-L2+ B1a cells and PD-L2− B1a cells was evaluated by flow cytometry. We find that both constitutive (Fig. 7H) and anti-Ig-stimulated (Fig. 7I) p-ERK in PD-L2+ B1a cells and PD-L2− B1a cells are similar. These results indicate that, since PD-L2+ B1a cells and PD-L2− B1a cells exhibit similar BCR signaling capability, preferential impairment of PD-L2+ B1a cells in RasGRP1-deficient mice is not due to differences in BCR signaling elements. PD-L2+ B1a cells are enriched with autoreactive BCR antigen receptors that produce weak BCR signals. RasGRP1-dependent signaling is indispensable for transduction of weak signals. Thus, RasGRP1 is specifically required for development of autoreactive B1a cells, whereas B1a cells without autoreactive antigen receptors are less affected.
DISCUSSION
B2 cells express mostly RasGRP3 and little RasGRP1 (18). Here we find that B1 cells express only RasGRP1. RasGRP1 and RasGRP3 are similar in structure in that they contain a REM-Cdc25 catalytic core domain, a pair of EF hands, and a C1 domain (32). The mechanism by which B1 cells express only RasGRP1 remains unknown. One explanation has to do with the molecular mechanism of signal propagation. Although the interaction between the C1 domain and DAG contributes to RasGRP1 translocation, the efficiency is low. A unique plasma membrane targeter (PT) domain at the RasGRP1 C terminal domain promotes its efficient plasma membrane targeting, particularly, in B cells (17). B1 cells express germline-like antigen receptors that initiate weak BCR signals in response to self-antigens. These weak signals are presumed to produce less DAG. In this circumstance, RasGRP1 but not RasGRP3 would readily translocate to the plasma membrane with the aid of the PT domain. In this way, RasGRP1 may perform a specialized role in B1 cells by mediating weak signals and thus has been retained in preference to RasGRP3 under evolutionary pressure. Another explanation relates to the fact that B1 cells originate from a different precursor compared with B2 cells (1). Our finding that B1 cells express only RasGRP1 while B2 cells predominantly express RasGRP3 lends strong support to the “lineage hypothesis” (31).
The fate of B cells is determined by BCR signaling. Deficiency of signalosome components (33), including BTK, Syk, BLNK, PI3-kinase, PLCγ2, and PKCβ, affects general signal strength and, therefore, influences all downstream signaling pathways. Consistent with this, deficiency of these BCR signaling molecules eliminates all B1 cells and significantly affects B2 cell as well. In contrast, deficiency of RasGRP1 specifically impairs the classical BCR signaling pathway (PLC-γ2-RasGRP1-Ras) for ERK activation while leaving other BCR signaling pathways untouched. In addition, B2 cells express both RasGRP1 and RasGRP3 with the latter as the dominant form, so RasGRP3 compensates for the loss of RasGRP1. Thus, RasGRP1 deficiency primarily affects B1a cells, and more specifically, PD-L2+ autoreactive B1a cells. In contrast, B2 cell development and function are normal(18).
B2 cells are negatively selected (34), while B1 cells are positively selected (35). However, molecular mechanisms for these different developmental pathways remain unclear. MAPK family members exhibit different activation thresholds, and ERK displays a lower activation threshold than p38 and JNK in T lymphocytes (36). These features are recapitulated in B1 cells. B1 cells exhibit sustained signaling that leads to constitutive p-ERK but not p-p38 and p-JNK (4). Lack of constitutive p-p38 and p-JNK is not due to disruption of BCR signaling as B1 cells do produce p-p38 and p-JNK in response to BCR crosslinking that triggers strong BCR signaling. In T lymphocytes, much evidence indicates that MAP kinases play critical roles in determining the fate of thymocytes. MAP kinase ERK regulates T cell positive selection (37, 38), while p38 and JNK govern T cell negative selection (39, 40). Our results suggest that this scheme may also apply to B cells. Autoreactive B1 cells express low affinity antigen receptors and produce weak signaling, which only activates ERK through RasGRP1-dependent signaling. JNK and p38 exhibit high activation thresholds and are not activated by weak signaling. Thus, autoreactive B1 cells undergo positive selection. In contrast, B2 cells express high affinity antigen receptors and produce relatively strong signaling, which activates the full range of MAP kinases, including JNK and p38. B2 cells then are negatively selected during development.
Adult RasGRP1-deficient mice demonstrate lupus-like autoimmune phenotypes, such as splenomegaly and production of anti-nuclear autoantibody (18). These phenotypes are recapitulated in RasGRP1d/d and RasGRP1Anaef mice (41, 42), which express RasGRP1 with a deleted tail domain or a mutated EF-hand, respectively. In humans, a high incidence of aberrant RasGRP1 splice variants exclusively occurs in SLE patients (43, 44). These observations suggest that RasGRP1 dysfunction is closely associated with autoimmune disease. Young RasGRP1-deficient mice exhibit T cell lymphopenia. However, the adult mice exhibit a substantial expansion of CD4+ T cells (41, 45). Thus, autoimmune phenotypes have been attributed to T cell lymphopenia in these animals. However, firstly, lymphopenia itself is not necessary for induction of autoimmune disease. It only plays a predisposing role. Development of autoimmune disease in a lymphopenic host needs a key “second hit” (46). Secondly, RasGRP1d/d (41) and RasGRP1Anaef (42) mice produce substantial antinuclear autoantibody, while T cell development in these mice is mildly impaired or normal, respectively. Thus, the mechanism responsible for the autoimmune disorder in RasGRP1-deficient mice remains unclear. T cells (45) and autoreactive B cells are activated and expanded suggesting an excess of self-antigen in adult RasGRP1-deficient mice (47). Autoreactive B1a cells are the major source of autoreactive natural IgM that is critical in facilitating self-antigen clearance (13, 30). Our results suggest an explanation that excess self-antigen may result from impaired autoreactive B1a cell development. However, the relationship between impaired autoreactive B1a cells and autoimmune phenotypes in RasGRP1-deficient mice remains to be determined.
In conclusion, in this study we find that B1 cells express only RasGRP1, which is required for weak signal transduction. RasGRP1 deficiency impairs B1a cell development and reduces natural IgM production. In particular, RasGRP1 is indispensable for development of B1a cells with autoantigen receptors, revealing a connection between a signaling molecule and development of a specific repertoire within the B1a cell population.
ABBREVIATIONS
- RasGRP1
Ras guanyl nucleotide releasing proteins 1
- BCR
B cell receptor
- PtC
Phosphatidylcholine
- DAG
diacylglycerol
- PMA
phorbol 12-myristate 13-acetate
Footnotes
This work was supported by USPHS grants AI029690 (to T.L.R) and 1 R03 AI115544-01 (to B.G) awarded by the National Institutes of Health.
The authors declare that they have no completing financial interests.
References
- 1.Montecino-Rodriguez E, Leathers H, Dorshkind K. Identification of a B-1 B cell-specified progenitor. Nature immunology. 2006;7:293–301. doi: 10.1038/ni1301. [DOI] [PubMed] [Google Scholar]
- 2.Herzenberg LA, Tung JW. B cell lineages: documented at last! Nat Immunol. 2006;7:225–226. doi: 10.1038/ni0306-225. [DOI] [PubMed] [Google Scholar]
- 3.Rothstein TL. Cutting edge commentary: two B-1 or not to be one. Journal of immunology. 2002;168:4257–4261. doi: 10.4049/jimmunol.168.9.4257. [DOI] [PubMed] [Google Scholar]
- 4.Wong SC, Chew WK, Tan JE, Melendez AJ, Francis F, Lam KP. Peritoneal CD5+ B-1 cells have signaling properties similar to tolerant B cells. The Journal of biological chemistry. 2002;277:30707–30715. doi: 10.1074/jbc.M202460200. [DOI] [PubMed] [Google Scholar]
- 5.Holodick NE, Tumang JR, Rothstein TL. Continual signaling is responsible for constitutive ERK phosphorylation in B-1a cells. Molecular immunology. 2009;46:3029–3036. doi: 10.1016/j.molimm.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karras JG, Wang Z, Huo L, Howard RG, Frank DA, Rothstein TL. Signal transducer and activator of transcription-3 (STAT3) is constitutively activated in normal, self-renewing B-1 cells but only inducibly expressed in conventional B lymphocytes. The Journal of experimental medicine. 1997;185:1035–1042. doi: 10.1084/jem.185.6.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morris DL, Rothstein TL. Abnormal transcription factor induction through the surface immunoglobulin M receptor of B-1 lymphocytes. J Exp Med. 1993;177:857–861. doi: 10.1084/jem.177.3.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baumgarth N, Herman OC, Jager GC, Brown L, Herzenberg LA. Innate and acquired humoral immunities to influenza virus are mediated by distinct arms of the immune system. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:2250–2255. doi: 10.1073/pnas.96.5.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhong X, Lau S, Bai C, Degauque N, Holodick NE, Steven SJ, Tumang J, Gao W, Rothstein TL. A novel subpopulation of B-1 cells is enriched with autoreactivity in normal and lupus-prone mice. Arthritis and rheumatism. 2009;60:3734–3743. doi: 10.1002/art.25015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhong X, Tumang JR, Gao W, Bai C, Rothstein TL. PD-L2 expression extends beyond dendritic cells/macrophages to B1 cells enriched for V(H)11/V(H)12 and phosphatidylcholine binding. European journal of immunology. 2007;37:2405–2410. doi: 10.1002/eji.200737461. [DOI] [PubMed] [Google Scholar]
- 11.Haas KM, Poe JC, Steeber DA, Tedder TF. B-1a and B-1b cells exhibit distinct developmental requirements and have unique functional roles in innate and adaptive immunity to S. pneumoniae. Immunity. 2005;23:7–18. doi: 10.1016/j.immuni.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 12.Baumgarth N, Herman OC, Jager GC, Brown LE, Herzenberg LA, Chen J. B-1 and B-2 cell-derived immunoglobulin M antibodies are nonredundant components of the protective response to influenza virus infection. The Journal of experimental medicine. 2000;192:271–280. doi: 10.1084/jem.192.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ehrenstein MR, Notley CA. The importance of natural IgM: scavenger, protector and regulator. Nature reviews Immunology. 2010;10:778–786. doi: 10.1038/nri2849. [DOI] [PubMed] [Google Scholar]
- 14.Silverman GJ. Regulatory natural autoantibodies to apoptotic cells: pallbearers and protectors. Arthritis and rheumatism. 2011;63:597–602. doi: 10.1002/art.30140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ebinu JO, Bottorff DA, Chan EY, Stang SL, Dunn RJ, Stone JC. RasGRP, a Ras guanyl nucleotide- releasing protein with calcium- and diacylglycerol-binding motifs. Science. 1998;280:1082–1086. doi: 10.1126/science.280.5366.1082. [DOI] [PubMed] [Google Scholar]
- 16.Carrasco S, Merida I. Diacylglycerol-dependent binding recruits PKCtheta and RasGRP1 C1 domains to specific subcellular localizations in living T lymphocytes. Molecular biology of the cell. 2004;15:2932–2942. doi: 10.1091/mbc.E03-11-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beaulieu N, Zahedi B, Goulding RE, Tazmini G, Anthony KV, Omeis SL, de Jong DR, Kay RJ. Regulation of RasGRP1 by B cell antigen receptor requires cooperativity between three domains controlling translocation to the plasma membrane. Molecular biology of the cell. 2007;18:3156–3168. doi: 10.1091/mbc.E06-10-0932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coughlin JJ, Stang SL, Dower NA, Stone JC. RasGRP1 and RasGRP3 regulate B cell proliferation by facilitating B cell receptor-Ras signaling. Journal of immunology. 2005;175:7179–7184. doi: 10.4049/jimmunol.175.11.7179. [DOI] [PubMed] [Google Scholar]
- 19.Dower NA, Stang SL, Bottorff DA, Ebinu JO, Dickie P, Ostergaard HL, Stone JC. RasGRP is essential for mouse thymocyte differentiation and TCR signaling. Nature immunology. 2000;1:317–321. doi: 10.1038/79766. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y, Zhu M, Nishida K, Hirano T, Zhang W. An essential role for RasGRP1 in mast cell function and IgE-mediated allergic response. The Journal of experimental medicine. 2007;204:93–103. doi: 10.1084/jem.20061598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen S, Chen Y, Gorentla BK, Lu J, Stone JC, Zhong XP. Critical roles of RasGRP1 for invariant NKT cell development. J Immunol. 2011;187:4467–4473. doi: 10.4049/jimmunol.1003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Priatel JJ, Teh SJ, Dower NA, Stone JC, Teh HS. RasGRP1 transduces low-grade TCR signals which are critical for T cell development, homeostasis, and differentiation. Immunity. 2002;17:617–627. doi: 10.1016/s1074-7613(02)00451-x. [DOI] [PubMed] [Google Scholar]
- 23.Mercolino TJ, Arnold LW, Hawkins LA, Haughton G. Normal mouse peritoneum contains a large population of Ly-1+ (CD5) B cells that recognize phosphatidyl choline. Relationship to cells that secrete hemolytic antibody specific for autologous erythrocytes. The Journal of experimental medicine. 1988;168:687–698. doi: 10.1084/jem.168.2.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berland R, Wortis HH. Normal B-1a cell development requires B cell-intrinsic NFATc1 activity. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:13459–13464. doi: 10.1073/pnas.2233620100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhattacharyya S, Deb J, Patra AK, Thuy Pham DA, Chen W, Vaeth M, Berberich-Siebelt F, Klein-Hessling S, Lamperti ED, Reifenberg K, Jellusova J, Schweizer A, Nitschke L, Leich E, Rosenwald A, Brunner C, Engelmann S, Bommhardt U, Avots A, Muller MR, Kondo E, Serfling E. NFATc1 affects mouse splenic B cell function by controlling the calcineurin–NFAT signaling network. The Journal of experimental medicine. 2011;208:823–839. doi: 10.1084/jem.20100945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murphy TP, Kolber DL, Rothstein TL. Elevated expression of Pgp-1 (Ly-24) by murine peritoneal B lymphocytes. European journal of immunology. 1990;20:1137–1142. doi: 10.1002/eji.1830200529. [DOI] [PubMed] [Google Scholar]
- 27.Tumang JR, Hastings WD, Bai C, Rothstein TL. Peritoneal and splenic B-1 cells are separable by phenotypic, functional, and transcriptomic characteristics. European journal of immunology. 2004;34:2158–2167. doi: 10.1002/eji.200424819. [DOI] [PubMed] [Google Scholar]
- 28.Yasuda T, Sanjo H, Pages G, Kawano Y, Karasuyama H, Pouyssegur J, Ogata M, Kurosaki T. Erk kinases link pre-B cell receptor signaling to transcriptional events required for early B cell expansion. Immunity. 2008;28:499–508. doi: 10.1016/j.immuni.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 29.Tung JW, Mrazek MD, Yang Y, Herzenberg LA, Herzenberg LA. Phenotypically distinct B cell development pathways map to the three B cell lineages in the mouse. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:6293–6298. doi: 10.1073/pnas.0511305103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gronwall C, Vas J, Silverman GJ. Protective Roles of Natural IgM Antibodies. Frontiers in immunology. 2012;3:66. doi: 10.3389/fimmu.2012.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baumgarth N. The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nature reviews Immunology. 2011;11:34–46. doi: 10.1038/nri2901. [DOI] [PubMed] [Google Scholar]
- 32.Stone JC. Regulation of Ras in lymphocytes: get a GRP. Biochemical Society transactions. 2006;34:858–861. doi: 10.1042/BST0340858. [DOI] [PubMed] [Google Scholar]
- 33.Niiro H, Clark EA. Regulation of B-cell fate by antigen-receptor signals. Nature reviews Immunology. 2002;2:945–956. doi: 10.1038/nri955. [DOI] [PubMed] [Google Scholar]
- 34.Hartley SB, Crosbie J, Brink R, Kantor AB, Basten A, Goodnow CC. Elimination from peripheral lymphoid tissues of self-reactive B lymphocytes recognizing membrane-bound antigens. Nature. 1991;353:765–769. doi: 10.1038/353765a0. [DOI] [PubMed] [Google Scholar]
- 35.Hayakawa K, Asano M, Shinton SA, Gui M, Allman D, Stewart CL, Silver J, Hardy RR. Positive selection of natural autoreactive B cells. Science. 1999;285:113–116. doi: 10.1126/science.285.5424.113. [DOI] [PubMed] [Google Scholar]
- 36.Gong Q, Cheng AM, Akk AM, Alberola-Ila J, Gong G, Pawson T, Chan AC. Disruption of T cell signaling networks and development by Grb2 haploid insufficiency. Nature immunology. 2001;2:29–36. doi: 10.1038/83134. [DOI] [PubMed] [Google Scholar]
- 37.Pages G, Guerin S, Grall D, Bonino F, Smith A, Anjuere F, Auberger P, Pouyssegur J. Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science. 1999;286:1374–1377. doi: 10.1126/science.286.5443.1374. [DOI] [PubMed] [Google Scholar]
- 38.Alberola-Ila J, Forbush KA, Seger R, Krebs EG, Perlmutter RM. Selective requirement for MAP kinase activation in thymocyte differentiation. Nature. 1995;373:620–623. doi: 10.1038/373620a0. [DOI] [PubMed] [Google Scholar]
- 39.Sugawara T, Moriguchi T, Nishida E, Takahama Y. Differential roles of ERK and p38 MAP kinase pathways in positive and negative selection of T lymphocytes. Immunity. 1998;9:565–574. doi: 10.1016/s1074-7613(00)80639-1. [DOI] [PubMed] [Google Scholar]
- 40.Dong C, Yang DD, Wysk M, Whitmarsh AJ, Davis RJ, Flavell RA. Defective T cell differentiation in the absence of Jnk1. Science. 1998;282:2092–2095. doi: 10.1126/science.282.5396.2092. [DOI] [PubMed] [Google Scholar]
- 41.Fuller DM, Zhu M, Song X, Ou-Yang CW, Sullivan SA, Stone JC, Zhang W. Regulation of RasGRP1 function in T cell development and activation by its unique tail domain. PloS one. 2012;7:e38796. doi: 10.1371/journal.pone.0038796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Daley SR, Coakley KM, Hu DY, Randall KL, Jenne CN, Limnander A, Myers DR, Polakos NK, Enders A, Roots C, Balakishnan B, Miosge LA, Sjollema G, Bertram EM, Field MA, Shao Y, Andrews TD, Whittle B, Barnes SW, Walker JR, Cyster JG, Goodnow CC, Roose JP. Rasgrp1 mutation increases naive T-cell CD44 expression and drives mTOR-dependent accumulation of Helios+ T cells and autoantibodies. eLife. 2013;2:e01020. doi: 10.7554/eLife.01020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yasuda S, Stevens RL, Terada T, Takeda M, Hashimoto T, Fukae J, Horita T, Kataoka H, Atsumi T, Koike T. Defective expression of Ras guanyl nucleotide-releasing protein 1 in a subset of patients with systemic lupus erythematosus. Journal of immunology. 2007;179:4890–4900. doi: 10.4049/jimmunol.179.7.4890. [DOI] [PubMed] [Google Scholar]
- 44.Rapoport MJ, Bloch O, Amit-Vasina M, Yona E, Molad Y. Constitutive abnormal expression of RasGRP-1 isoforms and low expression of PARP-1 in patients with systemic lupus erythematosus. Lupus. 2011;20:1501–1509. doi: 10.1177/0961203311418790. [DOI] [PubMed] [Google Scholar]
- 45.Priatel JJ, Chen X, Zenewicz LA, Shen H, Harder KW, Horwitz MS, Teh HS. Chronic immunodeficiency in mice lacking RasGRP1 results in CD4 T cell immune activation and exhaustion. Journal of immunology. 2007;179:2143–2152. doi: 10.4049/jimmunol.179.4.2143. [DOI] [PubMed] [Google Scholar]
- 46.Krupica T, Jr, Fry TJ, Mackall CL. Autoimmunity during lymphopenia: a two-hit model. Clinical immunology. 2006;120:121–128. doi: 10.1016/j.clim.2006.04.569. [DOI] [PubMed] [Google Scholar]
- 47.Bartlett A, Buhlmann JE, Stone J, Lim B, Barrington RA. Multiple checkpoint breach of B cell tolerance in Rasgrp1-deficient mice. Journal of immunology. 2013;191:3605–3613. doi: 10.4049/jimmunol.1202892. [DOI] [PubMed] [Google Scholar]